一种口服姜黄素-纳米金刚石复合物及其制备方法与流程
技术领域:
本发明属于药物制剂领域,涉及口服姜黄素-纳米金刚石复合物及其制备方法。
背景技术:
:
纳米金刚石(nanodiamonds,nds)是一种截断八面体结构的碳材料,单颗粒粒径为2-8nm。得益于nds尺寸小、比表面积大、化学性质稳定、生物相容性好、表面易修饰等诸多优点,使其成为一种极具应用前景前途的新型递药载体。nds在水溶液中会自发形成纳米尺寸的团簇,加之其表面富含大量含氧基团,如羟基、羧基、内酯等,药物可以通过物理吸附或者共价连接等方式结合到nds表面或嵌入其团簇中。众多证据显示,以nds作为药物载体,不仅能够增强药物的细胞摄取,延长体内滞留时间,还能提高药物的生物利用度和用药安全性。
虽然nds的聚集成簇有利于载药,但是过度聚集会降低制剂的胶体稳定性,阻碍载体的胃肠道转运,并引发一定的细胞毒性。虽然在溶剂中加入一定量表面活性剂后,通过超声、机械研磨等方式可以实现nds的再分散,但是在制剂的放置过程中,以及在体内复杂的生理环境中nds仍有重新聚集的风险。相对而言,表面化学修饰对提高nds分散稳定性更为行之有效。
聚乙二醇维生素e琥珀酸酯(d-α-tocopherylpolyethyleneglycolsuccinate,tpgs)是维生素e的水溶性衍生物,其两亲性和较大的分子表面积使之成为出色的非离子型表面活性剂,临界胶束浓度仅为0.02%。tpgs极性的peg头部和非极性tos尾部均具有庞大的表面积,这使其对油水两相的作用力都更强,既可溶于水又可溶于大多数有机溶剂。tpgs不仅能够提高药物的溶解度,还能够改善药物的化学稳定性。
姜黄素(curcumin,cur)是从姜科属植物根茎中提取得到的多酚类物质,其来源广泛,毒性低,具有抗炎、抗氧化、抗肿瘤等多种药理活性。然而,cur几乎不溶于水(在ph5.0缓冲液中溶解度仅为11ng/ml),在中性、碱性及光照条件下降解迅速,口服后大部分通过粪便排出体外,极少部分经胃肠道吸收后在肝脏和血液内快速代谢,首过效应明显,口服绝对生物利用度仅1%,属于生物药剂学分类系统(bcs)中的ⅳ类药物。如何有效提高cur的口服生物利用度是药剂学界亟待解决的关键科学问题。
因此,本发明首先将tpgs通过共价连接的方式接枝于nds表面,以解决nds的胶体分散性和分散稳定性问题,进而将cur以无定型状态包覆于nds-tpgs纳米团簇中,得到一种cur@nds-tpgs纳米复合物,从而极大地提升cur的溶解性、胃肠道粘附性、透过性和口服生物利用度,从而解决cur的成药性问题。
技术实现要素:
:
本发明的第一个目的在于提供一种nds-tpgs合成方法,通过合成条件的优选,使产物具有较高tpgs接枝率、较小的粒径和良好的分散性,从而得到一种具有广阔应用前景的递药载体。
本发明的第二个目的在于提供一种cur@nds-tpgs纳米复合物的处方及制备方法,通过对处方工艺的优选,将制剂的载药效率、粒径和粒度分布等控制在合理范围内,以提高cur的溶解性和口服生物利用度,解决cur的成药性问题。
本发明是通过如下技术方案实现的:
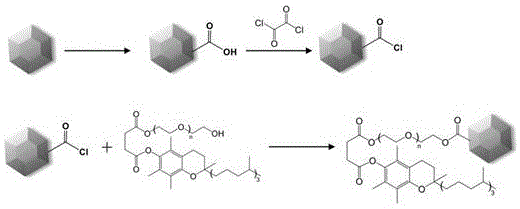
对nds先后进行羧基化和酰氯化处理,进而将酰氯化nds与tpgs反应,合成nds-tpgs纳米药物载体;将cur包载于nds-tpgs形成的纳米笼中,得到cur@nds-tpgs纳米复合物。
所述的nds-tpgs合成方法如下:
(1)将nds在一定温度下加入一定体积混酸中,搅拌一定时间,冷却至室温,水洗离心;以naoh和hcl调节ph值至5~6;水洗,真空干燥,得到nds-cooh。
(2)将nds-cooh超声分散于草酰氯中,然后加入二甲基甲酰胺,一定温度下搅拌反应一定时间,离心后将nds-cocl用无水四氢呋喃洗涤,真空干燥,得到nds-cocl。
(3)将nds-cocl超声分散于二甲基甲酰胺中,然后加入tpgs于一定温度下反应一定时间,将冷却后的反应液水洗后冷冻干燥,得到nds-tpgs。
步骤(1)所述的混酸为h2so4-hno3、h2so4-hclo4和h2so4-hcl中的一种,体积比为5:1~1:2,优选范围为5:1~2:1。
步骤(1)所述nds在混酸中的反应温度为25~100℃。
步骤(1)所述nds在混酸中的浓度为5~55mg/ml,优选浓度范围为5~40mg/ml。
步骤(1)所述nds在混酸中的反应时长为12~48h。
步骤(2)所述nds-cooh在草酰氯中的浓度为2~16mg/ml,优选浓度范围为2~8mg/ml。
步骤(2)所述二甲基甲酰胺的浓度为0.2~1mg/ml。
步骤(2)所述反应时长为6~48h。
