一种地黄饮子药物组合物、制备方法、检测方法与流程
本发明涉及药学领域,具体涉及一种地黄饮子药物组合物、制备方法、检测方法。
背景技术:
地黄饮子为国家中医药管理局2018年4月发布的《古代经典名方目录》(第一批)中的第53首处方。地黄饮子出自金·刘完素《黄帝素问宣明论方》。处方原文为:熟干地黄、巴戟(去心)、山茱萸、石斛、肉苁蓉(酒浸,焙)、附子(炮)、五味子、官桂、白茯苓、麦门冬(去心)、菖蒲、远志(去心)各等分。右为末,每服三钱,水一盏半,生姜五片、枣一枚、薄荷,同煎至八分,不计时候。原文中“一盏”考证为200~300ml,“一钱”合今3.75g使用,因此原文剂量换算成现代剂量为:熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志各1g,生姜5g(用时切厚片),大枣(用时破开)4.6g,薄荷1g。根据考证结果,“煎至八分”为液面高度的“八分”,故该方的煎煮方法为:加水300~450ml,煎煮至八分(液面高度)。该方具有滋肾阴,补肾阳,开窍化痰的功能,用于治疗下元虚衰,痰浊上泛之喑痱证,舌强不能言,足废不能用,口干不欲饮,足冷面赤,脉沉细弱。
地黄饮子具有一定的疗效,但存在以下缺陷:①由于处方原文中煎煮终点描述为“煎至八分”,根据考证结果为液面高度的“八分”,但是以提取液高度作为煎煮终点时,在试验过程中需要实时量取提取液高度,这样需要较长时间离开火源操作,导致煎煮不连续,在固定加热方式(明火)和控制火力条件下,利用提取液高度变化与煎煮时间之间的相关性,以达到煎煮终点提取液“八分”高度的时间节点作为煎煮时间。②由于地黄饮子为煮散剂,原文去滓服用,因此存在药渣和泥沙的情况。③未明确浓缩投料量与浓缩液质量比,导致干粉状态出现发泡不融化,粉末不疏松,④未明确浓缩温度,浓缩过程出现挂壁,莫诺苷和马钱苷提取总量、五味子醇甲提取总量受影响,浓缩效率低。⑤采取减压干燥,干膏粉状态呈颗粒状,较硬,有效成分莫诺苷和马钱苷以及五味子醇甲均有损失。⑥只明确了熟地黄要粉碎,但未明确粉碎成粗粉还是细粉,因此导致前处理时出现难以操作的现象。⑦现有的质量检测方法难以反映物质基准质量的优劣。
本发明团队为了解决上述问题了,将地黄饮子进行了物质基准工艺与质量检测研究。方中君药山萸肉中环烯醚萜苷类成分(莫诺苷、马钱苷)为其主要活性成分,臣药五味子中木脂素类成分(五味子醇甲)与其有效性相关,是其主要药效物质基础,可作为地黄饮子的指标成分;故本方以君药山萸肉中莫诺苷和马钱苷及臣药五味子中五味子醇甲为含量测定指标,考察处方工艺及质量的可控性。根据经典名方目录中记载的方法,结合卫生部、国家中医药管理局《医疗机构中药煎药室管理规范》(国中医药发〔2009〕3号)进行煎煮。采用砂锅煎煮,提取液经浓缩后冷冻干燥制备干膏,以出膏率、莫诺苷和马钱苷提取总量、五味子醇甲提取总量、指纹图谱为指标,对物质基准对应实物制备工艺中的煎煮时间、滤过、浓缩及干燥等相关参数进行研究,从而确定地黄饮子物质基准工艺。根据常规研究对质量标准的控制要求,并结合本品的制备工艺,建立了本品的常规的质量标准控制项:包括名称、处方、制法、性状、鉴别、双酯型生物碱限量、浸出物、指纹图谱、含量等。同时为了保证本品储存稳定,对对应实物(干膏粉)的水分纳入标准。为了更好的进行全成分的质量控制,建立了本品指纹图谱,以保证本品制备工艺的稳定,为后续复方制剂开发提供基础。
技术实现要素:
本发明的目的是提供一种地黄饮子药物组合物。
本发明的目的是提供一种地黄饮子药物组合物的制备方法。
本发明的目的是提供一种地黄饮子药物组合物的检测方法。
本发明的目的是提供一种地黄饮子药物组合物在滋肾阴,补肾阳,开窍化痰方面的应用。
本发明所述的一种地黄饮子药物组合物由以下配方组成:熟地黄0.5~1.5份、巴戟肉0.5~1.5份、山萸肉0.5~1.5份、石斛0.5~1.5份、酒苁蓉0.5~1.5份、炮附片0.5~1.5份、五味子0.5~1.5份、肉桂0.5~1.5份、茯苓0.5~1.5份、麦冬0.5~1.5份、石菖蒲0.5~1.5份、远志0.5~1.5份、生姜4~6份、大枣3~6份、薄荷0.5~1.5份。
优选的,本发明所述一种地黄饮子药物组合物由以下配方组成:熟地黄1份、巴戟肉1份、山萸肉1份、石斛1份、酒苁蓉1份、炮附片1份、五味子1份、肉桂1份、茯苓1份、麦冬1份、石菖蒲1份、远志1份、生姜5份、大枣4.6份、薄荷1份。
本发明所述地黄饮子药物组合物制备方法包括以下步骤:
(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志分别粉碎成粗粉,备用;生姜用时切2mm~4mm的厚片,大枣用时破开;备用;
(2)煎煮将前处理过后的药物、薄荷,置于600ml砂锅中,加水200~400ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮15~25分钟,得煎煮液;
(3)滤过煎煮液用200~300目滤布趁热常压滤过,得滤液;
(4)浓缩滤液在温度60℃~70℃、压力-0.08~-0.1mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:3g~1g:4g;
(5)冷冻干燥将浓缩液冷冻干燥,冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃,时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉加入药学上可接受的辅料制成药学上可接受的剂型。
优选的,本发明所述地黄饮子药物组合物制备方法包括以下步骤:
(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粉碎成粗粉,生姜用时切2mm~4mm的厚片,大枣用时破开;
(2)煎煮将前处理后的药物、薄荷,置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮20分钟,得煎煮液;
(3)滤过煎煮液用300目滤布趁热常压滤过;得滤液;
(4)浓缩滤液在温度70℃、压力-0.08~-0.1mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:4g;
(5)冷冻干燥冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃、时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉加入药学上可接受的辅料制成药学上可接受的剂型。
本发明所述药物组合物还包括用药学上可接受的辅料制成药学上可接受的剂型。
本发明所述剂型为颗粒剂、胶囊剂、片剂、丸剂、散剂、注射剂、泡腾片、膏剂等。
本发明所述的药物组合物的检测方法包括定性鉴别、指纹图谱、含量测定。
本发明所述定性鉴别、指纹图谱、含量测定具体检测方法为:
【鉴别】(1)取本品4g,加水40~60ml,超声使溶解,用二氯甲烷振摇提取1~3次,每次20~40ml,用正丁醇振摇提取1~3次,每次20~40ml,合并正丁醇液,用氨溶液(1→10)洗涤1~3次,每次20~30ml,弃去氨溶液,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液,另取山茱萸对照药材1g,加水50ml,煎煮15~25分钟,滤过,滤液用二氯甲烷振摇提取2次,每次30ml,弃去二氯甲烷液,水液自“用正丁醇振摇提取2次”起,同法制成对照药材溶液,再取莫诺苷对照品、马钱苷对照品,加80%甲醇制成每1ml各含1mg的混合溶液,作为对照品溶液,照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取供试品溶液5μl、对照药材溶液1μl、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以三氯甲烷-甲醇(4:1~3:1)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视,供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点;
(2)取本品1g,加70%甲醇25~35ml,加热回流0.5~1.5小时,滤过,滤液蒸干,残渣用0.1mol/l的氢氧化钠溶液15~25ml使溶解,用乙酸乙酯振摇提取1~3次,每次15~25ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液,另取或者15vb对照药材1g,加水50ml,煎煮20分钟,滤过,自“用乙酸乙酯振摇提取2次”起,同法制成对照药材溶液,再取五味子醇甲对照品,加80%甲醇制成每1ml含1mg的溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015年版通则)0502试验,吸取供试品溶液5μl、对照药材溶液1μl、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以石油醚(60~90℃)-乙酸乙酯-甲醇(5:3:0.2~15:5:2)为展开剂,展开,取出,晾干,置254nm紫外光灯下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;
【指纹图谱】照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.3%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长为254nm;理论板数按五味子醇甲计算应不低于3000;
参照物溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含0.15mg的溶液,即得;
供试品溶液的制备取本品2.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20~30ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,滤液用乙酸乙酯-正丁醇(1:1)混合溶液振摇提取2次,每次25ml,合并乙酸乙酯-正丁醇(1:1)混合溶液,蒸干,残渣加甲醇使溶解,并转移至5ml量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取参照物溶液与供试品溶液各5μl,注入液相色谱仪,测定,记录色谱图,即得;
供试品指纹图谱中应呈现与参照物色谱峰保留时间相同的色谱峰,供试品色谱图应与对照指纹图谱基本一致,有相对应的14个共有峰,按中药色谱指纹图谱相似度评价系统,以共有峰计算相似度,供试品指纹图谱与对照指纹图谱的相似度不得低于0.90。对照指纹图谱见图23。
【含量测定】山萸肉照高效液相色谱法(《中国药典》2015年版四部通则0512测定;
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.3%磷酸溶液为流动相b,按规定的流动相梯度洗脱程序进行梯度洗脱;检测波长为240nm;理论板数按莫诺苷峰计算应不低于3000;流动相梯度洗脱程序为:
对照品溶液的制备取莫诺苷、马钱苷对照品适量,精密称定,加80%甲醇制成每1ml含莫诺苷40μg、马钱苷20μg的混合溶液,即得;
供试品溶液的制备取本品1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20~30ml,密塞,称定重量,用功率250w、频率40khz超声处理30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得;
本品按干燥品计算,每1g含山萸肉以莫诺苷(c17h26o11)和马钱苷(c17h26o10)总量计,应为1.5mg~3.0mg;
五味子照高效液相色谱法(《中国药典》2015年版四部通则0502)测定;
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(45:55)为流动相;检测波长为250nm,理论板数按五味子醇甲峰计算应不低于3000;
对照品溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含20μg的溶液,即得;
供试品溶液的制备取本品1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20~30ml,密塞,称定重量,超声处理(功率250w,频率40khz)25~35分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得;
本品按干燥品计算,每1g含五味子以五味子醇甲(c24h32o7)计,应为0.4mg~0.8mg。
优选的,本发明所述定性鉴别、指纹图谱、含量测定具体检测方法为:
【鉴别】(1)取本品4g,加水50ml,超声使溶解,用二氯甲烷振摇提取2次,每次30ml,用正丁醇振摇提取2次,每次30ml,合并正丁醇液,用氨溶液(1→10)洗涤2次,每次25ml,弃去氨溶液,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液,另取山茱萸对照药材1g,加水50ml,煎煮20分钟,滤过,滤液用二氯甲烷振摇提取2次,每次30ml,弃去二氯甲烷液,水液自“用正丁醇振摇提取2次”起,同法制成对照药材溶液,再取莫诺苷对照品、马钱苷对照品,加80%甲醇制成每1ml各含1mg的混合溶液,作为对照品溶液,照中国药典2015年版四部通则0502薄层色谱法试验,吸取供试品溶液5μl、对照药材溶液1μl、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以三氯甲烷-甲醇(3:1)为展开剂,展开,取出,晾干,置254nm紫外光灯下检视,供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点;
(2)取本品1g,加70%甲醇30ml,加热回流1小时,滤过,滤液蒸干,残渣用0.1mol/l的氢氧化钠溶液20ml使溶解,用乙酸乙酯振摇提取2次,每次20ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液,另取五味子对照药材1g,加水50ml,煎煮20分钟,滤过,自“用乙酸乙酯振摇提取2次”起,同法制成对照药材溶液,再取五味子醇甲对照品,加80%甲醇制成每1ml含1mg的溶液,作为对照品溶液;照中国药典2015年版通则0502薄层色谱法试验,吸取供试品溶液5μl、对照药材溶液1μl、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以石油醚(60~90℃)-乙酸乙酯-甲醇(5:3:0.2)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;
【指纹图谱】照高效液相色谱法中国药典2015年版四部通则0512测定;
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.3%磷酸溶液为流动相b,按下表中的规定的程序进行梯度洗脱;检测波长为254nm;
理论板数按五味子醇甲计算应不低于3000;
参照物溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含0.15mg的溶液,即得;
