拷贝数变异预测方法、装置、计算机设备和存储介质与流程
本申请涉及生物技术领域,特别是涉及一种拷贝数变异预测方法、装置、计算机设备和存储介质。
背景技术:
随着生物技术的发展,出现了检测一个样本中是否存在染色体拷贝数异常的技术,拷贝数变异(copynumbervariation,cnv)是由基因组发生重排而导致的,一般指长度为1kb以上的基因组大片段的拷贝数增加或者减少,主要表现为亚显微水平的缺失和重复。cnv是基因组结构变异(structuralvariation,sv)的重要组成部分。cnv位点的突变率远高于snp(singlenucleotidepolymorphism),是肿瘤类疾病的重要致病因素之一。
然而,传统的技术方案是利用一个待检测的样本的基因组测序数据,通过数据分析的方法,判断样本中是否存在染色体拷贝数异常的问题,例如通过马尔代夫序列进行预测,但是马尔代夫序列因为是连续性有限、模式单一,这样就会造成检测不准确。
技术实现要素:
基于此,有必要针对上述技术问题,提供一种能够提高预测准确性的拷贝数变异预测方法、装置、计算机设备和存储介质。
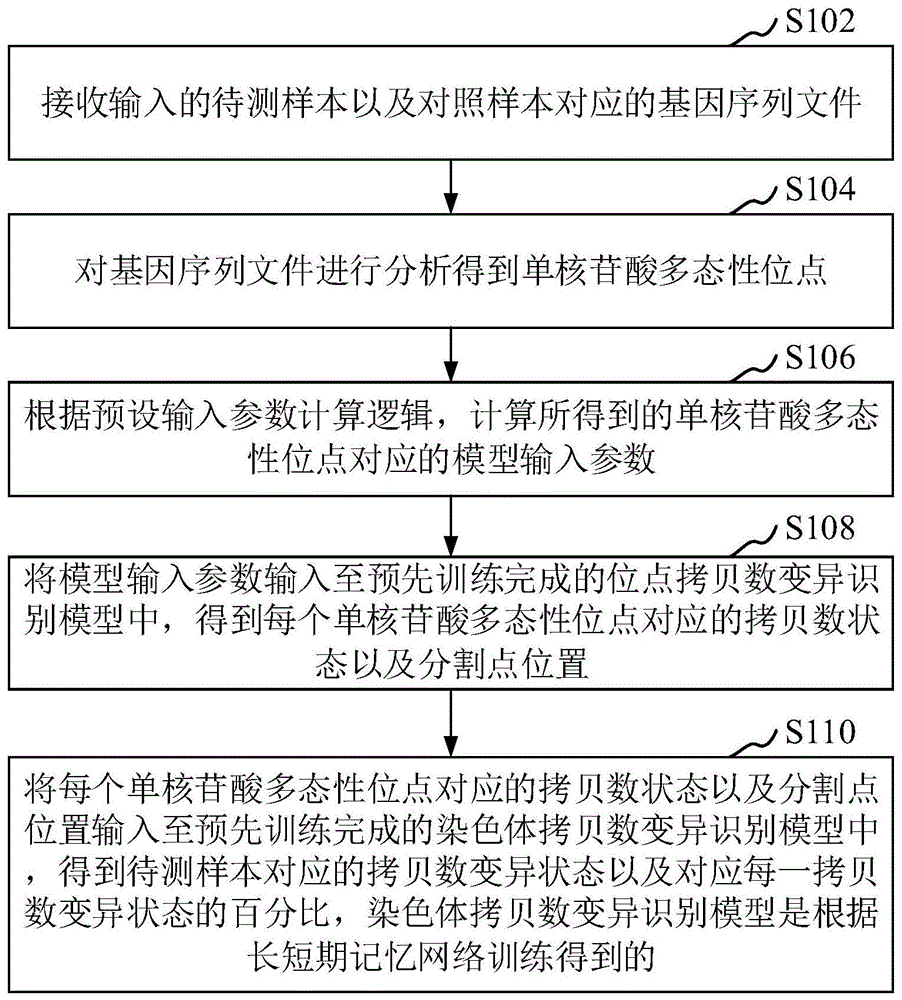
一种拷贝数变异预测方法,所述方法包括:
接收输入的待测样本以及对照样本对应的基因序列文件;
对所述基因序列文件进行分析得到单核苷酸多态性位点;
根据预设输入参数计算逻辑,计算所得到的单核苷酸多态性位点对应的模型输入参数;
将所述模型输入参数输入至预先训练完成的位点拷贝数变异识别模型中,得到每个所述单核苷酸多态性位点对应的拷贝数状态以及分割点位置;
将每个所述单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到所述待测样本对应的拷贝数变异状态以及对应每一所述拷贝数变异状态的百分比,所述染色体拷贝数变异识别模型是根据长短期记忆网络训练模型得到的。
在其中一个实施例中,所述将每个所述单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到所述待测样本对应的拷贝数变异状态以及对应每一所述拷贝数变异状态的百分比之后,还包括:
将所述待测样本对应的拷贝数变异状态以及对应每一所述拷贝数变异状态的百分比输入至肿瘤预测模型中,得到所述待测样本对应的肿瘤含量以及倍型,所述肿瘤预测模型是根据长短期记忆网络训练模型得到的。
在其中一个实施例中,所述对所述基因序列文件进行分析得到单核苷酸多态性位点,包括:
从所述待测样本和对照样本对应的基因序列文件中的提取基因序列,并将所提取的基因序列分别与标准基因组进行比对,得到单核苷酸多态性位点。
在其中一个实施例中,所述根据预设输入参数计算逻辑,计算所得到的单核苷酸多态性位点对应的模型输入参数,包括:
统计所述待测样本的基因序列与所述标准基因组相同的第一序列以及不同的第二序列;
读取所述第一序列中的目标位点的观测值、所述第二序列中的所述目标位点的观测值,以及所述待测样本中的基因序列中的观测值;
统计所述对照样本的基因序列与所述标准基因组相同的第三序列和不同的第四序列;
读取所述第三序列中的目标位点的观测值、所述第四序列中的所述目标位点的观测值,以及所述对照样本中的基因序列中的观测值;
根据所述目标位点前后预设长度区域内的点位计算得到所述目标点位对应的聚合酶链式反应亲和度;
