一种基于测序数据进行SSR分型的方法及系统与流程
本发明涉及基因测序技术领域,尤其涉及一种基于测序数据进行ssr分型的方法及系统。
背景技术:
目前市场上通用的基因测序软件主要集中在分析毛细管电泳的结果或者分析fasta格式(一种基于文本用于表示核酸序列或多肽序列的格式)的碱基序列的简单重复序列(simplesequencerepeats,简称ssr)分布,虽然设计有图形化界面,但是操作过程繁琐并且输入文件的数据类型有限制,自定义效果差。同时不适用于在大型服务器上,对于高通量、多位点数据的分析。此外,ssr分析的很重要一个目的就是要与以后的数据库进行比较,或者作为新的分型录入数据库,而目前没有能实现相关功能的系统或者软件。
现有的主流基因测序工具主要包括genemapper和genemarker。
genemapper是由abi公司开发的与荧光毛细管电泳仪捆绑销售的dna片段分析软件。该软件具有多种应用功能,包括扩增片段长度多态性、杂合性缺失、微卫星和单核苷酸多态性(singlenucleotidepolymorphism,snp)基因分型分析。可实现基于毛细管电泳数据结果,对样品ssr进行分型。genemapper以客户端形式发布,需要捆绑特定的荧光毛细管电泳仪,导致其可移植性差,同时复杂的操作界面和处理逻辑导致软件使用困难以及自定义效果不佳。在分析策略上,genemapper需要基于已有参考序列或者数据进行比对分型,无法实现从头分型。其分析对象以人类及马、牛、羊等动物为主,针对法庭科学和司法鉴定等应用需求,衍生出了人类dna鉴定专用定制版本。常见的动物拥有较全的微卫星指纹库,并且ssr标记成熟,尤其是对于人类的样品检测。但是对于其它非常规物种,比如玉米、水稻等农作物的检测能力不足。
genemarker是由softgenetics研发的,在国际生物信息学相关研究分析领域应用十分广泛的dna片断分析软件,旨在为领域科研人员、司法刑侦人员提供一个精确、快速、友好、自动化的数据分析平台。genemarker是dna片段数据分析领域的一个整体解决方案,集成了短重复序列(shorttandemrepeat,str)分析,遗传家系处理、峰图定量分析等十几个功能强大的分析应用模块。genemarker是一个开放的分析平台,可以支持多家厂商的毛细管电泳仪输出的dna片断峰图数据,并且支持市面上大部分主流试剂盒和特殊试剂。
基于上述两种软件工具,可以得出如下不足:
genemapper以客户端形式发布,需要捆绑特定的荧光毛细管电泳仪,导致其可移植性差,同时复杂的操作界面和处理逻辑导致软件使用困难以及自定义效果不佳。在分析策略上,genemapper需要基于已有参考序列或者数据进行比对分型,无法实现从头分型。其分析对象以人类及马、牛、羊等动物为主,针对法庭科学和司法鉴定等应用需求,衍生出了人类dna鉴定专用定制版本。常见的动物拥有较全的微卫星指纹库,并且ssr标记成熟,尤其是对于人类的样品检测。但是对于其它非常规物种,比如玉米、水稻等农作物的检测能力不足。此外需要配备专用的试剂盒,价格昂贵,切供货慢。软件开发较早,且版本更新慢,对于最新的电脑操作系统存在不兼容问题。
genemarker虽然兼容不同厂商的数据结果和市面上大多数主流试剂盒,但是其复杂的操作过程和较长的运行时间,让使用者的工作效率大打折扣。同时,面对日益增加的数据分析量,局限于客户端形式的软件无法胜任快速、便捷的分析。另外,genemarker同样需要基于已有参考序列或者数据进行比对分型,无法实现从头分型。与genemapper相似,genemarker同样集中被使用于动物样品,尤其是人类样品的分析,而植物样品以及其它非常规物种,比如玉米、水稻等农作物的检测能力不足。软件开发较早,且版本更新慢,对于最新的电脑操作系统存在不兼容问题。