步骤(2)所述反应温度为25~75℃,优选温度为40~75℃。
步骤(3)所述nds-cocl在二甲基甲酰胺中的浓度为0.1~10mg/ml,优选范围为0.1~5mg/ml。
步骤(3)所述tpgs在二甲基甲酰胺中的浓度为1~20mg/ml,优选范围为3~20mg/ml。
步骤(3)所述tpgs的种类为tpgs400、tpgs1000、tpgs2000、tpgs4000和tpgs6000中的一种。
步骤(3)所述反应时长为12~72h。
步骤(3)所述反应温度为40~120℃。
以接枝率、粒径和粒度分布为评价指标进行综合优化筛选,当步骤(1)nds浓度为5~30mg/ml;且步骤(2)nds-cooh浓度为2~8mg/ml;步骤(3)tpgs浓度为2~10mg/ml时,均可得到接枝率大于45%,粒径小于250nm,粒度分布小于0.25的优良纳米载体。
所述的cur@nds-tpgs纳米复合物制备方法如下:
(1)将一定量nds-tpgs加入适宜溶剂中,于一定温度下以一定功率探头超声分散一定时间。
(2)用适宜溶剂溶解处方量cur。
(3)将cur溶液滴入nds-tpgs悬液中,继续探头超声(功率与步骤(1)一致),减压干燥除去有机溶剂得cur@nds-tpgs。
(4)向cur@nds-tpgs中加入蒸馏水,探头超声进行再分散,即得。
步骤(1)所述nds-tpgs的浓度为1~30mg/ml,优选浓度范围为5~30。
步骤(1)所述nds-tpgs为nds-tpgs400、nds-tpgs1000、nds-tpgs2000、nds-tpgs4000中的一种。
步骤(1)所述分散溶剂为乙醇、50%乙醇、75%乙醇、二甲基亚砜、二甲基甲酰胺、丙酮、二氯甲烷中的一种。
步骤(1)所述超声功率为120~600w,优选超声功率为240~600w。
步骤(1)所述超声时长为5~60min,优选超声时长为15~60min。
步骤(1)所述的温度为0~50℃。
步骤(2)所述cur的浓度为0.5~3mg/ml,优选浓度范围为0.5~2mg/ml。
步骤(2)所述溶剂为乙醇、二甲基亚砜、二甲基甲酰胺、丙酮、二氯甲烷中的一种。
以载药效率、粒径和粒度分布为评价指标,对制剂处方进行综合优化,当步骤(1)nds-tpgs的浓度为5~20mg/ml,nds-tpgs为nds-tpgs400、nds-tpgs1000或者nds-tpgs2000中的一种;且步骤(2)cur浓度为0.5~2mg/ml时,所得制剂载药效率大于50%,粒径小于350nm,粒度分布小于0.3。
本发明具有以下有益效果:
1、本发明所合成的nds-tpgs能够显著改善nds的分散性和分散稳定性;与常规物理包覆方法相比,将tpgs通过共价连接法修饰nds能够进一步提高载体的体内外稳定性和递药效率。
2、与裸nds-cooh(cur@nds-cooh)以及tpgs物理包覆nds(cur@nds-cooh/tpgs)体系相比,nds-tpgs包载cur以后,递药系统的粒径减小,粒度分布更为均一,载药效率提高,肠道细胞摄取量明显增加,制剂在大鼠胃肠道的分布、滞留性和透过性进一步改善,口服生物利用度显著提高,并具备良好的缓释效果。
附图说明:
图1nds-tpgs合成路线图
图2cur@nds-tpgs透射电镜图和结构示意图
图3为按照实施例1制备cur@nds-tpgs的x射线衍射图
图4nds-tpgs的肠道滞留性研究(小动物活体成像图)(a)香豆素6(cou-6)溶液,(b)cou-6@nds-cooh/tpgs,(c)cou-6@nds-tpgs
图5为cur混悬液、cur@nds-cooh/tpgs和cur@nds-tpgs大鼠灌胃给药后的药时曲线图,给药剂量为75mg/kg。
具体实施方式
下面结合实例对本发明作进一步详细说明,但本发明的范围并不受这些实例的限制。
实施例1nds-tpgs合成条件的筛选
(1)将nds在一定温度下加入一定体积的混酸中,搅拌一定时间,冷却至室温,以蒸馏水稀释后离心;以0.1mnaoh和0.1mhcl调节ph值至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
(2)将100mgnds-cooh超声分散于25ml的草酰氯中,然后加入1ml二甲基甲酰胺,一定温度下搅拌反应24小时,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
(3)将10mgnds-cocl超声分散于10ml二甲基甲酰胺中,然后加入50mgtpgs1000于100℃下反应48小时,将冷却后的反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs。