根据所述待测样本中的基因序列中的观测值、所述对照样本中的基因序列中的观测值计算得到染色体扩增参考指标;
根据所述第二序列中的所述目标位点的观测值、所述待测样本中的基因序列中的观测值、所述第四序列中的所述目标位点的观测值以及所述对照样本中的基因序列中的观测值计算得到染色体缺失参考指标。
在其中一个实施例中,所述将所述模型输入参数输入至预先训练完成的位点拷贝数变异识别模型中,得到每个所述单核苷酸多态性位点对应的拷贝数状态以及分割点位置,包括:
将所述模型输入参数作为所述待测样本对应的基因序列的输入矩阵;
获取所述待测样本对应的基因序列的预设数量的点位作为初始序列,并将所述初始序列进行逆序拼接得到输入序列;
将所述输入序列输入至预先训练完成的位点拷贝数变异识别模型中,得到每个所述单核苷酸多态性位点对应的拷贝数状态以及分割点位置。
在其中一个实施例中,所述每个所述单核苷酸多态性位点对应的拷贝数状态包括父本染色体个数、母本染色体个数、细胞总数以及突变的细胞数;所述将每个所述单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到所述待测样本对应的拷贝数变异状态以及对应每一所述拷贝数变异状态的百分比,包括:
根据所述分割点位置对所述待测样本对应的基因序列进行分割;
根据所述细胞总数、突变的细胞数、父本染色体个数和母本染色体个数计算得到均一的父本染色体个数、均一母本染色体个数;
将分割后的所述基因序列、均一的父本染色体个数和均一母本染色体个数输入至预先训练完成的染色体拷贝数变异识别模型中,得到所述待测样本对应的拷贝数变异状态以及对应每一所述拷贝数变异状态的百分比。
一种拷贝数变异预测装置,所述装置包括:
接收模块,用于接收输入的待测样本以及对照样本对应的基因序列文件;
单核苷酸多态性位点提取模块,用于对所述基因序列文件进行分析得到单核苷酸多态性位点;
入参计算模块,用于根据预设输入参数计算逻辑,计算所得到的单核苷酸多态性位点对应的模型输入参数;
位点拷贝数变异预测模块,用于将所述模型输入参数输入至预先训练完成的位点拷贝数变异识别模型中,得到每个所述单核苷酸多态性位点对应的拷贝数状态以及分割点位置;
染色体拷贝数变异预测模块,用于将每个所述单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到所述待测样本对应的拷贝数变异状态以及对应每一所述拷贝数变异状态的百分比,所述染色体拷贝数变异识别模型是根据长短期记忆网络训练模型得到的。
在其中一个实施例中,所述装置还包括:
肿瘤含量预测模块,用于将所述待测样本对应的拷贝数变异状态以及对应每一所述拷贝数变异状态的百分比输入至肿瘤预测模型中,得到所述待测样本对应的肿瘤含量以及倍型,所述肿瘤预测模型是根据长短期记忆网络训练模型得到的。
一种计算机设备,包括存储器和处理器,所述存储器存储有计算机程序,所述处理器执行所述计算机程序时实现上述中任一项所述方法的步骤。
一种计算机可读存储介质,其上存储有计算机程序,所述计算机程序被处理器执行时实现上述中任一项所述的方法的步骤。