供试品溶液的制备取本品2.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,用超声(功率250w,频率40khz)处理30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,滤液用乙酸乙酯-正丁醇(1:1)混合溶液振摇提取2次,每次25ml,合并乙酸乙酯-正丁醇(1:1)混合溶液,蒸干,残渣加甲醇使溶解,并转移至5ml量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取参照物溶液与供试品溶液各5μl,注入液相色谱仪,测定,记录色谱图,即得;
供试品指纹图谱中应呈现与参照物色谱峰保留时间相同的色谱峰,供试品色谱图应与对照指纹图谱基本一致,有相对应的14个共有峰,按中药色谱指纹图谱相似度评价系统,以共有峰计算相似度,供试品指纹图谱与对照指纹图谱的相似度不得低于0.90;对照指纹图谱见图23。
【含量测定】山萸肉照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.3%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长为240nm;理论板数按莫诺苷峰计算应不低于3000;
对照品溶液的制备取莫诺苷、马钱苷对照品适量,精密称定,加80%甲醇制成每1ml含莫诺苷40μg、马钱苷20μg的混合溶液,即得;
供试品溶液的制备取本品1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,超声(功率250w、频率40khz)处理30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得;
本品按干燥品计算,每1g含山萸肉以莫诺苷(c17h26o11)和马钱苷(c17h26o10)总量计,应为1.5mg~3.0mg;
五味子照高效液相色谱法(《中国药典》2015年版四部通则0502)测定;
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(45:55)为流动相;检测波长为250nm,理论板数按五味子醇甲峰计算应不低于3000;
对照品溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含20μg的溶液,即得;
供试品溶液的制备取本品1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,用功率250w,频率40khz超声处理30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得;
本品按干燥品计算,每1g含五味子以五味子醇甲(c24h32o7)计,应为0.4mg~0.8mg。
所述重量份数可以是μg、mg、g、kg等医药领域公知的重量单位。
本发明所述药学上可接受的辅料是指药学领域常规的药物载体,选自填充剂、润滑剂、粘合剂、崩解剂、表面活性剂或矫味剂等中的一种或几种。
其中所述填充剂选自糊精、淀粉、山梨醇、乳糖、甘露醇、木糖醇、微晶纤维素等;
所述粘合剂选自纤维素衍生物、醋酸乙烯树脂、聚乙烯醇、明胶、藻酸盐等;
所述崩解剂选自干淀粉、碳酸轻纳、低取代纤维素、微晶纤维素、羧甲基淀粉钠、聚乙烯吡咯烷酮、低取代羟丙基纤维素或交联羧甲基纤维素钠等;
所述润滑剂选自硬脂酸、聚乙二醇、碳酸钙、碳酸氢钠、二氧化硅、滑石粉或硬脂酸镁等;
所述表面活性剂选自十二烷基苯磺酸钠、硬脂酸、聚氧乙烯-聚氧丙烯共聚物、脂肪酸山梨坦或聚山梨酯(吐温)等;
所述矫味剂选自甘油、山梨醇、甘露醇、阿斯巴甜、蔗糖素、柠檬、茴香、薄荷油等。
本发明所述的剂型为颗粒剂、胶囊剂、片剂、丸剂、散剂、注射剂、泡腾片、膏剂。
本发所述药物组合物在滋肾阴,补肾阳,开窍化痰方面的应用。具体用于治疗下元虚衰,痰浊上泛之喑痱证,舌强不能言,足废不能用,口干不欲饮,足冷面赤,脉沉细弱。
有益效果:
1、本发明与现有技术相比,具有以下有益效果:
现有技术国家中医药管理局2018年4月发布的《古代经典名方目录》(第一批)中的第53首处方地黄饮子,存在以下缺陷:①由于处方原文中煎煮终点描述为“煎至八分”,根据考证结果为液面高度的“八分”,但是以提取液高度作为煎煮终点时,在试验过程中需要实时量取提取液高度,这样需要较长时间离开火源操作,导致煎煮不连续。②由于地黄饮子为煮散剂,原文去滓服用,因此存在药渣和泥沙的情况。③未明确浓缩投料量与浓缩液质量比,导致干粉状态出现发泡不融化,粉末不疏松,④未明确浓缩温度,浓缩过程出现挂壁,莫诺苷和马钱苷提取总量、五味子醇甲提取总量受影响,浓缩效率低。⑤采取减压干燥,干膏粉状态呈颗粒状,较硬,有效成分莫诺苷和马钱苷以及五味子醇甲均有损失。⑥只明确了熟地黄要粉碎,但未明确粉碎成粗粉还是细粉,因此导致前处理时出现难以操作的现象。⑦现有的质量检测方法难以反映物质基准质量的优劣。
本发明克服了上述缺陷,解决了相应的技术问题,具体为:
①本发明在固定加热方式(明火)和控制火力条件下,利用提取液高度变化与煎煮时间之间的相关性,达到煎煮终点提取液“八分”高度的时间节点作为煎煮时间,明确了优选煎煮时间为20分钟;解决了现有技术处方原文中以提取液高度作为煎煮终点时,在试验过程中需要实时量取提取液高度,需要较长时间离开火源操作,导致煎煮不连续的技术问题;
②本发明明确了煎煮液优选用300目滤布趁热常压滤过,解决了现有技术为煮散剂,去滓服用,导致存在药渣和泥沙的技术问题;
③本发明明确了饮片投料量与浓缩液质量比优选为1g:4g,干粉状态融化未发泡,粉末疏松,解决了现有技术对比文件未明确浓缩投料量与浓缩液质量比,导致干粉状态出现发泡不融化,粉末不疏松的技术问题;
④本发明明确了优选浓缩条件为:温度70℃、压力-0.08~-0.1mpa条件下减压浓缩,浓缩过程未挂壁,莫诺苷和马钱苷提取总量、五味子醇甲提取总量未受影响,浓缩效率高;解决了现有技术未明确浓缩温度,浓缩过程出现挂壁,莫诺苷和马钱苷提取总量、五味子醇甲提取总量受影响,浓缩效率低的技术问题;
⑤本发明采取冷冻干燥并明确冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃、时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;冷冻干燥与干燥前比较,莫诺苷和马钱苷总量差别不大,五味子醇甲轻微损失,干膏粉状态疏松;解决了现有技术减压干燥(60℃)与干燥前相比,莫诺苷和马钱苷以及五味子醇甲均有损失,干膏粉状态呈颗粒状,较硬的技术问题。
⑥本发明明确了熟地黄粉碎成粗粉,解决了现有技术未明确粉碎成粗粉还是细粉,导致前处理时出现难以操作现象的技术问题。
⑦本发明确定了定性鉴别、指纹图谱、含量测定具体检测方法,良好的检测方法有效的反映物质基准质量的优劣,解决了现有技术质量检测方法难以反映物质基准质量优劣的技术问题。
2、对本发明建立的薄层色谱方法鉴别,通过对不同厂家的硅胶g薄层板以及不同展开温度、不同的相对湿度等影响因素的试验,结果耐用性好,主斑点清晰。
3、本发明建立的液相鉴别方法通过对不同柱温、不同流速、不同色谱柱进行耐用性考察,结果表明,不同条件下色谱图中松果菊苷峰型尖锐、对称,分离度良好,同时阴性无干扰,表明本方法对不同流速、不同柱温、不同色谱柱耐用性良好。
4、本发明建立了以指纹图谱为主的整体质量测定方法,克服了单纯的指标成分的定性鉴别和定量分析,难以反映物质基准质量的优劣的技术问题。
5、本发明建立的指纹图谱经方法学考察显示,专属性、整体性良、仪器精密度、重复性良好;不同流速、不同柱温、不同色谱柱、不同酸比例、不同仪器条件下测定结果基本一致,色谱图中各共有峰峰型尖锐,对称,分离度良好,相似度均不低于0.90,供试品指纹图谱中均呈现14个主要色谱峰,且与对照品参照物色谱峰中的特征峰相对应,表明该方法对不同流速、不同柱温、不同色谱柱、不同酸度、不同仪器耐用性良好;溶液在48小时内稳定性良好。
6、本发明建立的高效液相色谱法具有专属性,峰纯度检查显示莫诺苷和马钱苷色谱峰纯度符合要求;经过线性关系的考察表明马钱苷在0.02036μg~2.0360μg范围内线性良好;经过仪器精密度试验仪器精密度良好;重复性良好;供试品溶液在48小时内稳定性良好,准确度较好。
附图说明
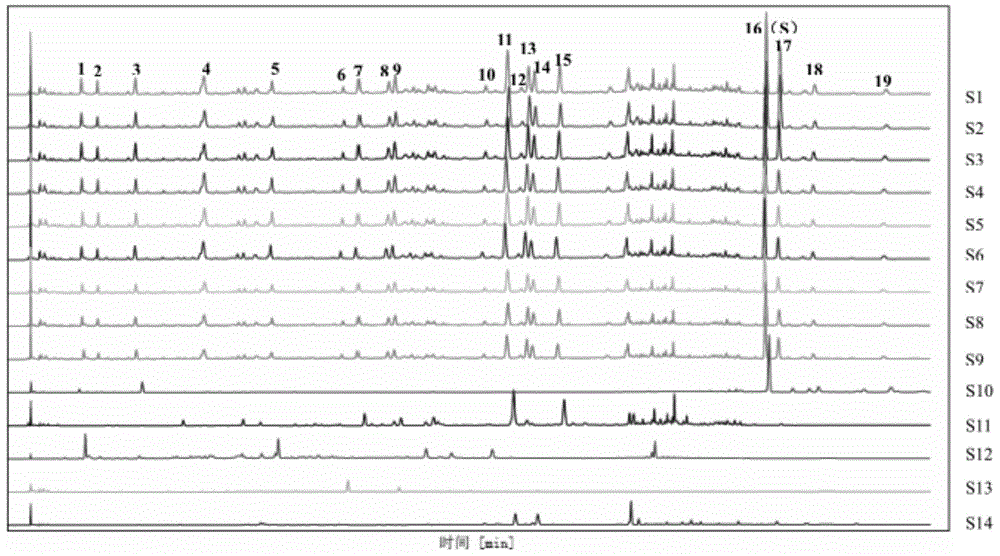
图1地黄饮子物质基准工艺验证指纹图谱(s1.提取液1、s2.提取液2、s3.提取液3、s4.浓缩液1、s5.浓缩液2、s6.浓缩液3、s7.干膏粉1、s8.干膏粉2、s9.干膏粉3、s10.五味子饮片、s11.远志饮片、s12.山萸肉饮片、s13.酒苁蓉饮片、s14.薄荷饮片)
图2山萸肉薄层鉴别图[硅胶gf254板(100*100mm)rt:23.5℃rh:37%;1、马钱苷对照品溶液(批号:11640-201707,中国食品药品检定研究院,5μl)、2、莫诺苷对照品溶液(批号:111998-201702,中国食品药品检定研究院,2μl)、3、山茱萸对照药材溶液(批号:121495-201303,中国食品药品检定研究院,2μl)、4、供试品溶液(d-181120-02,2μl)
5、供试品溶液(d-181120-02,5μl)、6、供试品溶液(d-181120-02,10μl)、7、缺山萸肉阴性样品溶液(d-181126-y-szy-02,10μl)]
图3五味子薄层鉴别摸索[青岛硅胶gf254板(100*100mm)rt:23.4℃rh:39%;1.五味子醇甲对照品溶液(批号:110857-201714,中国食品药品检定研究院,2μl)、2.五味子对照药材溶液(批号:120922-201610,中国食品药品检定研究院,1μl)、3~4.供试品溶液(d-181120-01,5μl)、5~6.缺五味子阴性样品溶液(d-181126-y-wwz-01,5μl)]
图4指纹图谱摸索
图5供试品制备方法考察[1、80%甲醇;2、乙酸乙酯振摇提取;3、乙酸乙酯-正丁醇(1:1)振摇提取]
图6药味归属hplc对比图谱
图7相关成分确认hplc对比图谱
图8地黄饮子物质基准供试液的uplc-uv色谱图和uplc-ms总离子流图
图9供试品、对照品、空白溶剂hplc对比图谱
图10仪器精密度试验hplc指纹图谱
图11稳定性试验试验hplc指纹图谱
图12重复性试验hplc指纹图谱
图13不同流速对比图
图14不同柱温耐用性对比图
图15不同酸比例耐用性对比图
图16不同仪器耐用性对比图
图17地黄饮子物质基准指纹图共有模式图-1[r.对照品图谱、s2.d-181212-01、s3.d-181212-03、s4.d-181212-05、s5.d-181212-07、s6.d-181213-09、s7.d-181213-11、s8.d-181213-13、s9.d-181213-15、s10.d-181214-17、s11.d-181214-19、s12.d-181214-21、s13.d-181214-23、s14.d-181215-25、s15.d-181215-27、s16.d-181215-29]
图18地黄饮子物质基准指纹图共有模式图-2[r.对照图谱、s2.d-181212-03、s3.d-181212-04、s4.d-181212-06、s5.d-181212-08、s6.d-181213-10、s7.d-181213-12、s8.d-181213-14、s9.d-181213-16、s10.d-181214-18、s11.d-181214-20、s12.d-181214-22、s13.d-181214-24、s14.d-181215-26、s15.d-181215-28、s16.d-181215-30]
图19对照指纹图谱[14个共有峰中峰1:没食子酸、峰4:马钱苷、峰5:松果菊苷、峰7:3,6’-二芥子酰基蔗糖、峰9:迷迭香酸、峰11(s):五味子醇甲]
图20莫诺苷和马钱苷含量测定摸索
图21对照品、供试品、阴性样品、空白溶剂hplc对比图
图22五味子醇甲全波长扫描图
图23对照指纹图谱(对应实物关键质量属性范围的确定)、[14个共有峰中峰1:没食子酸、峰4:马钱苷、峰5:松果菊苷、峰7:3,6’-二芥子酰基蔗糖、峰9:迷迭香酸、峰11(s):五味子醇甲
具体实施方式
下面通过具体实施例,对本发明的技术方案作进一步地具体说明。
实施例1处方:熟地黄0.5g、巴戟肉0.5g、山萸肉0.5g、石斛0.5g、酒苁蓉0.5g、炮附片0.5g、五味子0.5g、肉桂0.5g、茯苓0.5g、麦冬0.5g、石菖蒲0.5g、远志0.5g、生姜4g、大枣3g、薄荷0.5g。
制备方法:(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志分别粉碎成粗粉,备用;生姜用时切2mm~4mm的厚片,大枣用时破开;备用;
(2)煎煮将前处理过后的药物、薄荷,置于600ml砂锅中,加水200ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮15分钟,得煎煮液;
(3)滤过煎煮液用200目滤布趁热常压滤过;
(4)浓缩滤液在温度60℃、压力-0.08mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:3g;
(5)冷冻干燥将浓缩液冷冻干燥,冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃,时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉加入1/10的淀粉,混匀,制粒,得颗粒剂。
实施例2处方:熟地黄0.5g、巴戟肉0.5g、山萸肉0.5g、石斛0.5g、酒苁蓉0.5g、炮附片0.5g、五味子0.5g、肉桂0.5g、茯苓0.5g、麦冬0.5g、石菖蒲0.5g、远志0.5g、生姜4g、大枣3g、薄荷0.5g。
制备方法:(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志分别粉碎成粗粉,备用;生姜用时切2mm~4mm的厚片,大枣用时破开;备用;
(2)煎煮将前处理过后的药物、薄荷,置于600ml砂锅中,加水400ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮25分钟,得煎煮液;
(3)滤过煎煮液用300目滤布趁热常压滤过;得滤液;
(4)浓缩滤液在温度70℃、压力-0.08~-0.1mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:4g;
(5)冷冻干燥将浓缩液冷冻干燥,冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃,时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉加入1/12的淀粉,混匀,粉碎成细粉,装入胶囊,得胶囊剂。
实施例3处方:熟地黄0.5g、巴戟肉0.5g、山萸肉0.5g、石斛0.5g、酒苁蓉0.5g、炮附片0.5g、五味子0.5g、肉桂0.5g、茯苓0.5g、麦冬0.5g、石菖蒲0.5g、远志0.5g、生姜4g、大枣3g、薄荷0.5g。
制备方法:(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粉碎成粗粉,生姜用时切2mm~4mm的厚片,大枣用时破开;