合成结束后,以岛津tga-50热重分析仪分别检测nds-cooh、tpgs、和nds-tpgs在25~550℃温度范围内的失重情况,并计算接枝率;称取0.05gnds-tpgs,冰浴超声30min(超声功率300w),分散于10ml蒸馏水中,使用malvernzetasizer粒径仪测定粒径和粒度分布。以接枝率大于50%,粒径小于250nm,粒度分布小于0.25为最佳合成条件;以接枝率30~50%,粒径250~300nm,粒度分布0.25~0.3为次佳合成条件;以接枝率小于30%,粒径大于300nm,粒度分布大于0.3为一般合成条件筛选合成工艺。结果可见,nds在混酸中的浓度范围为5~25mg/ml时合成效果最佳,25~40mg/ml时次佳,40~55mg/ml时效果一般,55~70mg/ml时效果较差;当混酸为h2so4-hno3和h2so4-hcl时,合成效果最佳,h2so4-hclo4效果次之;当混酸比例为5:1~3:1时合成效果最佳,比例为3:1~2:1时次佳,2:1~1:2时效果一般;在混酸中的反应时间为12~48h时,效果均最佳;反应温度为40~100℃时效果最佳,25~40℃时效果次佳。
表1羧基化反应条件对nds-tpgs接枝率、粒度及粒度分布的影响
实施例2nds-tpgs合成条件的筛选
(1)将0.5gnds在60℃下加入20ml混酸(h2so4-hno3,体积比3:1)中,搅拌24小时,冷却至室温,以蒸馏水稀释后离心;以0.1mnaoh和0.1mhcl调节ph值至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
(2)将一定量nds-cooh超声分散于25ml的草酰氯中,然后加入一定量二甲基甲酰胺,一定温度下搅拌反应一定时间,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
(3)将10mgnds-cocl超声分散于10ml二甲基甲酰胺中,然后加入50mgtpgs1000于100℃下反应48小时,将冷却后的反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs。
合成结束后,以岛津tga-50热重分析仪分别检测nds-cooh、tpgs、和nds-tpgs在25~550℃温度范围内的失重情况,并计算接枝率;称取0.05gnds-tpgs,冰浴超声30min(超声功率300w),分散于10ml蒸馏水中,使用malvernzetasizer粒径仪测定粒径和粒度分布。以接枝率大于50%,粒径小于250nm,粒度分布小于0.25为最佳合成条件;以接枝率30~50%,粒径250~300nm,粒度分布0.25~0.3为次佳合成条件;以接枝率小于30%,粒径大于300nm,粒度分布大于0.3为一般合成条件筛选合成工艺。结果可见,nds-cooh在草酰氯中的浓度为2~8mg/ml时合成效果最佳,8~16mg/ml时次佳;二甲基甲酰胺的浓度为0.2~1mg/ml时合成效果最佳;反应时长为12~48h时效果最佳,6~12小时效果次之;反应温度为40~75℃效果最佳,25~40℃效果一般。
表2nds-cocl合成条件对nds-tpgs接枝率、粒度及粒度分布的影响
实施例3nds-tpgs合成条件的筛选
(1)将0.5gnds在60℃下加入20ml混酸(h2so4-hno3,体积比3:1)中,搅拌24小时,冷却至室温,以蒸馏水稀释后离心;以0.1mnaoh和0.1mhcl调节ph值至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
(2)将100mgnds-cooh超声分散于25ml的草酰氯中,然后加入1ml二甲基甲酰胺,一定温度下搅拌反应24小时,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
(3)将一定量的nds-cocl超声分散于10ml二甲基甲酰胺中,然后加入一定量tpgs于一定温度下反应一定时间,将冷却后的反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs。