因此,高通量,高速度,自定义灵活并且对接数据库的分析系统是目前急需开发的。
技术实现要素:
本发明实施例提供一种基于测序数据进行ssr分型的方法及系统,用以解决现有技术中基因分析软件可移植性差,自定义效果不佳和分型处理速度慢的缺陷。
第一方面,本发明实施例提供一种基于测序数据进行ssr分型的方法,包括:
获取待检测的基因序列,及所述基因序列每个检测位点的参考序列,生成配置文件;
将测序得到的测序序列回贴至所述参考序列上,使得所述配置文件转换成按照位点排序的预设格式文件;
对所述预设格式文件按照所述每个检测位点进行拆分,提取所述每个检测位点的测序片段以及所述测序片段对应的比对信息,基于所述测序片段和所述比对信息得到每一条序列对应的ssr分型。
优选的,该方法还包括:
根据不同的所述ssr分型,将所述测序片段进行分类,提取频率最高的序列作为所述ssr分型的高频序列,并对所述高频序列进行变异检测,得到所述高频序列对应的变异位点。
优选的,所述获取待检测的基因序列,及所述基因序列每个检测位点的参考序列,具体包括:
获取所述每个检测位点的参考序列及对应的模体;
设定所述模体的重复次数,根据所述重复次数生成所述参考序列;
对所述参考序列进行索引处理,生成所述配置文件,以供所述基因序列进行下游分析。
优选的,所述将测序得到的测序序列回贴至所述参考序列上,使得所述配置文件转换成按照位点排序的预设格式文件,具体包括:
所述将测序得到的测序序列回贴至所述参考序列上包括序列比对和ssr区域重排;
所述序列比对由预设工具完成,兼容单末端和双末端两种测序方式;
所述ssr区域重排包括将ssr区域的序列进行重排;
基于所述序列比对和所述ssr区域重排,将所述配置文件转换成按照位点排序的所述预设格式文件。
优选的,所述对所述预设格式文件按照所述每个监测位点进行拆分,提取所述每个检测位点的测序片段以及所述测序片段对应的比对信息,基于所述测序片段和所述比对信息得到每一条序列对应的ssr分型,具体包括:
对所述预设格式文件按照所述每个检测位点进行拆分;
提取每个所述测序片段以及所述比对信息;
计算所述比对信息对应序列的起始位置以及比对碱基的长度,得到ssr区域的序列变化结果;
根据所述序列变化结果,分析得到所述ssr分型。
优选的,所述预设格式文件包括头文件和比对信息;其中,所述比对信息包括11个域。
优选的,所述对所述高频序列进行变异检测,包括:
利用gatk工具包中的unifiedgenotyper工具进行所述变异检测。
优选的,所述对所述预设格式文件按照所述每个监测位点进行拆分,提取所述每个检测位点的测序片段以及所述测序片段对应的比对信息,基于所述测序片段和所述比对信息得到每一条序列对应的ssr分型,还包括:
所述每个检测位点对应若干个模体。
第二方面,本发明实施例提供一种基于测序数据进行ssr分型的系统,包括:
获取模块,用于获取待检测的基因序列,及所述基因序列每个检测位点的参考序列,生成配置文件;
测序模块,用于将测序得到的测序序列回贴至所述参考序列上,使得所述配置文件转换成按照位点排序的预设格式文件;
分型模块,用于对所述预设格式文件按照所述每个检测位点进行拆分,提取所述每个检测位点的测序片段以及所述测序片段对应的比对信息,基于所述测序片段和所述比对信息得到每一条序列对应的ssr分型。
第三方面,本发明实施例提供一种电子设备,包括:
存储器、处理器及存储在存储器上并可在处理器上运行的计算机程序,所述处理器执行所述程序时实现任一项所述一种基于测序数据进行ssr分型的方法的步骤。