(2)煎煮将前处理后的药物、薄荷,置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮20分钟,得煎煮液;
(3)滤过煎煮液用300目滤布趁热常压滤过;得滤液;
(4)浓缩滤液在温度70℃、压力-0.08~-0.1mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:4g;
(5)冷冻干燥冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃、时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉加入1/12的淀粉,混匀,粉碎成细粉,装入胶囊,得胶囊剂。
实施例4处方:熟地黄1.5g、巴戟肉1.5g、山萸肉1.5g、石斛1.5g、酒苁蓉1.5g、炮附片1.5g、五味子1.5g、肉桂1.5g、茯苓1.5g、麦冬1.5g、石菖蒲1.5g、远志1.5g、生姜6g、大枣6g、薄荷1.5g。
制备方法:(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志分别粉碎成粗粉,备用;生姜用时切2mm~4mm的厚片,大枣用时破开;备用;
(2)煎煮将前处理过后的药物、薄荷,置于600ml砂锅中,加水200ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮15分钟,得煎煮液;
(3)滤过煎煮液用200目滤布趁热常压滤过;
(4)浓缩滤液在温度60℃、压力-0.08mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:3g;
(5)冷冻干燥将浓缩液冷冻干燥,冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃,时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉加入1/10的淀粉,混匀,制粒,得颗粒剂。
实施例5处方:熟地黄1.5g、巴戟肉1.5g、山萸肉1.5g、石斛1.5g、酒苁蓉1.5g、炮附片1.5g、五味子1.5g、肉桂1.5g、茯苓1.5g、麦冬1.5g、石菖蒲1.5g、远志1.5g、生姜6g、大枣6g、薄荷1.5g。
制备方法:(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志分别粉碎成粗粉,备用;生姜用时切2mm~4mm的厚片,大枣用时破开;备用;
(2)煎煮将前处理过后的药物、薄荷,置于600ml砂锅中,加水400ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮25分钟,得煎煮液;
(3)滤过煎煮液用300目滤布趁热常压滤过;得滤液;
(4)浓缩滤液在温度70℃、压力-0.08~-0.1mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:4g;
(5)冷冻干燥将浓缩液冷冻干燥,冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃,时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉加入1/12的淀粉,混匀,粉碎成细粉,装入胶囊,得胶囊剂。
实施例6处方:熟地黄1.5g、巴戟肉1.5g、山萸肉1.5g、石斛1.5g、酒苁蓉1.5g、炮附片1.5g、五味子1.5g、肉桂1.5g、茯苓1.5g、麦冬1.5g、石菖蒲1.5g、远志1.5g、生姜6g、大枣6g、薄荷1.5g。
制备方法:(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粉碎成粗粉,生姜用时切2mm~4mm的厚片,大枣用时破开;
(2)煎煮将前处理后的药物、薄荷,置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮20分钟,得煎煮液;
(3)滤过煎煮液用300目滤布趁热常压滤过;得滤液;
(4)浓缩滤液在温度70℃、压力-0.08~-0.1mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:4g;
(5)冷冻干燥冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃、时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉加入1/12的淀粉,混匀,粉碎成细粉,装入胶囊,得胶囊剂。
实施例7处方:熟地黄1g、巴戟肉1g、山萸肉1g、石斛1g、酒苁蓉1g、炮附片1g、五味子1g、肉桂1g、茯苓1g、麦冬1g、石菖蒲1g、远志1g、生姜5g、大枣4.6g、薄荷1g。
制备方法:(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志分别粉碎成粗粉,备用;生姜用时切2mm~4mm的厚片,大枣用时破开;备用;
(2)煎煮将前处理过后的药物、薄荷,置于600ml砂锅中,加水200ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮15分钟,得煎煮液;
(3)滤过煎煮液用200目滤布趁热常压滤过;
(4)浓缩滤液在温度60℃、压力-0.08mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:3g;
(5)冷冻干燥将浓缩液冷冻干燥,冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃,时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉加入1/10的淀粉,混匀,制粒,得颗粒剂。
实施例8处方:熟地黄1g、巴戟肉1g、山萸肉1g、石斛1g、酒苁蓉1g、炮附片1g、五味子1g、肉桂1g、茯苓1g、麦冬1g、石菖蒲1g、远志1g、生姜5g、大枣4.6g、薄荷1g。
制备方法:(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志分别粉碎成粗粉,备用;生姜用时切2mm~4mm的厚片,大枣用时破开;备用;
(2)煎煮将前处理过后的药物、薄荷,置于600ml砂锅中,加水400ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮25分钟,得煎煮液;
(3)滤过煎煮液用300目滤布趁热常压滤过;得滤液;
(4)浓缩滤液在温度70℃、压力-0.08~-0.1mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:4g;
(5)冷冻干燥将浓缩液冷冻干燥,冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃,时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉加入1/12的淀粉,混匀,粉碎成细粉,装入胶囊,得胶囊剂。
实施例9处方:熟地黄1g、巴戟肉1g、山萸肉1g、石斛1g、酒苁蓉1g、炮附片1g、五味子1g、肉桂1g、茯苓1g、麦冬1g、石菖蒲1g、远志1g、生姜5g、大枣4.6g、薄荷1g。
制备方法:(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粉碎成粗粉,生姜用时切2mm~4mm的厚片,大枣用时破开;
(2)煎煮将前处理后的药物、薄荷,置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮20分钟,得煎煮液;
(3)滤过煎煮液用300目滤布趁热常压滤过;得滤液;
(4)浓缩滤液在温度70℃、压力-0.08~-0.1mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:4g;
(5)冷冻干燥冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃、时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉加入1/12的淀粉,混匀,粉碎成细粉,装入胶囊,得胶囊剂。
实施例10
处方:熟地黄1g、巴戟肉1g、山萸肉1g、石斛1g、酒苁蓉1g、炮附片1g、五味子1g、肉桂1g、茯苓1g、麦冬1g、石菖蒲1g、远志1g、生姜5g、大枣4.6g、薄荷1g。
制备方法
(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粉碎成粗粉,生姜用时切2mm~4mm的厚片,大枣用时破开;
(2)煎煮将前处理后的药物、薄荷,置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮20分钟,得煎煮液;
(3)滤过煎煮液用300目滤布趁热常压滤过;得滤液;
(4)浓缩滤液在温度70℃、压力-0.08~-0.1mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:4g;
(5)冷冻干燥冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃、时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉加入1/7的糊精,混匀,干燥,制成丸剂。
实施例11
处方:熟地黄1g、巴戟肉1g、山萸肉1g、石斛1g、酒苁蓉1g、炮附片1g、五味子1g、肉桂1g、茯苓1g、麦冬1g、石菖蒲1g、远志1g、生姜5g、大枣4.6g、薄荷1g。
制备方法:(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粉碎成粗粉,生姜用时切2mm~4mm的厚片,大枣用时破开;
(2)煎煮将前处理后的药物、薄荷,置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮20分钟,得煎煮液;
(3)滤过煎煮液用300目滤布趁热常压滤过;得滤液;
(4)浓缩滤液在温度70℃、压力-0.08~-0.1mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:4g;
(5)冷冻干燥冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃、时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)加入1/6糊精,压片,干燥,得片剂。
实施例12
处方:熟地黄1g、巴戟肉1g、山萸肉1g、石斛1g、酒苁蓉1g、炮附片1g、五味子1g、肉桂1g、茯苓1g、麦冬1g、石菖蒲1g、远志1g、生姜5g、大枣4.6g、薄荷1g。
制备方法:(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粉碎成粗粉,生姜用时切2mm~4mm的厚片,大枣用时破开;
(2)煎煮将前处理后的药物、薄荷,置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮20分钟,得煎煮液;
(3)滤过煎煮液用300目滤布趁热常压滤过;得滤液;
(4)浓缩滤液在温度70℃、压力-0.08~-0.1mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:4g;
(5)冷冻干燥冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃、时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)加入1/12的碳酸氢钠,制粒,压片,制成泡腾片。
实施例13处方:熟地黄1g、巴戟肉1g、山萸肉1g、石斛1g、酒苁蓉1g、炮附片1g、五味子1g、肉桂1g、茯苓1g、麦冬1g、石菖蒲1g、远志1g、生姜5g、大枣4.6g、薄荷1g。
制备方法:(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粉碎成粗粉,生姜用时切2mm~4mm的厚片,大枣用时破开;
(2)煎煮将前处理后的药物、薄荷,置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮20分钟,得煎煮液;
(3)滤过煎煮液用300目滤布趁热常压滤过;得滤液;
(4)浓缩滤液在温度70℃、压力-0.08~-0.1mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:4g;
(5)冷冻干燥冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃、时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉粉碎,用0.8倍量的乙醇,调匀,得膏剂。
实施例14处方:熟地黄1g、巴戟肉1g、山萸肉1g、石斛1g、酒苁蓉1g、炮附片1g、五味子1g、肉桂1g、茯苓1g、麦冬1g、石菖蒲1g、远志1g、生姜5g、大枣4.6g、薄荷1g。
制备方法:(1)前处理称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粉碎成粗粉,生姜用时切2mm~4mm的厚片,大枣用时破开;
(2)煎煮将前处理后的药物、薄荷,置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮20分钟,得煎煮液;
(3)滤过煎煮液用300目滤布趁热常压滤过;得滤液;
(4)浓缩滤液在温度70℃、压力-0.08~-0.1mpa条件下减压浓缩,至饮片投料量与浓缩液质量比为1g:4g;
(5)冷冻干燥冷冻干燥条件为:预冻温度:-20~-48℃、冷阱温度:-40~-80℃、时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时,得干膏粉;
(6)将干膏粉粉碎,加入6倍量注射用水,过滤,灭菌,得注射剂。
实施例1-14按实施例15-17检测方法中任一检测方法检测
实施例15检测方法
【鉴别】(1)取本品4g,加水40ml,超声使溶解,用二氯甲烷振摇提取1次,每次20ml,用正丁醇振摇提取1次,每次2ml,合并正丁醇液,用氨溶液(1→10)洗涤1次,每次20ml,弃去氨溶液,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液,另取山茱萸对照药材1g,加水50ml,煎煮15分钟,滤过,滤液用二氯甲烷振摇提取2次,每次30ml,弃去二氯甲烷液,水液自“用正丁醇振摇提取2次”起,同法制成对照药材溶液,再取莫诺苷对照品、马钱苷对照品,加80%甲醇制成每1ml各含1mg的混合溶液,作为对照品溶液,照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取供试品溶液5μl、对照药材溶液1μl、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以三氯甲烷-甲醇(4:1~3:1)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视,供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