合成结束后,以岛津tga-50热重分析仪分别检测nds-cooh、tpgs、和nds-tpgs在25~550℃温度范围内的失重情况,并计算接枝率;称取0.05gnds-tpgs,冰浴超声30min(超声功率300w),分散于10ml蒸馏水中,使用malvernzetasizer粒径仪测定粒径和粒度分布。以接枝率大于50%,粒径小于250nm,粒度分布小于0.25为最佳合成条件;以接枝率30~50%,粒径250~300nm,粒度分布0.25~0.3为次佳合成条件;以接枝率小于30%,粒径大于300nm,粒度分布大于0.3为一般合成条件筛选合成工艺。结果可见,nds-cocl在二甲基甲酰胺中的浓度为0.1~5mg/ml时合成效果最佳,5~10mg/ml时效果一般;tpgs在二甲基甲酰胺中的浓度为3~20mg/ml时效果最佳,1~3mg/ml是效果次佳;tpgs的种类为tpgs400、tpgs1000和tpgs2000时,效果最佳,为tpgs4000时效果次佳,为tpgs6000时效果一般;反应时长为12~72h时结果均佳;反应温度为60~120℃时效果最佳,40~60℃时效果次佳。
表3nds-tpgs合成条件对接枝率、粒度及粒度分布的影响
实施例4nds-tpgs合成条件的优化
由单因素考察,确定影响nds-tpgs合成的三个关键因素为步骤(1)nds浓度、步骤(2)nds-cooh浓度和步骤(3)tpgs浓度,对合成条件进一步优化:
(1)将一定量nds在60℃下加入20ml混酸(h2so4-hno3,体积比3:1)中,搅拌24小时,冷却至室温,以蒸馏水稀释后离心;以0.1mnaoh和0.1mhcl调节ph值至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
(2)将一定量nds-cooh超声分散于25ml草酰氯中,然后加入0.5ml二甲基甲酰胺,60℃下搅拌反应24h,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
(3)将10mgnds-cocl超声分散于10ml二甲基甲酰胺中,然后加入一定量tpgs2000于100℃下反应48小时,将冷却后的反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs。
合成结束后,以岛津tga-50热重分析仪分别检测nds-cooh、tpgs、和nds-tpgs在25~550℃温度范围内的失重情况,并计算接枝率;称取0.05gnds-tpgs,冰浴超声30min(超声功率300w),分散于10ml蒸馏水中,使用malvernzetasizer粒径仪测定粒径和粒度分布。计算不同因素不同水平指标平均值,并计算平均值最低值与最高值之间的差值(极差),评价因素的显著程度(见表5)。
由考察结果可见,各因素对合成产物nds-tpgs接枝率、粒径和粒度分布影响显著程度依次为:步骤(3)tpgs浓度>步骤(2)nds-cooh浓度>步骤(1)nds浓度。当步骤(1)nds浓度为5~30mg/ml;且步骤(2)nds-cooh浓度为2~8mg/ml;步骤(3)tpgs浓度为2~10mg/ml时,均可得到接枝率大于45%,粒径小于250nm,粒度分布小于0.25的优良纳米载体。
表4nds-tpgs合成条件优化
表5优化结果分析
实施例5cur@nds-tpgs纳米复合物的处方筛选
1.nds-tpgs合成方法
1.1将0.5gnds在60℃下加入20ml混酸(h2so4-hno3,体积比3:1)中,搅拌24小时,冷却至室温,以蒸馏水稀释后离心;以0.1mnaoh和0.1mhcl调节ph值至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
1.2将100mgnds-cooh超声分散于25ml的草酰氯中,然后加入1ml二甲基甲酰胺,一定温度下搅拌反应24小时,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
1.3将10mgnds-cocl超声分散于10ml二甲基甲酰胺中,然后加入50mgtpgs1000于100℃下反应48小时,将冷却后的反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs。
2.cur@nds-tpgs纳米复合物的制备方法
2.1将一定量nds-tpgs1000加入10ml适宜溶剂中,探头超声(360w,0℃)分散30min。
2.2用2ml适宜溶剂溶解处方量cur。
2.3将cur溶液滴入nds-tpgs1000悬液中,继续探头超声30min(功率与步骤2.1一致),减压干燥除去有机溶剂。
2.4向cur@nds-tpgs1000中加入10ml蒸馏水,探头超声(360w,0℃)10min进行再分散,即得。