本发明实施例提供的一种基于测序数据进行ssr分型的方法及系统,通过基于测序数据实现待检测基因序列的ssr分型,实现了快速对多位点、多样品的高通量测序检测,很好地兼容各物种的ssr分型需求,可自定义输出文件格式,大大提高了分型效率。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为本发明实施例提供的一种基于测序数据进行ssr分型的方法流程图;
图2为本发明实施例提供的侧翼变异位点检测示意图;
图3为本发明实施例提供的参考序列生成示意图;
图4为本发明实施例提供的序列回贴示意图;
图5为本发明实施例提供的ssr分型示意图;
图6为本发明实施例提供的ssr分型中比对信息的域构成示意图;
图7为本发明实施例提供的ssr分型中序列比对结果示意图;
图8为本发明实施例提供的一种基于测序数据进行ssr分型的系统结构图;
图9为本发明实施例提供的电子设备的结构框图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
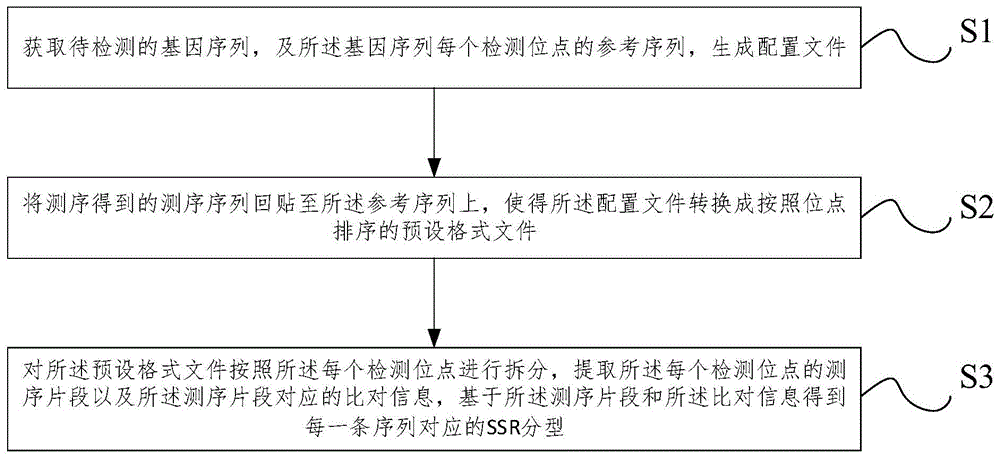
图1为本发明实施例提供的一种基于测序数据进行ssr分型的方法流程图,如图1所示,包括:
s1,获取待检测的基因序列,及所述基因序列每个检测位点的参考序列,生成配置文件;
s2,将测序得到的测序序列回贴至所述参考序列上,使得所述配置文件转换成按照位点排序的预设格式文件;
s3,对所述预设格式文件按照所述每个检测位点进行拆分,提取所述每个检测位点的测序片段以及所述测序片段对应的比对信息,基于所述测序片段和所述比对信息得到每一条序列对应的ssr分型。
具体地,在步骤s1中,首先获取要检测的原始基因序列,例如玉米、水稻等农作物的基因序列,同时还获取该基因序列中每个检测位点的参考序列,进一步生成配置文件,用于基因序列的下游分析;在步骤s2中,进一步进行序列回贴的操作,具体为将测序得到的序列回贴到参考序列上,使得生成的配置文件转换成按照位点排序的预设格式文件,这里预设格式文件为bam文件;在步骤s3中,对bam文件再按照每个检测位点进行拆分,提取出每个检测位点的测序片段(reads)以及对应的比对信息,最终得到每一条序列对应的ssr分型结果。
本发明实施例通过基于测序数据实现待检测基因序列的ssr分型,实现了快速对多位点、多样品的高通量测序检测,很好地兼容各物种的ssr分型需求,可自定义输出文件格式,大大提高了分型效率。
基于上述实施例的内容,作为一种可选实施例,该方法还包括:
根据不同的所述ssr分型,将所述测序片段进行分类,提取频率最高的序列作为所述ssr分型的高频序列,并对所述高频序列进行变异检测,得到所述高频序列对应的变异位点。
其中,所述对所述高频序列进行变异检测,包括:
利用gatk工具包中的unifiedgenotyper工具进行所述变异检测。
具体地,图2为本发明实施例提供的侧翼变异位点检测示意图,如图2所示,根据不同的ssr分型,将每个位点的reads进行分类,并从中提取频率最高的序列作为该分型的高频序列(hfsequence,high-frequentsequence),再利用gatk(genomeanalysistoolkit)工具包中的unifiedgenotyper进行变异检测,从而报告每个位点的高频序列上的变异位点,这里unifiedgenotyper是gatk中一个主要工具,用于variantcalling,能对单个或多个样本进行snp和indel(插入缺失标记)calling,使用beyesiangenotypelikelihoodmodel来对n个sample进行基因型的判断和等位基因频率的计算。