(2)取本品1g,加70%甲醇25ml,加热回流0.5小时,滤过,滤液蒸干,残渣用0.1mol/l的氢氧化钠溶液15ml使溶解,用乙酸乙酯振摇提取1次,每次15ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液,另取五味子对照药材1g,加水50ml,煎煮20分钟,滤过,自“用乙酸乙酯振摇提取2次”起,同法制成对照药材溶液,再取五味子醇甲对照品,加80%甲醇制成每1ml含1mg的溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015年版通则0502)试验,吸取供试品溶液5μl、对照药材溶液1μl、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以(60~90℃)石油醚-乙酸乙酯-甲醇(5:3:0.2~15:5:2)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点。
【指纹图谱】照高效液相色谱法(《中国药典》2015年版四部通则0512)测定。
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.3%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长为254nm;理论板数按五味子醇甲计算应不低于3000;
参照物溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含0.15mg的溶液,即得。
供试品溶液的制备取本品2.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20~30ml,密塞,称定重量,用超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,滤液用比例为1:1的乙酸乙酯-正丁醇混合溶液振摇提取2次,每次25ml,合并乙酸乙酯-正丁醇(1:1)混合溶液,蒸干,残渣加甲醇使溶解,并转移至5ml量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取参照物溶液与供试品溶液各5μl,注入液相色谱仪,测定,记录色谱图,即得。
供试品指纹图谱中应呈现与参照物色谱峰保留时间相同的色谱峰,供试品色谱图应与对照指纹图谱基本一致,有相对应的14个共有峰,按中药色谱指纹图谱相似度评价系统,以共有峰计算相似度,供试品指纹图谱与对照指纹图谱的相似度不得低于0.90。对照指纹图谱见图23。
【含量测定】山萸肉照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.3%磷酸溶液为流动相b,按规定的梯度洗脱程序进行梯度洗脱;检测波长为240nm;理论板数按莫诺苷峰计算应不低于3000;流动相梯度洗脱程序为:
对照品溶液的制备取莫诺苷、马钱苷对照品适量,精密称定,加80%甲醇制成每1ml含莫诺苷40μg、马钱苷20μg的混合溶液,即得;
供试品溶液的制备取本品1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20~30ml,密塞,称定重量,用超声处理(功率250w、频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得;
本品按干燥品计算,每1g含山萸肉以莫诺苷(c17h26o11)和马钱苷(c17h26o10)总量计,应为1.5mg~3.0mg;
五味子照高效液相色谱法(《中国药典》2015年版四部通则0502)测定;
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(45:55)为流动相;检测波长为250nm,理论板数按五味子醇甲峰计算应不低于3000;
对照品溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含20μg的溶液,即得;
供试品溶液的制备取本品1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20~30ml,密塞,称定重量,用超声处理(功率250w,频率40khz)25分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得;
本品按干燥品计算,每1g含五味子以五味子醇甲(c24h32o7)计,应为0.4mg~0.8mg。
实施例16检测方法
【鉴别】(1)取本品4g,加水60ml,超声使溶解,用二氯甲烷振摇提取3次,每次40ml,用正丁醇振摇提取1~3次,每次20~40ml,合并正丁醇液,用氨溶液(1→10)洗涤3次,每次30ml,弃去氨溶液,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液,另取山茱萸对照药材1g,加水50ml,煎煮25分钟,滤过,滤液用二氯甲烷振摇提取2次,每次30ml,弃去二氯甲烷液,水液自“用正丁醇振摇提取2次”起,同法制成对照药材溶液,再取莫诺苷对照品、马钱苷对照品,加80%甲醇制成每1ml各含1mg的混合溶液,作为对照品溶液,照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取供试品溶液5μl、对照药材溶液1μl、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以三氯甲烷-甲醇(4:1~3:1)为展开剂,展开,取出,晾干,置254nm紫外光灯下检视,供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
(2)取本品1g,加70%甲醇25~35ml,加热回流1.5小时,滤过,滤液蒸干,残渣用0.1mol/l的氢氧化钠溶液15~25ml使溶解,用乙酸乙酯振摇提取3次,每次25ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液,另取五味子对照药材1g,加水50ml,煎煮20分钟,滤过,自“用乙酸乙酯振摇提取2次”起,同法制成对照药材溶液,再取五味子醇甲对照品,加80%甲醇制成每1ml含1mg的溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015年版通则0502)试验,吸取供试品溶液5μl、对照药材溶液1μl、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以(60~90℃)石油醚-乙酸乙酯-甲醇(5:3:0.2~15:5:2)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点。
【指纹图谱】照高效液相色谱法(《中国药典》2015年版四部通则0512)测定。
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.3%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长为254nm;
理论板数按五味子醇甲计算应不低于3000;
参照物溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含0.15mg的溶液,即得。
供试品溶液的制备取本品2.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇30ml,密塞,称定重量,用超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,滤液用比例为1:1的乙酸乙酯-正丁醇混合溶液振摇提取2次,每次25ml,合并乙酸乙酯-正丁醇(1:1)混合溶液,蒸干,残渣加甲醇使溶解,并转移至5ml量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取参照物溶液与供试品溶液各5μl,注入液相色谱仪,测定,记录色谱图,即得。
供试品指纹图谱中应呈现与参照物色谱峰保留时间相同的色谱峰,供试品色谱图应与对照指纹图谱基本一致,有相对应的14个共有峰,按中药色谱指纹图谱相似度评价系统,以共有峰计算相似度,供试品指纹图谱与对照指纹图谱的相似度不得低于0.90。对照指纹图谱见图23。
【含量测定】山萸肉照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.3%磷酸溶液为流动相b,按规定的流动相梯度洗脱程序进行梯度洗脱;检测波长为240nm;理论板数按莫诺苷峰计算应不低于3000;梯度洗脱程序为:
对照品溶液的制备取莫诺苷、马钱苷对照品适量,精密称定,加80%甲醇制成每1ml含莫诺苷40μg、马钱苷20μg的混合溶液,即得;
供试品溶液的制备取本品1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20~30ml,密塞,称定重量,用超声处理(功率250w、频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得;
本品按干燥品计算,每1g含山萸肉以莫诺苷(c17h26o11)和马钱苷(c17h26o10)总量计,应为1.5mg~3.0mg;
五味子照高效液相色谱法(《中国药典》2015年版四部通则0502)测定;
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(45:55)为流动相;检测波长为250nm,理论板数按五味子醇甲峰计算应不低于3000;
对照品溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含20μg的溶液,即得;
供试品溶液的制备取本品1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20~30ml,密塞,称定重量,用超声处理(功率250w,频率40khz)25~35分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得;
本品按干燥品计算,每1g含五味子以五味子醇甲(c24h32o7)计,应为0.4mg~0.8mg。
实施例17检测方法
【鉴别】(1)取本品4g,加水50ml,超声使溶解,用二氯甲烷振摇提取2次,每次30ml,用正丁醇振摇提取2次,每次30ml,合并正丁醇液,用氨溶液(1→10)洗涤2次,每次25ml,弃去氨溶液,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液,另取山茱萸对照药材1g,加水50ml,煎煮20分钟,滤过,滤液用二氯甲烷振摇提取2次,每次30ml,弃去二氯甲烷液,水液自“用正丁醇振摇提取2次”起,同法制成对照药材溶液,再取莫诺苷对照品、马钱苷对照品,加80%甲醇制成每1ml各含1mg的混合溶液,作为对照品溶液,照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取供试品溶液5μl、对照药材溶液1μl、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以三氯甲烷-甲醇(3:1)为展开剂,展开,取出,晾干,置254nm紫外光灯下检视,供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
(2)取本品1g,加70%甲醇30ml,加热回流1小时,滤过,滤液蒸干,残渣用0.1mol/l的氢氧化钠溶液20ml使溶解,用乙酸乙酯振摇提取2次,每次20ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液,另取五味子对照药材1g,加水50ml,煎煮20分钟,滤过,自“用乙酸乙酯振摇提取2次”起,同法制成对照药材溶液,再取五味子醇甲对照品,加80%甲醇制成每1ml含1mg的溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015年版通则0502)试验,吸取供试品溶液5μl、对照药材溶液1μl、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以石油醚(60~90℃)-乙酸乙酯-甲醇(5:3:0.2)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点。
【指纹图谱】照高效液相色谱法(《中国药典》2015年版四部通则0512)测定。
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.3%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长为254nm。理论板数按五味子醇甲计算应不低于3000;
参照物溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含0.15mg的溶液,即得;
供试品溶液的制备取本品2.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,超声(功率250w,频率40khz)处理30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,滤液用乙酸乙酯-正丁醇(1:1)混合溶液振摇提取2次,每次25ml,合并乙酸乙酯-正丁醇(1:1)混合溶液,蒸干,残渣加甲醇使溶解,并转移至5ml量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取参照物溶液与供试品溶液各5μl,注入液相色谱仪,测定,记录色谱图,即得;
供试品指纹图谱中应呈现与参照物色谱峰保留时间相同的色谱峰,供试品色谱图应与对照指纹图谱基本一致,有相对应的14个共有峰,按中药色谱指纹图谱相似度评价系统,以共有峰计算相似度,供试品指纹图谱与对照指纹图谱的相似度不得低于0.90;对照指纹图谱见图23。
【含量测定】山萸肉照高效液相色谱法(《中国药典》2015年版四部通则0512)测定。
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.3%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长为240nm;理论板数按莫诺苷峰计算应不低于3000;
对照品溶液的制备取莫诺苷、马钱苷对照品适量,精密称定,加80%甲醇制成每1ml含莫诺苷40μg、马钱苷20μg的混合溶液,即得。
供试品溶液的制备取本品1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,超声处理(功率250w、频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得。
本品按干燥品计算,每1g含山萸肉以莫诺苷(c17h26o11)和马钱苷(c17h26o10)总量计,应为1.5mg~3.0mg。
五味子照高效液相色谱法(《中国药典》2015年版四部通则0502)测定。