采用紫外可见分光光度法测定制剂的载药效率,使用malvernzetasizer测定粒径和粒度分布。以载药效率大于70%,粒径小于300nm,粒度分布小于0.3为最佳条件;载药效率50~70%,粒径300~400nm,粒度分布0.3~0.35为次佳条件;载药效率小于50%,粒径大于400nm,粒度分布大于0.35为一般条件筛选处方。结果表明,nds-tpgs的浓度5~20mg/ml时效果最佳,浓度在3~5mg/ml和20~30mg/ml时效果次佳,1~3mg/ml时效果一般;cur的浓度为0.5~1mg/ml时效果最佳,1~2mg/ml时效果次佳,2~3mg/ml时效果一般;步骤2.1中nds-tpgs分散溶剂为丙酮、二甲基甲酰胺、乙醇、75%乙醇中的一种时效果最佳,为二甲基亚砜或者50%乙醇时效果次佳,为二氯甲烷时效果一般;步骤2.2的溶剂和丙酮或者乙醇时效果最佳,为二甲基亚砜或者二甲基甲酰胺时效果次佳,为二氯甲烷时效果一般。
表6cur@nds-tpgs纳米复合物的处方筛选
实施例6cur@nds-tpgs纳米复合物的工艺筛选
1.nds-tpgs合成方法
1.1将0.5gnds在60℃下加入20ml混酸(h2so4-hno3,体积比3:1)中,搅拌24小时,冷却至室温,以蒸馏水稀释后离心;以0.1mnaoh和0.1mhcl调节ph值至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
1.2将100mgnds-cooh超声分散于25ml的草酰氯中,然后加入1ml二甲基甲酰胺,一定温度下搅拌反应24小时,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
1.3将10mgnds-cocl超声分散于10ml二甲基甲酰胺中,然后加入50mgtpgs1000于100℃下反应48小时,将冷却后的反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs。
2.cur@nds-tpgs纳米复合物的制备方法
2.1将50mgnds-tpgs1000加入10ml乙醇中,于一定温度下以一定功率探头超声分散一定时间。
2.2将3mgcur溶解于2ml丙酮。
2.3将cur溶液滴入nds-tpgs1000悬液中,继续于一定功率(功率与步骤2.1一致)探头超声30min,减压干燥除去有机溶剂。
2.4向cur@nds-tpgs1000中加入10ml蒸馏水,探头超声(360w,0℃)10min进行再分散,即得。
采用紫外可见分光光度法测定制剂的载药效率,使用malvernzetasizer测定粒径和粒度分布。以载药效率大于70%,粒径小于300nm,粒度分布小于0.3为最佳条件;载药效率50~70%,粒径300~400nm,粒度分布0.3~0.35为次佳条件;载药效率小于50%,粒径大于400nm,粒度分布大于0.35为一般条件筛选工艺条件。结果表明,超声功率为360~600w时效果最佳,240~360w时效果次之,120~240w时效果一般;超声温度为0~25℃是效果最佳,25~50℃时效果次之;超声时长为15~60min时效果最佳,5~15min时效果次之。
表7cur@nds-tpgs纳米复合物的工艺筛选
实施例7tpgs型号对纳米复合物制剂学性质的影响
1.nds-tpgs合成方法
1.1将0.5gnds在60℃下加入20ml混酸(h2so4-hno3,体积比3:1)中,搅拌24小时,冷却至室温,以蒸馏水稀释后离心;以0.1mnaoh和0.1mhcl调节ph值至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
1.2将100mgnds-cooh超声分散于25ml的草酰氯中,然后加入1ml二甲基甲酰胺,一定温度下搅拌反应24小时,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
1.3将10mgnds-cocl超声分散于10ml二甲基甲酰胺中,然后加入50mgtpgs(tpgs400或tpgs1000或tpgs2000或tpgs2000)于100℃下反应48小时,将冷却后的反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs。
2.cur@nds-tpgs纳米复合物的制备方法
2.1将50mgnds-tpgs加入10ml乙醇中,探头超声(360w,0℃)分散30min。
2.2将3mgcur溶解于2ml丙酮。
2.3将cur溶液滴入nds-tpgs悬液中,继续于一定功率(功率与步骤2.1一致)探头超声30min,减压干燥除去有机溶剂。
2.4向cur@nds-tpgs中加入10ml蒸馏水,探头超声(360w,0℃)10min进行再分散,即得。