本发明实施例通过对侧翼变异位点进行检测,非常清晰、准确地展现样品的分型结果,有助于位点的筛选,同时若侧翼保守性不好,就考虑舍弃对应的位点,实现了有效地发现高频序列上的变异位点。
基于上述实施例的内容,作为一种可选实施例,该方法中步骤s1具体包括:
获取所述每个检测位点的参考序列及对应的模体;
设定所述模体的重复次数,根据所述重复次数生成所述参考序列;
对所述参考序列进行索引处理,生成所述配置文件,以供所述基因序列进行下游分析。
具体地,图3为本发明实施例提供的参考序列生成示意图,如图3所示,首先获取待检测的基因序列的每个检测位点的参考序列以及对应的模体(motif),ref-preparation模块自动根据设定的motif重复次数生成对应的参考序列文件,同时进行索引(index)处理,以及生成相关的配置文件,例如intervals,bed文件等,用于生成含有固定重复次数ssr区域的参考序列用于下游的比对分析。
本发明实施例通过对原始基因序列的位点进行检测,并根据设置的模体重复次数自动生成对应的参考序列文件,实现从头分型的策略,不依赖已有ssr分型结果,从而可以很好的兼容各物种的ssr分型需求。
基于上述实施例的内容,作为一种可选实施例,该方法中步骤s2具体包括:
所述将测序得到的测序序列回贴至所述参考序列上包括序列比对和ssr区域重排;
所述序列比对由预设工具完成,兼容单末端和双末端两种测序方式;
所述ssr区域重排包括将ssr区域的序列进行重排;
基于所述序列比对和所述ssr区域重排,将所述配置文件转换成按照位点排序的所述预设格式文件。
具体地,图4为本发明实施例提供的序列回贴示意图,如图4所示,序列回贴是指将测序得到的序列回贴到由ref-preparation模块生成的参考序列上的过程,主要分为序列比对(mapping)和ssr区域重排(localrealignment)两个步骤。序列比对主要由预设工具完成,主要是由bwamem工具来完成,bwamem工具是一种常用的比对工具,可以兼容单末端和双末端两种测序方式,ssr区域重排是将ssr区域的序列进行重排,以保证排序正确,最终生成按照位点排序的预设格式文件,即bam文件。
本发明实施例通过对参考序列进行序列回贴操作,由比对工具完成序列比对,根据需求自定义输出文件格式,方便基因序列的下游分析使用。
基于上述实施例的内容,作为一种可选实施例,该方法中步骤s3具体包括:
对所述预设格式文件按照所述每个检测位点进行拆分;
提取每个所述测序片段以及所述比对信息;
计算所述比对信息对应序列的起始位置以及比对碱基的长度,得到ssr区域的序列变化结果;
根据所述序列变化结果,分析得到所述ssr分型。
其中,所述预设格式文件包括头文件和比对信息;其中,所述比对信息包括11个域。
具体地,图5为本发明实施例提供的ssr分型示意图,如图5所示,
通过对预设格式文件,即bam文件按照检测位点进行拆分后,预设格式文件主要由头文件和比对信息构成,bam文件是sam文件的二进制格式,通过文件的压缩实现减小文件体积,便于存储的目的。sam/bam文件的比对信息部分主要有11个域构成,如图6所示,cigar域包含有每条reads的简要比对信息,由数字、字母间隔组成,其中“m”表示和参考序列匹配序列的长度,“i”和“d”分别表示与参考序列相比插入和缺失的序列长度,“s”表示reads中由于无法比对被省略的序列长度,例如在图6中列出了read1的11个域的值,cigar域的值为“48m13s”表示。