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(45:55)为流动相;检测波长为250nm,理论板数按五味子醇甲峰计算应不低于3000;
对照品溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含20μg的溶液,即得。
供试品溶液的制备取本品1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
本品按干燥品计算,每1g含五味子以五味子醇甲(c24h32o7)计,应为0.4mg~0.8mg。
为了进一步验证本发明的可行性,发明人进行了一系列的试验。
实验例1:前处理研究
由于地黄饮子为煮散剂,根据原文记载,熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志十二味需要粉碎,考证结果为粉碎过三号筛(50目),经试验发现粉碎过三号筛难以操作,故确定粉碎成粗粉。生姜为块状根茎,不宜煮透,故参考《中国药典》2015年版一部“生姜”项下饮片处理方法,用时切厚片,因此将生姜切成2mm~4mm的厚片,备用;大枣为果实类中药,不宜煮透,故参考《中国药典》2015年版一部“大枣”项下饮片处理方法,用时破开,备用;薄荷饮片呈不规则的短段,原文没有前处理要求,因此可以直接投料。饮片粉碎结果见表1。
表1饮片粉碎结果表
试验结果:熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志十二味药粉碎成粗粉收率均大于80%。
实验例2煎煮研究
1、煎煮终点考察及煎煮时间的确定
由于处方原文中煎煮终点描述为“煎至八分”,根据考证结果为液面高度的“八分”,但是以提取液高度作为煎煮终点时,在试验过程中需要实时量取提取液高度,这样需要较长时间离开火源操作,导致煎煮不连续,在固定加热方式(明火)和控制火力条件下,利用提取液高度变化与煎煮时间之间的相关性,以达到煎煮终点提取液“八分”高度的时间节点作为煎煮时间。过程如下:称取每服量饮片(熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粗粉各1g、生姜(用时切厚片)5g、大枣(用时破开)4.6g、薄荷1g)共3份,置于600ml砂锅中,加水300ml,量取液面至锅底高度h1,采用明火(燃气灶)加热,加盖煎煮,武火煮沸后改文火煎煮,煮沸后每隔5分钟进行高度量取,直至液面高度达到0.8h1,记录液面高度及煎煮时间,分别测定提取液中莫诺苷和马钱苷含量、五味子醇甲含量、出膏率,并计算莫诺苷和马钱苷总量、五味子醇甲总量。结果见表2。
提取液出膏率的测定方法:精密量取提取液20ml,置已干燥至恒重的蒸发皿w1中,在水浴上蒸干后,于105℃干燥3小时,置干燥器中冷却30分钟,迅速精密称定重量w2。
表2地黄饮子物质基准煎煮终点考察结果表
试验结果表明:地黄饮子煎煮工艺中以提取液高度的八分为煎煮终点,液面高度为1.46cm,最佳煎煮时间为20分钟。
2、煎煮工艺总结综上,暂定地黄饮子物质基准煎煮工艺为:取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粗粉各1g、生姜(用时切厚片)5g、大枣(用时破开)4.6g、薄荷1g,置于600ml砂锅中,加水300ml,采用明火(燃气灶)加热,加盖煎煮,武火煮沸,文火煎煮20分钟,得煎煮液。
实验例3滤过工艺研究
取“煎煮终点考察及煎煮时间的确定”项下提取液,分别趁热常压200目滤布滤过、300目滤布滤过、200目滤布滤过后离心(转速为每分钟4000转)5分钟、300目滤布滤过后离心(转速为每分钟4000转)5分钟,观察滤液澄清度和静置30分钟后滤液状态。
试验结果:趁热常压200目、300目滤过澄清,静置30分钟后均有沉淀生成,200目、300目过滤后离心有沉淀,但沉淀经确定不是药渣和泥沙,可能为脂溶性成分。由于地黄饮子为煮散剂,原文去滓服用,故综合考虑,确定最佳滤过方式为300目滤布趁热常压滤过。
实验例4浓缩工艺研究
由于提取液体积较大,故进行浓缩比例、浓缩温度、浓缩方式的考察。
(1)浓缩比例考察
称取每服量饮片(熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粗粉各1g、生姜5g(用时切厚片)、大枣4.6g(用时破开)、薄荷1g)共2份,分别置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸后,改用文火煎煮20分钟,300目滤布趁热常压滤过,滤液分别减压浓缩(60℃)至饮片投料量与浓缩液质量比约为1:3(g:g)、1:4(g:g),记录浓缩时间。将浓缩液置低温冷阱(温度:-20~-48℃)进行预冻,观察预冻时物料的挂瓶状态,记录预冻时间。将预冻好的浓缩液在冷冻干燥机上进行干燥,观察干膏粉状态。结果见表3。
表3地黄饮子物质基准浓缩比例考察结果表
试验结果表明,滤液浓缩至饮片投料量与浓缩液质量比约为1:3(g:g)、1:4(g:g)时,预冻挂瓶均良好,冻干粉不发泡不融化,质地疏松,由于投料量与浓缩液质量比约为1:4(g:g)时浓缩时间较短,综合考虑,故选择浓缩至投料量与浓缩液质量比约为1:4(g:g)。
(2)浓缩温度考察
称取每服量饮片(熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粗粉各1g、生姜5g(用时切厚片)、大枣4.6g(用时破开)、薄荷1g)共4份,分别置于600ml砂锅中,加水300ml,采用明火加盖煎煮,武火煮沸,文火煎煮20分钟,趁热常压300目滤布滤过,滤液合并,混匀,留样(需折合投料量)进行莫诺苷和马钱苷含量、五味子醇甲含量测定,剩余滤液平均分成四份,两份进行60℃减压浓缩,两份进行70℃减压浓缩,分别浓缩至投料量与浓缩液质量为1:4(g:g),浓缩液样品进行莫诺苷和马钱苷含量、五味子醇甲含量测定,并计算莫诺苷和马钱苷总量、五味子醇甲总量。结果见表4。
表4地黄饮子物质基准浓缩温度考察表
试验结果表明,地黄饮子物质基准在不同浓缩温度时莫诺苷和马钱苷提取总量、五味子醇甲提取总量基本一致,由于70℃浓缩时间较短,考虑到浓缩效率,故浓缩温度确定为70℃。
(3)单锅浓缩与合并浓缩对比考察
称取每服量饮片(熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粗粉各1g、生姜5g(用时切厚片)、大枣4.6g(用时破开)、薄荷1g)共4份,分别置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸后,改用文火煎煮20分钟300目滤布趁热常压滤过,滤液合并,混匀,分为四份,两份合并减压浓缩(70℃)、两份单独进行减压浓缩(70℃),分别浓缩至饮片投料量与浓缩液质量为1:4(g:g),考察多锅合并浓缩和单锅(日处方)浓缩时浓缩液中莫诺苷和马钱苷总量、五味子醇甲总量的差异。结果见表5。
表5地黄饮子物质基准浓缩方式考察结果表
试验结果,单锅(日处方)浓缩和多锅合并浓缩时浓缩液中莫诺苷和马钱苷总量、五味子醇甲总量基本一致,表明多锅合并浓缩对指标含量无影响。因此浓缩既可以单锅浓缩,也可多锅合并浓缩,故工艺验证及不同批物质基准制备可以采取多锅合并浓缩方式。
实验例5干燥工艺研究
取实验例4项下两份单锅浓缩液,混匀,留样(需折合投料量),浓缩液平均分为两份,分别进行冷冻干燥(预冻温度:预冻温度:-20~-48℃,冷阱温度:-40~-80℃,冷阱温度:-60~-70℃,真空度:<100pa,冷冻干燥时间至少17小时)和减压干燥(60℃)。记录预冻时间与干燥时间。分别测定水分(快速水分测定仪)、莫诺苷和马钱苷含量、五味子醇甲含量,并计算莫诺苷和马钱苷总量、五味子醇甲总量。结果见表6。
表6地黄饮子物质基准干燥工艺研究结果表
试验结果,冷冻干燥与干燥前比较,莫诺苷和马钱苷总量差别不大,五味子醇甲轻微损失;而减压干燥(60℃)与干燥前相比,莫诺苷和马钱苷以及五味子醇甲均有损失,故确定干燥方式为冷冻干燥。
经上述研究,暂定地黄饮子物质基准制备工艺为:称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粗粉各1g、生姜(用时切厚片)5g、大枣(用时破开)4.6g、薄荷1g,置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸,文火煎煮20分钟,趁热常压300目滤布滤过,滤液经减压浓缩(70℃,-0.08~-0.1mpa)至饮片投料量与浓缩液质量比约为1:4(g:g),冷冻干燥(预冻温度:-20~-48℃,冷阱温度:-40~-80℃,时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa,冻干时间:至少17小时),即得。
实验例6关键工艺步骤和中间体
1、物质基准工艺验证
按照上述确定的工艺,进行三批工艺验证,每批包含14份每服量:称取每服量饮片(熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粗粉各1g、生姜(用时切厚片)5g、大枣(用时破开)4.6g、薄荷1g)共42份,置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸后,改用文火煎煮20分钟,趁热常压300目滤布滤过,每批14份提取液混匀,分别留样100ml,并平均分为2份,分别减压浓缩(70℃,-0.09~-0.1mpa)至投料量与浓缩液质量比约为1:4(g:g),浓缩液混匀,分别留样50ml,并平均分为10份,冷冻干燥(预冻温度:-20~-48℃,冷阱温度:-40~-80℃,时间:至少30分钟;干燥:冷阱温度:-60~-70℃,真空度:<100pa),收集对应实物(干膏粉),计算出膏率。测定提取液、浓缩液、干膏粉中莫诺苷和马钱苷含量、五味子醇甲含量和指纹图谱以及干膏粉水分、浸出物,并分别计算提取液、浓缩液、干膏粉中莫诺苷和马钱苷总量、五味子醇甲总量。另取药材和饮片进行指纹图谱测定。结果见表7、表8,图1。
表7地黄饮子物质基准工艺验证结果
表8地黄饮子物质基准工艺验证相对保留时间结果
由以上结果可知,从中间体(提取液)体积上分析,三批物质基准(每批包括14份每服量)体积均值为2187ml,按每服量计,则为156ml,与原文中“煎至八分”(八分高度药液160ml)接近,证明工艺煎煮时间确定合理。
从莫诺苷和马钱苷提取总量、五味子醇甲提取总量上分析,三批验证中间体(提取液)与对应实物(干膏粉)数据均基本平行,且工艺过程中物质传递损失较小;从出膏率、浸出物、水分上分析,三批验证对应实物(干膏粉)无明显差异。
从指纹图谱分析,峰1、4、5、10为山萸肉色谱峰,峰3、16、18、19为五味子色谱峰,峰6为酒苁蓉色谱峰,峰7、8、9、11、12、15为远志色谱峰,峰14为薄荷色谱峰。且三批验证提取液、浓缩液、干膏粉相应位置均有相同的共有峰,其相对保留时间基本一致,初步确定的物质基准制备工艺稳定可行。
综上,确定物质基准优选工艺为:称取熟地黄、巴戟肉、山萸肉、石斛、酒苁蓉、炮附片、五味子、肉桂、茯苓、麦冬、石菖蒲、远志粗粉各1g,生姜(用时切厚片)5g,大枣(用时破开)4.6g,薄荷1g,置于600ml砂锅中,加水300ml,采用明火加热,加盖煎煮,武火煮沸后,文火煎煮20分钟,300目滤布趁热常压滤过,滤液减压浓缩(70℃,-0.08~-0.1mpa)至饮片投料量与浓缩液质量比约为1:4(g:g),冷冻干燥(预冻温度:-20~-48℃,冷阱温度:-40~-80℃,时间:至少30分钟;干燥:冷阱温度:-60~-70℃真空度:<100pa,冻干时间:至少17小时),得干膏粉,包装,即得对应实物,或加入药学上的辅料制成药学上可接受的剂型。
多批次物质基准的制备
每个药材收集2~3个产地,包括道地产区和主产区,每个产地不少于3批,总收集批次15批,其中山萸肉收集河南西峡共4批、浙江淳安共7批、陕西丹凤3批;五味子收集辽宁清原、吉林蛟河、黑龙江木兰各5批;将目前收集到的饮片按浸出物高低顺序排序,从低到高的顺序一一对应组合,制备15批物质基准。15批饮片排序组合结果见下表9。
按照上述确定工艺,制备15批物质基准对应实物(干膏粉),每批次饮片信息见9,每组实验设计两个平行样,每个平行样包括10份每服量,浓缩液混合,再分开冷冻干燥,包装,即得。15批物质基准出膏率结果见表10。
表9批地黄饮子物质基准组合结果
表1015批地黄饮子物质基准出膏率结果
试验结果表明:15批地黄饮子物质基准对应实物(干膏粉)出膏率范围为23.69%~31.12%,均值为27.30%,考虑到实际生产过程中的各种影响因素等,暂定对应实物(干膏粉)出膏率范围为20.0%~40.0%。
实验例7定性鉴别方法摸索
本品由十五味中药经水煎煮制成,选用了专属性强,快速、简便、重现性好的薄层鉴别方法以及专属性强、快速、检测灵敏度高、重现性好的液相鉴别方法,对方中山萸肉、五味子的主要特征成分进行鉴别试验研究,并将其鉴别方法列入检测方法正文。又对处方中熟地黄、石菖蒲、肉桂、茯苓、麦冬、远志、大枣进行鉴别试验研究,因其专属性差,故未列入检测方法正文,具体研究过程如下。
1、安全性相关的化学成分及其限度汇总表
处方中君药熟地黄、山萸肉、酒苁蓉、巴戟肉,臣药炮附片、肉桂、麦冬、石斛、五味子,佐药石菖蒲、远志、茯苓,使药生姜、大枣、薄荷,以上药味除炮附片、远志、大枣外,目前暂未发现安全性相关的化学成分,炮附片内源性毒性成分含量限度、远志和大枣外源性成分含量限度详见下表11。
表11安全性相关的化学成分及其限度
除炮附片有限量要求外,其他药味均无内源性毒性成分,安全性良好。
2、山萸肉山萸肉其主要化学成分有多糖类、环烯醚萜苷类、有机酸类(齐墩果酸)、鞣质类等。其中环烯醚萜苷类包括马钱苷、莫诺苷、山茱萸新苷,而莫诺苷和马钱苷为其主要有效成分。根据这些化学成分对山萸肉进行如下薄层摸索实验。
①建立薄层色谱方法参考《中国药典》2015年版一部“山茱萸”薄层鉴别方法。
取本品4g,加水50ml,超声使溶解,用二氯甲烷振摇提取2次,每次30ml,用正丁醇振摇提取2次,每次25ml,合并正丁醇液,用氨溶液(1→10)洗涤2次,每次30ml,弃去氨溶液,合并正丁醇液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取缺山萸肉阴性样品4g,加水50ml使溶解,用二氯甲烷振摇提取2次,每次30ml,弃去二氯甲烷液,水液自“用正丁醇振摇提取2次“起,同法制成阴性样品溶液。再取山茱萸对照药材1g,加水50ml,煎煮20分钟,滤过,滤液用二氯甲烷振摇提取2次,每次30ml,弃去二氯甲烷液,水液自“用正丁醇振摇提取2次”起,同法制成对照药材溶液。另取莫诺苷对照品,加80%甲醇制成每1ml含0.5mg的溶液。继取马钱苷对照品,加80%甲醇制成每1ml含1mg的对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述五种溶液适量,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇(4:1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置紫外光灯(365nm)下检视。
试验结果:供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点,但是有背景干扰。