采用紫外可见分光光度法测定制剂的载药效率,使用malvernzetasizer测定粒径和粒度分布。以载药效率大于70%,粒径小于300nm,粒度分布小于0.3为最佳条件;载药效率50~70%,粒径300~400nm,粒度分布0.3~0.35为次佳条件;载药效率小于50%,粒径大于400nm,粒度分布大于0.35为一般条件筛选tpgs型号。结果可见,在相同工艺和处方条件下,tpgs1000和tpgs2000的效果最佳,tpgs400的效果次之,tpgs4000的效果一般。
表8tpgs型号对纳米复合物制剂学性质的影响
实施例8cur@nds-tpgs纳米复合物处方优化
1.nds-tpgs合成方法
1.1将一定量nds在60℃下加入20ml混酸(h2so4-hcl,体积比3:1)中,搅拌24小时,冷却至室温,以蒸馏水稀释后离心;以0.1mnaoh和0.1mhcl调节ph值至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
1.2将100mgnds-cooh超声分散于25ml的草酰氯中,然后加入1ml二甲基甲酰胺,一定温度下搅拌反应24小时,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
1.3将10mgnds-cocl超声分散于10ml二甲基甲酰胺中,然后加入50mgtpgs(tpgs400或tpgs1000或tpgs2000或tpgs2000)于100℃下反应48小时,将冷却后的反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs。
2.cur@nds-tpgs纳米复合物的制备方法
2.1将一定量nds-tpgs加入10ml乙醇中,探头超声(360w,0℃)分散30min。
2.2将一定量cur溶解于2ml丙酮。
2.3将cur溶液滴入nds-tpgs悬液中,继续探头超声30min,减压干燥除去有机溶剂。
2.4向cur@nds-tpgs中加入10ml蒸馏水,探头超声(360w,0℃)10min进行再分散,即得。
采用紫外可见分光光度法测定制剂的载药效率,使用malvernzetasizer测定粒径和粒度分布。由优化结果可见,各因素对载药效率、粒径和粒度分布影响显著程度依次为步骤2.1nds-tpgs种类>步骤2.2cur浓度>步骤2.1nds-tpgs浓度。当步骤(1)nds-tpgs的浓度为5~20mg/ml,nds-tpgs为nds-tpgs400、nds-tpgs1000或者nds-tpgs2000中的一种;且步骤(2)cur浓度为0.5~2mg/ml时,所得制剂载药效率大于50%,粒径小于350nm,粒度分布小于0.3。
表9cur@nds-tpgs纳米复合物处方优化
表10优化结果分析
实施例9cur@nds-tpgs口服纳米复合物的制备
(1)nds-tpgs的合成方法
1)将0.5gnds在60℃下加入20ml混酸(h2so4-hno3,体积比3:1)中,搅拌24小时,冷却至室温,以蒸馏水稀释后在10,000rpm下离心15min;将nds颗粒在0.1mnaoh中于90℃加热2小时,在0.1mhcl中于90℃加热2小时,使ph值降至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
2)将0.1gnds-cooh超声分散于20ml草酰氯中,然后加入0.5ml的二甲基甲酰胺,60℃下搅拌反应24小时,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
3)将10mgnds-cocl超声分散于10ml二甲基甲酰胺中,然后加入50mgtpgs1000于100℃下反应48小时,将反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs1000。
(2)cur@nds-tpgs的制备方法
1)将50mgnds-tpgs1000以探头超声(360w,0℃)分散于10ml乙醇中,超声时长30min。
2)将3mg的cur溶解于2ml丙酮。
3)将cur溶液超声下滴加于nds-tpgs1000乙醇分散液中,超声30min,减压干燥。
4)将50mgcur@nds-tpgs1000超声下再分散于10ml蒸馏水中,超声时长10min。
所得cur@nds-tpgs1000平均粒径为196.33nm,粒度分布为0.19,电位-24.52mv,载药效率87.19%。
实施例10cur@nds-tpgs口服纳米复合物的制备
(1)nds-tpgs的合成方法