上述拷贝数变异预测方法、装置、计算机设备和存储介质,首先将待测样本以及对照样本对应的基因序列文件进行分析得到单核苷酸多态性位点,并计算对应的模型输入参数,将所述模型输入参数输入至预先训练完成的位点拷贝数变异识别模型中,得到每个所述单核苷酸多态性位点对应的拷贝数状态以及分割点位置,这样将原始数据进行碎片化,识别是是每一个片段的拷贝数,且根据结果进行重新编码,再通过根据长短期记忆网络训练模型得到的染色体拷贝数变异识别模型进行处理得到所述待测样本对应的拷贝数变异状态以及对应每一所述拷贝数变异状态的百分比,充分利用了长短期记忆网络的优势,保证不会像马尔可夫序列因为序列连续性有限、模式单一造成的误差,提高了拷贝数变异预测的准确性。
附图说明
图1为一个实施例中拷贝数变异预测方法的流程示意图;
图2为一个实施例中的位点拷贝数变异识别模型的模型输入参数的示意图;
图3为一个实施例中的位点拷贝数变异识别模型的模型输出参数的示意图;
图4为一个实施例中的拷贝数变异预测方法的框架图;
图5为一个实施例中的染色体拷贝数变异识别模型的示意图;
图6为一个实施例中的肿瘤预测模型的结构示意图;
图7为一个实施例中的滑窗模型的结构示意图;
图8为一个实施例中的位点拷贝数变异识别模型的结构示意图;
图9为一个实施例中拷贝数变异预测装置的结构框图;
图10为一个实施例中计算机设备的内部结构图。
具体实施方式
为了使本申请的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本申请进行进一步详细说明。应当理解,此处描述的具体实施例仅仅用以解释本申请,并不用于限定本申请。
在一个实施例中,如图1所示,提供了一种拷贝数变异预测方法,本实施例以该方法应用于终端进行举例说明,可以理解的是,该方法也可以应用于服务器,还可以应用于包括终端和服务器的系统,并通过终端和服务器的交互实现。本实施例中,该方法包括以下步骤:
s102:接收输入的待测样本以及对照样本对应的基因序列文件。
具体地,待测样本可以是指肿瘤样本,对照样本是正常的组织的样本,其中终端可以首先对待测样本以及对照样本进行二代测序得到基因序列文件,例如bam文件,该bam文件中存放了所有测序获得的碱基序列,并且以150个碱基的序列的文件形式进行保存,该文件中数据已经和标准基因组进行比对,文件中包含每一条150个基因的序列的内容,包括染色体位置、详细的碱基序列、碱基对应标准基因组的位置、突变碱基的信息还有每一个碱基的质量,bam是sam的二进制文件格式,bam格式中的b是binary的意思,由于sam格式文件大小通常是十分巨大的,所以为了减少存储量等因素而将sam转换为二进制格式以便于分析。
s104:对基因序列文件进行分析得到单核苷酸多态性位点。
具体地,单核苷酸多态性(singlenucleotidepolymorphism,snp),即snp位点主要是指在基因组水平上由单个核苷酸的变异所引起的dna序列多态性。它是可遗传的变异中最常见的一种。占所有已知多态性的90%以上。snp在基因组中广泛存在,平均每500~1000个碱基对中就有1个,估计其总数可达300万个甚至更多。
在其中一个实施例中,对基因序列文件进行分析得到单核苷酸多态性位点,包括:从待测样本和对照样本对应的基因序列文件中的提取基因序列,并将所提取的基因序列分别与标准基因组进行比对,得到单核苷酸多态性位点。具体地,终端可以将基因序列文件输入至预先训练的模型中得到单核苷酸多态性位点,例如从待测样本和对照样本对应的基因序列文件中的提取基因序列,并将所提取的基因序列分别与标准基因组进行比对,得到单核苷酸多态性位点。实际应用中终端可以利用gatk软件来识别得到单核苷酸多态性位点,并获取到待测样本以及对照样本所有基因组的与标准基因序列不一致的read个数(每一条read包括150个碱基)和一致的read个数。