方法二:在“方法一”基础上,将薄层板更换为gf254硅胶板,将展开剂调整为三氯甲烷-甲醇(3:1),展开,取出,晾干,置紫外光灯(254nm)下检视,见图2:
试验结果:供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点,且阴性无干扰。确定点样量为:供试品溶液5μl、马钱苷对照品溶液5μl、莫诺苷对照品溶液2μl,对照药材溶液1μl。
初步确定山萸肉优选薄层鉴别方法为:取本品4g,加水50ml,超声使溶解,用二氯甲烷振摇提取2次,每次30ml,用正丁醇振摇提取2次,每次30ml,合并正丁醇液,用氨溶液(1→10)洗涤2次,每次25ml,弃去氨溶液,合并正丁醇液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取山茱萸对照药材1g,加水50ml,煎煮20分钟,滤过,滤液用二氯甲烷振摇提取2次,每次30ml,弃去二氯甲烷液,水液自“用正丁醇振摇提取2次”起,同法制成对照药材溶液。继取莫诺苷对照品,加80%甲醇制成每1ml含0.5mg的溶液,作为对照品溶液。继取马钱苷对照品,加80%甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取供试品溶液5μl、对照药材溶液1μl、马钱苷对照品溶液5μl和莫诺苷对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以三氯甲烷-甲醇(3:1)为展开剂,展开,取出,晾干,在紫外光灯(254nm)下检视。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
②耐用性考察
按照确定的方法进行耐用性试验,不同厂家的硅胶gf254薄层板(烟台市化学工业研究所、青岛海洋化工厂和德国macherey-nagel(mn))以及不同展开温度(23.5℃和6.6℃)、不同的相对湿度(38%和87%)等影响因素,结果此展开条件耐用性好,主斑点清晰。
③方法确定
取本品4g,加水50ml,超声使溶解,用二氯甲烷振摇提取2次,每次30ml,用正丁醇振摇提取2次,每次30ml,合并正丁醇液,用氨溶液(1→10)洗涤2次,每次25ml,弃去氨溶液,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取山茱萸对照药材1g,加水50ml,煎煮20分钟,滤过,滤液用二氯甲烷振摇提取2次,每次30ml,弃去二氯甲烷液,水液自“用正丁醇振摇提取2次”起,同法制成对照药材溶液。再取莫诺苷对照品、马钱苷对照品,加80%甲醇制成每1ml各含1mg的混合溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取供试品溶液5μl、对照药材溶液1μl、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以三氯甲烷-甲醇(3:1)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。
(2)五味子
五味子主要化学成分有木脂素类、三萜类化合物、挥发油、多糖等。其中木脂素包括五味子醇甲、五味子甲素、五味子乙素等。根据这些化学成分对五味子进行如下薄层摸索实验。
①建立薄层色谱方法
方法一:参考《中国药典》2015年版一部“五味子”薄层鉴别方法
取本品1g,加70%甲醇30ml,加热回流1小时,滤过,滤液蒸干,残渣用0.1mol/l的氢氧化钠溶液20ml使溶解,用乙酸乙酯振摇提取2次,每次20ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取缺五味子阴性样品1g,同法制成阴性样品溶液。再取五味子对照药材1g,加水50ml,煎煮20分钟,滤过,自“用乙酸乙酯振摇提取2次”起,同法制成对照药材溶液。继取五味子甲素对照品,加80%甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版通则0502)试验,吸取供试品溶液、阴性样品溶液和对照药材溶液适量、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以石油醚(60~90℃)-甲酸乙酯-甲醇(15:5:0.3)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。
试验结果:供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点,但是rf值较低;在与对照品色谱相应的位置的斑点较浅,需要更换对照品。
方法二:将对照品更换为五味子醇甲,展开剂调整为石油醚(60℃~90℃)-甲酸乙酯-甲醇(15:5:2)。
试验结果:供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点,但是阴性相应的位置上有阴性干扰。
方法三:在“方法二”基础上,将展开剂调整为石油醚(60~90℃)-乙酸乙酯-甲醇(5:3:0.2),试验结果如下图3:
试验结果:供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点,且阴性无干扰,方法可行。
初步确定方法为:取本品1g,加70%甲醇30ml,加热回流1小时,滤过,滤液蒸干,残渣用0.1mol/l的氢氧化钠溶液20ml使溶解,用乙酸乙酯振摇提取2次,每次20ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取五味子对照药材1g,加水50ml,煎煮20分钟,滤过,自“用乙酸乙酯振摇提取2次”起,同法制成对照药材溶液。再取五味子醇甲对照品,加80%甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版通则0502)试验,吸取供试品溶液5μl、对照药材溶液1μl、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以石油醚(60~90℃)-乙酸乙酯-甲醇(5:3:0.2)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点。
②耐用性考察
按照确定的方法进行耐用性试验,不同厂家的硅胶gf254薄层板(烟台市化学工业研究所、青岛海洋化工厂和德国macherey-nagel(mn))以及不同展开温度(22.7℃和2.6℃)、不同的相对湿度(50%和90%)等影响因素,结果此展开条件耐用性好,主斑点清晰。
③方法确定取本品1g,加70%甲醇30ml,加热回流1小时,滤过,滤液蒸干,残渣用0.1mol/l的氢氧化钠溶液20ml使溶解,用乙酸乙酯振摇提取2次,每次20ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取五味子对照药材1g,加水50ml,煎煮20分钟,滤过,自“用乙酸乙酯振摇提取2次”起,同法制成对照药材溶液。再取五味子醇甲对照品,加80%甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版通则0502)试验,吸取供试品溶液5μl、对照药材溶液1μl、对照品溶液2μl,分别点于同一硅胶gf254薄层板上,以石油醚(60℃~90℃)-乙酸乙酯-甲醇(5:3:0.2)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点。
实验例8指纹图谱方法摸索
由于单纯的指标成分的定性鉴别和定量分析,难以反映物质基准质量的优劣。作为衡量物质基准对应实物(干膏粉)是否与经典名方汤剂基本一致的标准参照物,其质量应加强专属性鉴别和多成分、整体质量控制。因而物质基准质量控制要重视以指纹图谱为主的整体质量控制。
(1)仪器与试药液相色谱仪:岛津lc-20a,安捷伦1260ⅱ;色谱柱:glscienceswondasilc18(250×4.6mm,5μm);电子分析天平:常熟市双杰测试仪器厂jj1000(百分之一),赛多利斯科学仪器(北京)有限公司bsa224s-cw(万分之一)梅特勒-托利多仪器(上海)有限公司me503t(千分之一),乙腈为色谱纯,oceanpak生产;磷酸为色谱纯,西亚试剂有限公司生产;甲醇为分析纯,广州化学试剂厂生产;甲酸为色谱纯,天津科密欧化学试剂有限公司;水为超纯水,上海和泰仪器有限公司。莫诺苷对照品(批号:111998-201703,纯度:97.4%)、马钱苷对照品(批号:111640-201707,纯度:99.2%),五味子醇甲对照品(批号:110857-201714,纯度:99.9%),3,6’-二芥子酰基蔗糖对照品(批号:111848-201604,纯度96.7%),购自中国食品药品检定研究院,迷迭香酸对照品(批号:111871-201706,纯度90.5%),购自中国食品药品检定研究院,松果菊苷对照品(批号:111670-201706,纯度89.7%),购自中国食品药品检定研究院。
(2)流动相的选择
方法一:参考文献“大柴胡汤和桂附地黄方的化学成分指纹图谱研究”中指纹图谱方法,在此基础上调整流动相比例:
色谱条件以glscienceswondasilc18(4.6×250mm,5μm)为色谱柱,以乙腈为流动相a,以0.1%甲酸为流动相b,按表12中的规定进行梯度洗脱;检测波长为254nm。
表12流动相梯度洗脱程序
供试品溶液的制备取冻干粉2g,精密称定,置具塞锥形瓶中,精密量取70%甲醇50ml,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,滤液蒸干,残渣加水25ml使溶解,溶液用乙酸乙酯-正丁醇(1:1)混合溶液振摇提取3次,每次25ml,合并乙酸乙酯-正丁醇(1:1)混合溶液,蒸干,残渣加甲醇使溶解,并转移至5ml量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取供试品溶液各5μl,注入液相色谱仪,测定,记录色谱图,即得。
试验结果:20分钟~48分钟、54分钟~56分钟峰分离度不佳。
方法二:在“方法一”基础上将流动相梯度洗脱程序调整,见表13:
表13流动相梯度洗脱程序
试验结果:在箭头处色谱峰分离度不佳,需要优化。
方法三:在“方法二”基础上进行流动相比例优化,同时将流动相b更换为0.3%磷酸,调整流动相梯度洗脱程序如表14:
表14流动相梯度洗脱程序
试验结果:该条件下色谱峰型良好,分离度较佳。可以用于后续研究,见图4。
供试品制备方法的选择
80%甲醇超声取本品约2g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,蒸干,残渣加甲醇使溶解,并转移至5ml量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
乙酸乙酯振摇提取取本品约2g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,蒸至近干,残渣加水25ml使溶解,用乙酸乙酯振摇提取2次,每次25ml,合并乙酸乙酯溶液,蒸干,残渣加甲醇使溶解,并转移至5ml量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
乙酸乙酯与正丁醇(1:1)混合溶液振摇提取取本品约2g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,称定重量,超声处理(功率2500w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,蒸至近干,残渣加水25ml使溶解,用乙酸乙酯与正丁醇(1:1)混合溶液振摇提取2次,每次25ml,合并乙酸乙酯与正丁醇(1:1)混合振摇提取溶液,蒸干,残渣加甲醇使溶解,并转移至5ml量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
试验结果如图5:
试验结果:乙酸乙酯样峰信息较少,乙酸乙酯-正丁醇(1:1)和80%甲醇样中峰信息较多,二者基本一致,但是供试品经80%甲醇处理后粘度比经乙酸乙酯-正丁醇(1:1)处理后粘度大,综合考虑选择优选乙酸乙酯-正丁醇(1:1)进行供试品制备。
(3)检测波长的选择
采用二极管阵列检测器,供试品溶液在190~400nm波长下进行扫描,通过等吸收图谱进行分析,结果显示后50分钟色谱峰在230~280nm波长范围内,信息量较大。选取波长230nm、254nm、280nm进行分析,在254nm波长下,各色谱峰信息量较大,响应值适中,分离度良好,基线较平稳。故选用254nm作为检测波。
(4)峰归属及参照物的选择
①药味峰归属
饮片样品溶液的制备取处方下各药味粉末(过二号筛)约0.5g,精密称定,置锥形瓶中,加水50ml,煎煮30分钟,放冷,滤过,滤液用乙酸乙酯-正丁醇(1:1)混合溶液振摇提取2次,每次25ml,合并乙酸乙酯-正丁醇(1:1)混合溶液层,蒸干,残渣加甲醇使溶解,并转移至5ml量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
按照确定的色谱条件进行测定,同时通过质谱检测,确定物质基准指纹图谱中各个峰的归属(见图6和表15)。其中峰1、3、11、13、14为五味子色谱峰、峰2、4为山萸肉色谱峰,峰5为酒苁蓉色谱峰,峰6、7、8、10为远志色谱峰,9、12为薄荷色谱峰。
表15药味归属表
②色谱峰指认
通过文献调研选择各药味相关的主要成分:莫诺苷、马钱苷、松果菊苷、3,6-二芥子酰基蔗糖、迷迭香酸、五味子醇甲进行峰定位研究。(见图7)。
通过对照品比对7个已知成分(没食子酸、莫诺苷、马钱苷、松果菊苷、3,6′-二芥子酰基蔗糖、五味子醇甲),分析结果与质谱结果一致。19个色谱峰中,最终共指认了其中7个已知成分。质谱分析结果与对照品指认结果对应关系见表16。
同时通过质谱检测地黄饮子物质基准对应实物(干膏粉)指纹图谱中的色谱峰进行初步指认,uplc-uv色谱图和总离子流图(正模式)见图8,由于液质联用仪的液相部分为uplc,其色谱图与hplc指纹图谱的色谱图有些差异,具体峰对应关系见下表16
表16质谱分析结果与共有峰指认结果对应关系
③参照物的选择
色谱图中五味子醇甲色谱峰保留时间适中,响应值较高,达到基线分离,故选择五味子醇甲作为对照品参照峰,并将对五味子醇甲标记为峰s,同时将五味子醇甲对照品作为参照物。
(5)方法的初步确定
照高效液相色谱法(《中国药典》2015年版四部通则0512)测定。
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,0.3%磷酸溶液为流动相b,按表17中的规定进行梯度洗脱;检测波长为254nm;理论板数按五味子醇甲计算应不低于3000。
表17流动相梯度洗脱程序表
参照物溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含0.15mg的溶液,即得。
供试品溶液的制备取本品约2.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,滤液用乙酸乙酯-正丁醇(1:1)混合溶液振摇提取2次,每次25ml,合并乙酸乙酯-正丁醇(1:1)混合溶液层,蒸干,残渣加甲醇使溶解,并转移至5ml量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取参照物溶液与供试品溶液各5μl,注入液相色谱仪,测定,记录色谱图,即得。