1)将0.1gnds在25℃下加入20ml混酸(h2so4-hcl,体积比2:1)中,搅拌48小时,冷却至室温,以蒸馏水稀释后在10,000rpm下离心15min;将nds颗粒在0.1mnaoh中于90℃加热2小时,在0.1mhcl中于90℃加热2小时,使ph值降至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
2)将0.32gnds-cooh超声分散于20ml草酰氯中,然后加入1ml的二甲基甲酰胺,75℃下搅拌反应48小时,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
3)将100mgnds-cocl超声分散于10ml二甲基甲酰胺中,然后加入30mgtpgs2000于120℃下反应72小时,将反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs2000。
(2)cur@nds-tpgs的制备方法
1)将300mgnds-tpgs2000以探头超声(600w,25℃)分散于10ml75%乙醇中,超声时长60min。
2)将4mg的cur溶解于2ml二甲基亚砜。
3)将cur溶液超声下滴加于nds-tpgs2000分散液中,超声30min,减压干燥。
4)将50mgcur@nds-tpgs400超声下再分散于10ml蒸馏水中,超声时长10min。
所得cur@nds-tpgs2000平均粒径为368.07nm,粒度分布为0.37,载药效率38.25%。
实施例11cur@nds-tpgs口服纳米复合物的制备
(1)nds-tpgs的合成方法
1)将1.1gnds在60℃下加入20ml混酸(h2so4-hcl,体积比5:1)中,搅拌12小时,冷却至室温,以蒸馏水稀释后在10,000rpm下离心15min;将nds颗粒在0.1mnaoh中于90℃加热2小时,在0.1mhcl中于90℃加热2小时,使ph值降至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
2)将0.04gnds-cooh超声分散于20ml草酰氯中,然后加入0.2ml的二甲基甲酰胺,40℃下搅拌反应6小时,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
3)将1mgnds-cocl超声分散于10ml二甲基甲酰胺中,然后加入200mgtpgs400于100℃下反应12小时,将反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs400。
(2)cur@nds-tpgs的制备方法
1)将10mgnds-tpgs400以探头超声(120w,50℃)分散于10ml75%乙醇中,超声时长10min。
2)将1mg的cur溶解于2ml乙醇。
3)将cur溶液超声下滴加于nds-tpgs400分散液中,超声30min,减压干燥。
4)将50mgcur@nds-tpgs400超声下再分散于10ml蒸馏水中,超声时长10min。
所得cur@nds-tpgs400平均粒径为287.35nm,粒度分布为0.28,载药效率60.27%。
实施例12cur@nds-tpgs口服纳米复合物的制备
(1)nds-tpgs的合成方法
1)将0.3gnds在100℃下加入20ml混酸(h2so4-hclo4,体积比3:1)中,搅拌48小时,冷却至室温,以蒸馏水稀释后在10,000rpm下离心15min;将nds颗粒在0.1mnaoh中于90℃加热2小时,在0.1mhcl中于90℃加热2小时,使ph值降至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
2)将0.16gnds-cooh超声分散于20ml草酰氯中,然后加入0.5ml的二甲基甲酰胺,90℃下搅拌反应36小时,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
3)将50mgnds-cocl超声分散于10ml二甲基甲酰胺中,然后加入100mgtpgs4000于120℃下反应72小时,将反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs4000。
(2)cur@nds-tpgs的制备方法
1)将100mgnds-tpgs4000以探头超声(240w,36℃)分散于10ml二甲基甲酰胺中,超声时长20min。
2)将5mg的cur溶解于2ml二甲基甲酰胺。
3)将cur溶液超声下滴加于nds-tpgs4000分散液中,超声30min,减压干燥。
4)将50mgcur@nds-tpgs4000超声下再分散于10ml蒸馏水中,超声时长10min。
所得cur@nds-tpgs4000平均粒径为435.09nm,粒度分布为0.35,载药效率30.46%。