通过对sam/bam文件按照检测位点进行拆分后,可以提取每个检测位点的reads以及对应的比对信息(特别是cigar域)。通过计算序列对比的起始位置(pos域)以及cigar域中比对碱基的长度,找到ssr区域的序列变化情况,从而分析出每一条read的ssr的分型。例如在图7中query序列的cigar域的值为“15m10d15m”,表示与参考序列相比有10bp的缺失,并且左右两侧有15bp的序列匹配。参考序列的ssr类型为ssr15(ssr区域的长度为15bp,motif为tgctg,重复次数为3次)。query序列与ref序列相比在ssr区域存在10bp的碱基缺失,由此可知query序列的ssr类型为ssr5。
本发明实施例通过实现快速对多位点、多样品的高通量测序数据的ssr分型,操作简单,可以实现多样品、多位点同时分型,大大提高分型效率。
在上述实施例的基础上,所述对所述预设格式文件按照所述每个监测位点进行拆分,提取所述每个检测位点的测序片段以及所述测序片段对应的比对信息,基于所述测序片段和所述比对信息得到每一条序列对应的ssr分型,还包括:
所述每个检测位点对应若干个模体。
具体地,对于现有的ssr分型技术,一般只能处理一个检测位点,对应一个motif的场景,本发明实施例的ssr分型技术,不仅能实现一个检测位点对应一个motif的场景,还能实现一个检测位点对应多个motif的场景,即一个位点能分型出多种ssr。
本发明实施例通过处理一个检测位点包含多个模体的ssr分型,能有效处理复杂的检测位点情形,具有更好的分型效果。
图8为本发明实施例提供的一种基于测序数据进行ssr分型的系统结构图,如图8所示,包括:获取模块20、测序模块21和分型模块22;其中:
获取模块20用于获取待检测的基因序列,及所述基因序列每个检测位点的参考序列,生成配置文件;测序模块21用于将测序得到的测序序列回贴至所述参考序列上,使得所述配置文件转换成按照位点排序的预设格式文件;分型模块22用于对所述预设格式文件按照所述每个检测位点进行拆分,提取所述每个检测位点的测序片段以及所述测序片段对应的比对信息,基于所述测序片段和所述比对信息得到每一条序列对应的ssr分型。
本发明实施例提供的系统用于执行上述对应的方法,其具体的实施方式与方法的实施方式一致,涉及的算法流程与对应的方法算法流程相同,此处不再赘述。
本发明实施例通过基于测序数据实现待检测基因序列的ssr分型,实现了快速对多位点、多样品的高通量测序检测,很好地兼容各物种的ssr分型需求,可自定义输出文件格式,大大提高了分型效率。
进一步地,该系统还包括变异检测模块23,变异检测模块23用于根据不同的所述ssr分型,将所述测序片段进行分类,提取频率最高的序列作为所述ssr分型的高频序列,并对所述高频序列进行变异检测,得到所述高频序列对应的变异位点。
本发明实施例通过对侧翼变异位点进行检测,非常清晰、准确地展现样品的分型结果,有助于位点的筛选,同时若侧翼保守性不好,就考虑舍弃对应的位点,实现了有效地发现高频序列上的变异位点。
其中,所述对所述高频序列进行变异检测,包括:
利用gatk工具包中的unifiedgenotyper工具进行所述变异检测。
进一步地,获取模块20具体用于获取所述每个检测位点的参考序列及对应的模体;设定所述模体的重复次数,根据所述重复次数生成所述参考序列;对所述参考序列进行索引处理,生成所述配置文件,以供所述基因序列进行下游分析。
本发明实施例通过对原始基因序列的位点进行检测,并根据设置的模体重复次数自动生成对应的参考序列文件,实现从头分型的策略,不依赖已有ssr分型结果,从而可以很好的兼容各物种的ssr分型需求。