(6)方法学考察
①专属性试验与整体性考察
通过专属性与整体性试验考察,空白溶剂无干扰,专属性良好;在延长的30分钟内没有色谱峰,整体性良好。具体见图9。
②仪器精密度试验
取本品约2.0g(批号:d-181120-03),精密称定,按“方法的初步确定”项下供试品溶液的制备和测定法操作,连续进样6次,计算各特征峰与s峰的相对保留时间、相似度,测定结果见表18、19,图10。
表18指纹图谱精密度试验相对保留时间结果(n=6)
表19指纹图谱仪器精密度试验相似度结果(n=6)
r对照图谱、s2仪器精密度1、s3仪哭器精密度2、s4仪器精密度3、s5仪精密度4、s6仪器精密度5、s7仪器精密度6
结果表明,供试品指纹图谱呈现与参照物色谱峰保留时间相同的色谱峰,并呈现14个主要色谱峰。其相对保留时间小于3.0%、相似度大于0.90,说明仪器精密度良好。
③稳定性试验
取本品(批号:d-181112-03)约2.0g,精密称定,按“方法初步确定”项下供试品溶液的制备和测定法操作,分别在制备后放置0、3、6、9、12、15、24、36、48小时进样测定,计算各特征峰与s峰的相对保留时间、与对照图谱相似度,测定结果见表20-表21,图11
表20稳定性试验相对保留时间结果(n=6)
表21稳定性试验相似度结果
结果表明,指纹图谱相对保留时间rsd小于3.0%,相似度大于0.90,说明溶液在48小时内稳定性良好。
④重复性试验
取本品(批号:d-181120-03)约2.0g(共6份),精密称定,按“方法初步确定”项下供试品溶液的制备和测定法操作,计算各特征峰与s峰的相对保留时间、相似度,测定结果见表22、表23、图12。
表22重复性试验相对保留时间结果(n=6)
表23重复性试验相似度结果(n=6)
r对照图谱、s2重复性1、s3重复性2、s4重复性3、s5重复性4、s6重复性5、s7重复性6
结果表明,指纹图谱相对保留时间rsd小于3.0%,相似度大于0.90,说明指纹图谱方法重复性良好。
⑤耐用性试验
考察了不同流速、不同柱温、不同色谱柱、不同酸比例、不同仪器对本色谱条件的耐用性,与对照图谱r(采用国家药典委员会颁布的《中药色谱指纹图谱相似度评价软件系统2012年版》,以重复性hplc图谱色谱峰进行自动匹配,形成共有模式图,并建立对照图谱r),计算相似度,测定结果见图13~图16,表24~27。
表24不同流速耐用性相似度结果
r对照图谱s21.1ml/mins31.0ml/mins40.9ml/min
表25不同色谱柱耐用性相似度结果
r对照图谱、s2aglient5tc、s3glscienceswondasilc18、s4xtimatec18、s5inerstilods-sp
表26不同仪器耐用性相似度结果
r对照图谱s20.25%磷酸s30.3%磷酸s40.35%磷酸
表27不同仪器耐用性相似度结果
r对照图谱s2agilent1260iis3岛津lc20a
结果表明,不同流速、不同柱温、不同色谱柱、不同酸比例、不同仪器条件下测定结果基本一致,色谱图中各共有峰峰型尖锐,对称,分离度良好,相似度均不低于0.90。供试品指纹图谱中均呈现14个主要色谱峰,且与对照品参照物色谱峰中的特征峰相对应。表明该方法对不同流速、不同柱温、不同色谱柱、不同酸度、不同仪器耐用性良好。
(8)指纹图谱测定方法的确定
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.3%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长为254nm;理论板数按五味子醇甲计算应不低于3000。
表28流动相梯度洗脱程序
参照物溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含0.15mg的溶液,即得。
供试品溶液的制备取本品约2.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,滤液用乙酸乙酯-正丁醇(1:1)混合溶液振摇提取2次,每次25ml,合并乙酸乙酯-正丁醇(1:1)混合溶液,蒸干,残渣加甲醇使溶解,并转移至5ml量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取参照物溶液与供试品溶液各5μl,注入液相色谱仪,测定,记录色谱图,即得。
(7)指纹图谱的建立
①共有峰的标定
按照确定的方法对15批(30份)供试品进行测定,并对所得指纹图谱进行分析,选择15批(30份)样品指纹图谱中稳定性较好、响应值适宜的色谱峰为共有峰,共标定了14个共有峰,结果见图17~图18。
②对照图谱的建立
采用国家药典委员会颁布的《中药色谱指纹图谱相似度评价软件系统2012年版》,以15批(30份)地黄饮子物质基准对应实物(干膏粉)hplc图谱色谱峰进行自动匹配,形成共有模式图,并建立对照图谱,结果见图19。
③相似度的计算
采用中药色谱指纹图谱相似度评价软件系统,以共有峰对15批(30份)物质基准对应实物(干膏粉)与对照指纹图谱的相似度进行计算,结果见表29-表30。按指纹图谱计算的相似度,15批(30份)物质基准与地黄饮子对照指纹图谱相似度比较,实测范围为0.902~0.997,均大于0.90,故选择指纹图谱作为地黄饮子物质基准的评价标准。供试品指纹图谱中应呈现与参照物色谱峰保留时间相同的色谱峰,供试品色谱图应与对照指纹图谱基本一致,有相对应的14个共有峰,按中药色谱指纹图谱相似度评价系统,以共有峰计算相似度,供试品指纹图谱与对照指纹图谱的相似度不得低于0.90。
表29相似度结果-1
r.对照品图谱s2.d-181212-01、s3.d-181212-03、s4.d-181212-05、s5.d-181212-07、s6.d-181213-09、s7.d-181213-11、s8.d-181213-13、s9.d-181213-15、s10.d-181214-17、s11.d-181214-19、s12.d-181214-21、s13.d-181214-23、s14.d-181215-25、s15.d-181215-27、s16.d-181215-29
表30相似度结果-2
r.对照图谱s2.d-181212-03、s3.d-181212-04、s4.d-181212-06、s5.d-181212-08、s6.d-181213-10、s7.d-181213-12、s8.d-181213-14、s9.d-181213-16、s10.d-181214-18s11.d-181214-20、s12.d-181214-22、s13.d-181214-24、s14.d-181215-26、s15.d-181215-28s16.d-181215-3
实验例9含量测定
地黄饮子处方中熟地黄、巴戟肉、山萸肉、酒苁蓉为君药,石斛、炮附片、五味子、肉桂、麦冬、为臣药,茯苓、石菖蒲、远志为佐药,生姜、大枣、薄荷为使药。方中君药山萸肉中环烯醚萜苷(莫诺苷、马钱苷)成分为其主要活性成分,臣药五味子中木脂素类(五味子醇甲)与其有效性相关,是其主要药效物质基础,可作为地黄饮子的指标成分;故选择君药山萸肉中莫诺苷和马钱苷总量及臣药五味子中五味子醇甲含量进行研究。
(1)山萸肉照高效液相色谱法(《中国药典》2015年版四部通则0512)测定。
①仪器与试药
②色谱条件的选择
a流动相及流动相比例的选择
方法一:参照《中国药典》2015年版一部“山茱萸”项下[467]莫诺苷、马钱苷含量测定方法进行摸索:
色谱条件以kromasil100-5-c18(4.6*250mm,5um)为色谱柱,以乙腈为流动相a,0.3%磷酸为流动相b,按表31中的规定梯度洗脱;检测波长为240nm;柱温为35℃。
表31流动相梯度洗脱程序
对照品溶液的配制取莫诺苷对照品、马钱苷对照品适量,精密称定,加80%甲醇制成每1ml各含50μg的混合溶液,即得。
供试品溶液的制备取本品约1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
结果分析:该条件下马钱苷分离度不佳且阴性有干扰,需要优化流动相比例。马钱苷对照品与供试品峰面积相差较大,需调整供试品峰面积与对照品峰面积约为1:1。
方法二:在“方法一”基础上,将流动相梯度洗脱程序调整如表32,试验结果如图20。
表32流动相梯度洗脱程序
结果分析:阴性无干扰,莫诺苷、马钱苷峰型、峰分离度良好,可以用于后续研究。
故初步确定莫诺苷和马钱苷含测测定方法为:
色谱条件与系统适用性试验以kromasil100-5-c18(4.6*250mm,5um)为色谱柱,以乙腈为流动相a,以0.3%磷酸为流动相b,按表33中的规定梯度洗脱,检测波长为240nm;柱温为35℃。
表33流动相梯度洗脱程序
对照品溶液的配制取莫诺苷、马钱苷对照品适量,精密称定,加80%甲醇制成每1ml含莫诺苷40μg、马钱苷20μg的混合溶液,即得。
供试品溶液的制备取本品约1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得。
b检测波长的确定
取莫诺苷对照品、马钱苷对照品适量,精密称定,加80%甲醇制成每1ml各含莫诺苷40μg、马钱苷20μg的混合溶液,在190~400nm波长下进行扫描,全波长扫描图见图6.3.3-132~图6.3.3-133,可见莫诺苷在240nm处有最大吸收峰且无杂质干扰,马钱苷在238nm处有最大吸收峰且无杂质干扰,与《中国药典》2015年版一部中“山茱萸”含量测定波长240nm基本一致,故选用240nm作为检测波长。
③供试品溶液制备方法考察
a提取溶媒的考察
取本品(批号:d-181120-01)约1.0g(共3组),精密称定,置具塞锥形瓶中,分别精密加入80%乙醇、80%甲醇、甲醇25ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,取出,放冷,再称定重量,分别用80%乙醇、80%甲醇、甲醇补足减失的重量,摇匀,滤过,取续滤液,即得,精密吸取续滤液5μl,注入液相色谱仪,进行测定。结果见表34。
表34提取溶媒考察结果
结果表明,不同提取溶媒提取率基本一致,与《中国药典》2015年版一部“山茱萸”含量测定项下保持一致,故选择80%甲醇作为提取溶媒。
b提取方式的考察
取本品(批号:d-181120-01)约1.0g(共3组),精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,分别超声处理(功率250w,频率40khz)30分钟、回流提取30分钟、振摇提取30分钟,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得,精密吸取续滤液5μl,注入液相色谱仪,进行测定。结果见表35。
表35提取方式考察结果
实验结果:不同提取方式莫诺苷和马钱苷含量基本一致,考虑操作方便,故选择超声作为提取方式。
c提取时间的考察
取本品(批号:d-181120-01)约1.0g(共3组),精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,分别超声处理(功率250w,频率40khz)15分钟、30分钟、45分钟,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得,精密吸取续滤液5μl,注入液相色谱仪,进行测定。结果见表36。
表36提取时间考察结果
结果表明,不同提取时间,莫诺苷和马钱苷含量差异不大,考虑不同批次饮片含量存在差异,选择提取时间为30分钟。
④含量测定方法的初步确定色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.3%磷酸溶液为流动相b,按表37中的规定进行梯度洗脱;检测波长为240nm。理论板数按莫诺苷峰计算应不低于3000。
表37流动相梯度洗脱程序
对照品溶液的制备取莫诺苷、马钱苷对照品适量,精密称定,加80%甲醇制成每1ml含莫诺苷40μg、马钱苷20μg的混合溶液,即得。
供试品溶液的制备取本品约1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液和供试品溶液各5μl,注入液相色谱仪,测定,即得。
⑤方法学考察
a专属性试验
山萸肉中含有莫诺苷、马钱苷,为考察其它药味是否干扰莫诺苷、马钱苷的测定,按处方比例称取饮片同法制成缺山萸肉阴性样品,并按照供试品的处理方法制备缺山萸肉的阴性样品溶液进样,记录色谱图。结果表明,莫诺苷、马钱苷色谱峰分离效果佳,且阴性色谱中在与莫诺苷、马钱苷相应的保留时间没有色谱峰,表明山萸肉以外的其他饮片对莫诺苷、马钱苷的测定无干扰,以本法同时测定本品中莫诺苷、马钱苷的含量具有专属性。结果见图21。
b峰纯度检查
取上述供试品溶液,注入液相色谱仪,采用上述色谱条件以pda检测器进行全波长检测,计算峰纯度,结果供试品色谱中莫诺苷和马钱苷均未检测到杂质峰;最小峰纯度阈值分别为995.03和992.00,说明在该色谱条件下,莫诺苷和马钱苷色谱峰纯度符合要求。
c线性关系的考察
精密称取莫诺苷对照品10.29mg(批号:111998-201703,纯度:97.4%),马钱苷对照品10.18mg(批号:111640-201707,纯度:99.2%),置10ml量瓶中,加80%甲醇溶解并定容至刻线,摇匀,分别配制成浓度为1.0022mg/ml的莫诺苷对照品溶液①、1.0180mg/ml的马钱苷对照品溶液②;精密量取对照品①2ml、②1ml置5ml量瓶中,用80%甲醇稀释至刻度,摇匀,配制成浓度0.4009mg/ml的莫诺苷和0.2036mg/ml的马钱苷对照品溶液③;精密量取对照品①2ml、②1ml置100ml量瓶中,用80%甲醇稀释至刻度,摇匀,配制成浓度20.04μg/ml的莫诺苷和10.18μg/ml的马钱苷对照品溶液④。分别精密吸取上述对照品溶液④2、5、10、15μl,对照品溶液③2、5、10μl注入液相色谱仪,照液相色谱法(《中国药典》2015年版四部通则0512)测定。以进样量(μg)为横坐标,峰面积为纵坐标,绘制标准曲线。见表38-表39。
表38莫诺苷线性关系
结果表明,莫诺苷在0.04008μg~4.0090μg范围内线性良好。
表39马钱苷线性关系
结果表明,马钱苷在0.02036μg~2.0360μg范围内线性良好。
d仪器精密度试验
取莫诺苷和马钱苷混合对照品溶液,莫诺苷浓度为40.09μg/ml,马钱苷浓度为20.36μg/ml,连续进样6次,测定结果见表40。
表40仪器精密度试验结果(n=6)
结果表明,rsd小于2.0%,说明仪器精密度良好。
e重复性试验
实验人员(a)操作,取本品(批号:d-181120-01)约1.0g(共6份),精密称定,按“方法的初步确定”项下供试品溶液制备和测定法进行操作。结果见表41。
表41重复性试验结果(n=6)
结果表明,rsd小于3.0%,说明本方法的重复性良好。
f稳定性试验
取本品(批号:d-181120-01)约1g,精密称定,按“方法的初步确定”项下供试品溶液的制备和测定法项下操作,分别在制备后放置0、2、4、8、12、24、28、36、48小时进样测定,测定结果见表42。
表42稳定性试验结果
结果表明,rsd<3%,供试品溶液在48小时内稳定性良好。
g准确度试验