实施例13cur@nds-tpgs口服纳米复合物的制备
(1)nds-tpgs的合成方法
1)将0.4gnds在70℃下加入20ml混酸(h2so4-hcl,体积比3:1)中,搅拌36小时,冷却至室温,以蒸馏水稀释后在10,000rpm下离心15min;将nds颗粒在0.1mnaoh中于90℃加热2小时,在0.1mhcl中于90℃加热2小时,使ph值降至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
2)将0.08gnds-cooh超声分散于20ml草酰氯中,然后加入0.2ml的二甲基甲酰胺,70℃下搅拌反应24小时,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
3)将8mgnds-cocl超声分散于10ml二甲基甲酰胺中,然后加入60mgtpgs2000于100℃下反应36小时,将反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs2000。
(2)cur@nds-tpgs的制备方法
1)将40mgnds-tpgs2000以探头超声(480w,10℃)分散于10ml二甲基甲酰胺中,超声时长45min。
2)将2mg的cur溶解于2ml乙醇。
3)将cur溶液超声下滴加于nds-tpgs2000分散液中,超声60min,减压干燥。
4)将50mgcur@nds-tpgs2000超声下再分散于10ml蒸馏水中,超声时长10min。
所得cur@nds-tpgs2000平均粒径为164.27nm,粒度分布为0.17,载药效率91.47%。
实施例14cur@nds-tpgs口服纳米复合物的制备
(1)nds-tpgs的合成方法
1)将0.6gnds在80℃下加入20ml混酸(h2so4-hcl,体积比3:1)中,搅拌48小时,冷却至室温,以蒸馏水稀释后在10,000rpm下离心15min;将nds颗粒在0.1mnaoh中于90℃加热2小时,在0.1mhcl中于90℃加热2小时,使ph值降至ph5~6;所得nds-cooh用蒸馏水冲洗三次,真空干燥24小时备用。
2)将0.15gnds-cooh超声分散于20ml草酰氯中,然后加入0.8ml的二甲基甲酰胺,60℃下搅拌反应30小时,离心后将nds-cocl用无水四氢呋喃洗涤三次,真空干燥24小时备用。
3)将6mgnds-cocl超声分散于10ml二甲基甲酰胺中,然后加入40mgtpgs1000于100℃下反应40小时,将反应液用0.22μm微孔滤膜过滤,水洗后冷冻干燥,得到nds-tpgs1000。
(2)cur@nds-tpgs的制备方法
1)将40mgnds-tpgs1000以探头超声(600w,0℃)分散于10ml二甲基甲酰胺中,超声时长30min。
2)将1mg的cur溶解于2ml丙酮。
3)将cur溶液超声下滴加于nds-tpgs1000分散液中,超声30min,减压干燥。
4)将50mgcur@nds-tpgs2000超声下再分散于10ml蒸馏水中,超声时长10min。
所得cur@nds-tpgs2000平均粒径为141.44nm,粒度分布为0.18,载药效率93.33%。
实施例15大鼠体内药动学研究
选取按照实施例9制备的cur@nds-tpgs1000为受试制剂,选取cur的0.5%羧甲基纤维素钠混悬液和cur@nds-cooh/tpgs1000(物理修饰组)为参比制剂。将雄性wistar大鼠(体重220±20g)随机分为三组,每组六只,给药前12h禁食,自由饮水。以75mg/kg的剂量口服灌胃给药,分别于给药后0.25、0.5、0.75、1、1.5、2、3、4、5、6、8和12小时眼眶取血,置于涂有肝素离心管中,10,000×g离心5min,分离血浆,置于-20℃冰箱待用。血浆处理时精密量取200μl血浆样品,加入50μl内标大黄素,以200μl乙酸乙酯萃取,涡旋3min,10,000×g离心5min,分离有机层至另一离心管,氮气吹干,以200μl流动相溶解,涡旋3min,10,000×g离心5min,取上清液注入高效液相色谱仪。记录色谱图和峰面积,计算各时间点样品中血药浓度和主要药动学参数(见表7)。
表11大鼠口服给予cur制剂的药动学参数
结果可见,cur@nds-tpgs的cmax是cur混悬剂和cur@nds-cooh/tpgs组的5.71和1.21倍,auc0-t是cur混悬剂和cur@nds-cooh/tpgs组的16.15和1.37倍,显著提高了cur的口服生物利用度,并显示出良好的缓释效果。
- 还没有人留言评论。精彩留言会获得点赞!