进一步地,测序模块21具体用于所述将测序得到的测序序列回贴至所述参考序列上包括序列比对和ssr区域重排;所述序列比对由预设工具完成,兼容单末端和双末端两种测序方式;所述ssr区域重排包括将ssr区域的序列进行重排;基于所述序列比对和所述ssr区域重排,将所述配置文件转换成按照位点排序的所述预设格式文件。
本发明实施例通过对参考序列进行序列回贴操作,由比对工具完成序列比对,根据需求自定义输出文件格式,方便基因序列的下游分析使用。
进一步地,分型模块22具体用于对所述预设格式文件按照所述每个检测位点进行拆分;提取每个所述测序片段以及所述比对信息;计算所述比对信息对应序列的起始位置以及比对碱基的长度,得到ssr区域的序列变化结果;根据所述序列变化结果,分析得到所述ssr分型。
其中,所述预设格式文件包括头文件和比对信息;其中,所述比对信息包括11个域。
本发明实施例通过实现快速对多位点、多样品的高通量测序数据的ssr分型,操作简单,可以实现多样品、多位点同时分型,大大提高分型效率。
图9示例了一种电子设备的实体结构示意图,如图9所示,该电子设备可以包括:处理器(processor)910、通信接口(communicationsinterface)920、存储器(memory)930和通信总线940,其中,处理器910,通信接口920,存储器930通过通信总线940完成相互间的通信。处理器910可以调用存储器930中的逻辑指令,以执行如下方法:获取待检测的基因序列,及所述基因序列每个检测位点的参考序列,生成配置文件;将测序得到的测序序列回贴至所述参考序列上,使得所述配置文件转换成按照位点排序的预设格式文件;对所述预设格式文件按照所述每个检测位点进行拆分,提取所述每个检测位点的测序片段以及所述测序片段对应的比对信息,基于所述测序片段和所述比对信息得到每一条序列对应的ssr分型。
此外,上述的存储器930中的逻辑指令可以通过软件功能单元的形式实现并作为独立的产品销售或使用时,可以存储在一个计算机可读取存储介质中。基于这样的理解,本发明的技术方案本质上或者说对现有技术做出贡献的部分或者该技术方案的部分可以以软件产品的形式体现出来,该计算机软件产品存储在一个存储介质中,包括若干指令用以使得一台计算机设备(可以是个人计算机,服务器,或者网络设备等)执行本发明各个实施例所述方法的全部或部分步骤。而前述的存储介质包括:u盘、移动硬盘、只读存储器(rom,read-onlymemory)、随机存取存储器(ram,randomaccessmemory)、磁碟或者光盘等各种可以存储程序代码的介质。
以上所描述的装置实施例仅仅是示意性的,其中所述作为分离部件说明的单元可以是或者也可以不是物理上分开的,作为单元显示的部件可以是或者也可以不是物理单元,即可以位于一个地方,或者也可以分布到多个网络单元上。可以根据实际的需要选择其中的部分或者全部模块来实现本实施例方案的目的。本领域普通技术人员在不付出创造性的劳动的情况下,即可以理解并实施。
通过以上的实施方式的描述,本领域的技术人员可以清楚地了解到各实施方式可借助软件加必需的通用硬件平台的方式来实现,当然也可以通过硬件。基于这样的理解,上述技术方案本质上或者说对现有技术做出贡献的部分可以以软件产品的形式体现出来,该计算机软件产品可以存储在计算机可读存储介质中,如rom/ram、磁碟、光盘等,包括若干指令用以使得一台计算机设备(可以是个人计算机,服务器,或者网络设备等)执行各个实施例或者实施例的某些部分所述的方法。
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!