精密称取莫诺苷11.49mg(批号:111998-201703,纯度:97.4%)置10ml量瓶中,加80%甲醇溶液溶解并稀释至刻度,摇匀,即得浓度为1.1191mg/ml对照品溶液①;精密称取马钱苷对照品12.14mg(批号:111640-201707,纯度:99.2%)置20ml量瓶中,加80%甲醇溶液溶解并稀释至刻度,摇匀,即得浓度为0.6070mg/ml溶液②;分别精密量取对照品溶液①、②各4ml至200ml的量瓶中,加80%甲醇溶液稀释至刻度,摇匀,即得浓度为22.38μg/ml莫诺苷、12.14μg/ml马钱苷混合对照品溶液③。
取本品约0.5g(批号:d-181120-01)(共6份),精密称定,置具塞锥形瓶中,分别精密加入对照品溶液③25ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,精密吸取供试品溶液5μl,注入液相色谱仪,测定,即得结果见表43~表44。地黄饮子物质基准对应实物(干膏粉)(批号:d-181120-01)中莫诺苷含量为1.1135mg/g,马钱苷含量为0.5863mg/g。
表43莫诺苷准确度试验结果(n=6)
表44马钱苷准确度试验结果(n=6)
结果表明,结果,莫诺苷平均回收率为101.26%,rsd<3%,马钱苷平均回收率为97.10%,rsd<3%,表明本方法准确度较好。
h中间精密度试验
选择不同的测定时间、不同的高效液相色谱仪、不同的实验人员(b),取本品(批号:d-181120-01)约1.0g,精密称定,按“方法的初步确定”项下供试品溶液制备和测定法进行操作。结果见表45~表46。
表45莫诺苷中间精密度试验结果(n=6)
表46马钱苷中间精密度试验结果(n=6)
结果,a和b测定的样品中莫诺苷平均含量为1.0948mg/g,rsd<2.0%;马钱苷平均含量为0.5798mg/g,rsd<2.0%,表明本方法中间精密度良好。
i耐用性试验
考察不同流速、不同柱温、不同酸比例、不同色谱柱对本色谱条件的耐用性,结果见表47。
表47莫诺苷和马钱苷耐用性试验结果
结果表明,各个条件下测定结果基本一致,rsd<5%,色谱图中莫诺苷、马钱苷峰型尖锐,对称,分离度良好,表明本方法不同流速、不同色谱柱、不同柱温、不同酸度耐用性良好。
以上的研究结果表明,地黄饮子物质基准对应实物(干膏粉)中山萸肉的含量测定方法具有专属性,稳定性、重复性、中间精密度、准确度等均符合规定,且色谱条件的耐用性较好,故此液相色谱条件可用于地黄饮子物质基准对应实物(干膏粉)中山萸肉的含量测定。
⑥含量测定方法的确定
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,0.3%磷酸溶液为流动相b,按表48中的规定进行梯度洗脱;检测波长为240nm。理论板数按莫诺苷峰计算应不低于3000。
表48流动相梯度洗脱程序
对照品溶液的制备取莫诺苷、马钱苷对照品适量,精密称定,加80%甲醇制成每1ml含莫诺苷40μg、马钱苷20μg的混合溶液,即得。
供试品溶液的制备取本品约1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得。
⑦不同批次对应实物(干膏粉)含量确定
按“含量测定方法的确定”项下供试品溶液的制备和测定法操作,测定15批(30份)对应实物(干膏粉)中莫诺苷、马钱苷含量,结果见表49。
表4915批(30份)对应实物(干膏粉)中莫诺苷和马钱苷含量测定结果
结论:15批(30份)对应实物(干膏粉)中莫诺苷和马钱苷含量为1.8189mg/g~6614mg/g,平均值为2.21mg/g,考虑到实际生产过程中的各种影响因素等,暂定本品按干燥品计算,每1g含山萸肉以莫诺苷(c17h26o11)和马钱苷(c17h26o10)总量计,应为1.5mg~3.0mg。
(2)五味子
照高效液相色谱法(《中国药典》2015年版四部通则0512)测定。
①仪器与试药
②色谱条件的选择
a流动相及流动相比例的选择
方法一:参照《中国药典》2015年版一部“五味子”项下[467]五味子醇甲含量测定方法进行摸索:
色谱条件及系统适用性试验以ultimatelpc18(4.6×250mm,5μl);以甲醇-水(65:35)为流动相;检测波长为250nm。
对照品溶液的配制取五味子醇甲对照品适量,精密称定,加甲醇制成每1ml含0.3mg的溶液,即得。
供试品溶液的制备取本品1g,精密称定,置具塞锥形瓶中,精密加入甲醇10ml,称定重量,超声处理20分钟(250w,40khz),取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液与供试品溶液各10μl,注人液相色谱仪,测定,记录色谱图,即得。
试验结果:阴性有干扰,方法不可行。
方法二:在方法一基础上,将流动相更换为乙腈-水(45:55),
试验结果:阴性无干扰,五味子醇甲峰型、峰分离度良好。
b检测波长的确定
取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含五味子醇甲20μg的溶液,在190~400nm波长下进行扫描,全波长扫描图见图22,可见五味子醇甲在252nm处有最大吸收峰且无杂质干扰,与《中国药典》2015年版一部“五味子”含量测定波长250nm基本一致,故选用250nm作为检测波长。
③供试品溶液制备方法考察
a提取溶媒的考察
取本品(批号:d-181120-01)约1.0g(共3组),精密称定,置具塞锥形瓶中,分别精密加入80%乙醇、80%甲醇、甲醇25ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,取出,放冷,再称定重量,分别用80%乙醇、80%甲醇、甲醇补足减失的重量,摇匀,滤过,取续滤液,即得,精密吸取续滤液10μl,注入液相色谱仪,进行测定。结果见表50。
表50提取溶媒考察结果
实验结果:不同提取溶媒五味子醇甲含量基本一致,为与莫诺苷和马钱苷含量测定共用一份供试品溶液,故选择80%甲醇作为提取溶媒。
b提取方式的考察取本品(批号:d-181120-01)约1g(共3组),精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,分别超声处理(功率250w,频率40khz)30分钟、回流提取30分钟、振摇提取30分钟,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得,精密吸取续滤液10μl,注入液相色谱仪,进行测定。结果见表51。
表51提取方式考察结果
实验结果:不同提取方式五味子醇甲含量基本一致,考虑操作方便,故选择超声作为提取方式。
c提取时间的考察
取本品(批号:d-181120-01)约1.0g(共3组),精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,分别超声处理(功率250w,频率40khz)15分钟、30分钟、45分钟,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得,精密吸取续滤液10μl,注入液相色谱仪,进行测定。结果见表52。
表52提取时间考察结果
结果表明,不同提取时间,五味子醇甲含量差异不大,考虑不同批次饮片含量存在差异,选择提取时间为30分钟。
④含量测定方法的初步确定
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(45:55)为流动相;检测波长为250nm。理论板数按五味子醇甲峰计算应不低于3000。对照品溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含20μg的溶液,即得。
供试品溶液的制备取本品约1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
⑤方法学考察
a专属性试验
五味子中含有五味子醇甲,为考察其它药味是否干扰五味子醇甲的测定,按处方比例称取饮片同法制成缺五味子阴性样品,并按照供试品的处理方法制备缺五味子的阴性样品溶液进样,记录色谱图。结果表明,阴性色谱中在与五味子醇甲相应的保留时间没有色谱峰,表明五味子以外的其他饮片对五味子醇甲的测定无干扰,以本法同时测定本品中五味子醇甲的含量具有专属性。
b峰纯度检查
取上述供试品溶液,注入液相色谱仪,采用上述色谱条件以pda检测器进行全波长检测,计算峰纯度,结果供试品色谱中五味子醇甲均未检测到杂质峰;最小峰纯度阈值为999.99,说明在该色谱条件下,五味子醇甲色谱峰纯度符合要求。
c线性关系的考察
精密称取五味子醇甲对照品9.96mg(批号:110857-201714,纯度:99.9%),置10ml量瓶中,加甲醇适量,超声处理使溶解,取出,放冷,加甲醇稀释至刻度,摇匀,即得0.9960mg/ml的五味子醇甲对照品溶液①;精密量取对照品溶液1ml,置5ml量瓶中,用甲醇稀释至刻度,摇匀,配制成浓度0.1992mg/ml的五味子醇甲对照品溶液②;精密量取对照品溶液①1ml置50ml量瓶中,用甲醇稀释至刻度,摇匀,配制成浓度19.92μg/ml的五味子醇甲对照品溶液③。分别精密吸取上述对照品溶液③2、5、10、15μl,对照品溶液②2、5、10μl注入液相色谱仪,照液相色谱法(《中国药典》2015年版四部通则0512)测定。以进样量(μg)为横坐标,峰面积为纵坐标,绘制标准曲线。见表53。
表53五味子醇甲线性关系
结果表明,五味子醇甲在0.03984μg~1.9920μg范围内线性良好。
d仪器精密度试验
取五味子醇甲对照品溶液,浓度为19.92μg/ml,连续进样6次,测定结果见表54。
表54仪器精密度试验结果(n=6)
试验结果:rsd<2.0%,仪器精密度良好。
e重复性试验
实验人员(a)操作,取本品(批号:d-181120-01)约1.0g(共6份),精密称定,按“方法的初步确定”项下供试品溶液制备和测定法进行操作。结果见表55。
表55重复性试验结果(n=6)
结果表明,rsd小于3.0%,说明本方法的重复性良好。
f稳定性试验
取本品(批号:d-181120-01)约1g,精密称定,按“方法的初步确定”项下供试品溶液制备和测定法进行操作,分别在制备后放置0、2、4、8、12、24、28、36、48小时进样测定,测定结果见表56。
表56稳定性试验结果
结果表明,rsd<3.0%,供试品溶液在48小时内稳定性良好。
g准确度试验
精密称取五味子醇甲10.37mg(批号:110857-201714,纯度:99.9%)置20ml量瓶中,加80%甲醇溶液溶解并稀释至刻度,摇匀,即得浓度为0.5185mg/ml对照品溶液①;精密量取对照品溶液①1ml至100ml的量瓶中,加80%甲醇溶液稀释至刻度,摇匀,即得浓度为5.19μg/ml对照品溶液②;精密量取对照品溶液①2ml至100ml的量瓶中,加80%甲醇溶液稀释至刻度,摇匀,即得浓度为10.37μg/ml对照品溶液③;精密量取对照品溶液①3ml至100ml的量瓶中,加80%甲醇溶液稀释至刻度,摇匀,即得浓度为15.56μg/ml对照品溶液④
取本品约0.5g(批号:d-181120-01)(共9份),精密称定,置具塞锥形瓶中,分别精密加入对照品溶液②、③、④各25ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,精密吸取供试品溶液10μl,注入液相色谱仪,测定,即得,结果见表57。
地黄饮子物质基准(批号:d-181120-01)中:五味子醇甲苷含量为0.4937mg/g。
表57五味子醇甲准确度试验结果(n=9)
结果表明,五味子醇甲平均回收率为98.44%,rsd≤3.0%,表明本方法准确度较好。
h中间精密度试验
选择不同的测定时间、不同的高效液相色谱仪、不同的实验人员(b),取本品(批号:d-181120-01)约1.0g,精密称定,按“方法的初步确定”项下供试品溶液制备和定法进行操作。结果见表58。
表58五味子醇甲中间精密度试验结果(n=6)
结果,a和b测定的样品中五味子醇甲平均含量为0.4888mg/g,rsd<2.0%,表明本方法中间精密度良好。
j耐用性试验
考察不同流速、不同色谱柱、不同柱温、不同流动相比例对本色谱条件的耐用性,结果见表59。
表59五味子醇甲耐用性试验结果
结果表明,各个条件下测定结果基本一致,rsd<5.0%,色谱图中五味子醇甲峰型尖锐,对称,分离度良好,表明本方法不同流速、不同色谱柱、不同柱温、不同流动相比例耐用性良好。
以上的研究结果表明,地黄饮子物质基准对应实物(干膏粉)中五味子醇甲的含量测定方法具有专属性,稳定性、重复性、中间精密度、准确度等均符合规定,且色谱条件的耐用性较好,故此液相色谱条件可用于地黄饮子物质基准对应实物(干膏粉)中五味子醇甲的含量测定。
⑥含量测定方法的确定
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(45:55)为流动相;检测波长为250nm。理论板数按五味子醇甲峰计算应不低于3000。
对照品溶液的制备取五味子醇甲对照品适量,精密称定,加80%甲醇制成每1ml含20μg的溶液,即得。
供试品溶液的制备取本品约1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
⑦不同批次对应实物(干膏粉)含量确定
按“含量测定方法的确定”项下供试品溶液的制备和测定法操作,测定15批(30份)对应实物(干膏粉)中五味子醇甲含量,结果见表60。
表6015批(30份)对应实物(干膏粉)中五味子醇甲含量测定结果
结论:15批(30份)对应实物(干膏粉)中五味子醇甲含量为0.5307mg/g~0.6776mg/g,平均值为0.58mg/g,考虑到实际生产过程中的各种影响因素等,暂定本品按干燥品计算,每1g含五味子以五味子醇甲(c24h32o7)计,应为0.4mg~0.8mg。
k对应实物的质量分析
15批物质基准对应实物结果汇总
对15批物质基准主要检测结果汇总表61。
表6115批物质基准对应实物数据汇总
对应实物关键质量属性范围的确定
表62物质基准对应实物关键质量属性范围
(1)指标成分的含量
15批(30份)对应实物(干膏粉)莫诺苷和马钱苷含量为为1.8189mg/g~2.6614mg/g,平均值为2.21mg/g;五味子醇甲含量为0.5307mg/g~0.6776mg/g,平均值为0.58mg/g,考虑到实际生产过程中的各种影响因素等,暂定本品按干燥品计,每1g山萸肉以莫诺苷(c17h26o11)和马钱苷(c17h26o10)总量计,应为1.5mg~3.0mg;每1g含五味子以五味子醇甲(c24h32o7)计,应为0.4mg~0.8mg。
(2)指纹图谱
采用15批(30份)对应实物(干膏粉)的色谱图,生成了对照指纹图谱(见图23)。供试品指纹图谱中应呈现与参照物色谱峰保留时间相同的色谱峰,按中药色谱指纹图谱相似度评价系统计算,以共有峰计算相似度,供试品指纹图谱与对照指纹图谱的相似度不得低于0.90。
本发明通过实验得出:15批(30份)对应实物(干膏粉)莫诺苷和马钱苷含量为为1.8189mg/g~2.6614mg/g,平均值为2.21mg/g;五味子醇甲含量为0.5307mg/g~0.6776mg/g,平均值为0.58mg/g,考虑到实际生产过程中的各种影响因素等,暂定本品按干燥品计,每1g山萸肉以莫诺苷(c17h26o11)和马钱苷(c17h26o10)总量计,应为1.5mg~3.0mg;每1g含五味子以五味子醇甲(c24h32o7)计,应为0.4mg~0.8mg。
虽然,上文中已经用一般性说明、具体实施方式及试验,对本发明作了详尽的描述,但在本发明基础上,可以对之作出一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!