s106:根据预设输入参数计算逻辑,计算所得到的单核苷酸多态性位点对应的模型输入参数。
具体地,预设输入参数计算逻辑是终端中预先存储的,其可以包括但不限于每个单核苷酸多态性位点对应的聚合酶链式反应亲和度、染色体扩增参考指标以及染色体缺失参考指标的计算逻辑。
上述的模型输入参数可以包括但不限于:待测样本与标准基因组不一致的read数量中的第t个read的第j个位点目标位点的观测值mtj*、待测样本与标准基因组一致的read数量的第t个read的的第j个位点目标位点的观测值ptj*、待测样本总的read数量中第t个read的第j个位点目标位点的观测值dtj*、对照样本与标准基因组不一致的read中的第t个read的第j个位点目标位点的观测值mnj*、与对照样本与标准基因组一致的read中的第t个read的第j个位点目标位点的观测值pnj*、与对照样本总的read数量中第t个read的第j个位点目标位点的观测值dnj*、在第j个snp位点前后长度150bp的区域内,碱基c及g所占个数比例gcp_j(该参数反馈了该点做pcr(聚合酶链式反应)时候亲和度,模型中作为校准系数)、在第j个snp位点的染色体扩增参考指标rpj、在第j个snp位点的染色体缺失参考指标orpj,其中j表示snp位点位置。
其中
rpj=log((dtj*-median(dt))/(dnj*-median(dn)))(1)
orpj=log((ptj*/dtj*)/(pnj*/dnj*))(2)
其中,dt表示肿瘤组织上所有观测到的所有位点reads深度的观察量,median(dt)表示dt这些深度的中位数;dn表示对照(正常)组织上所有观测到的所有位点reads深度的观察量,median(dt)表示dt这些深度的中位数。
s108:将模型输入参数输入至预先训练完成的位点拷贝数变异识别模型中,得到每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置。
具体地,拷贝数变异(copynumbervariation,cnv)是由基因组发生重排而导致的,一般指长度为1kb以上的基因组大片段的拷贝数增加或者减少,主要表现为亚显微水平的缺失和重复。cnv是基因组结构变异(structuralvariation,sv)的重要组成部分。cnv位点的突变率远高于snp(singlenucleotidepolymorphism),是肿瘤疾病的重要致病因素之一。
具体地,结合图2和图3所示,其中图2为一个实施例中的位点拷贝数变异识别模型的模型输入参数的示意图,图3为一个实施例中的位点拷贝数变异识别模型的模型输出参数的示意图。
其中模型输入参数是每个snp位点的参数,模型的输入是一个序列,序列中包括若干snp位点,每个snp位点通过模型输入参数来进行表征,如图2所示,终端可以将一个基因中所有snp位点作为一个单元,形成一个序列,然后序列中的每个snp位点通过模型输入参数来进行表征。终端将所得到的序列输入至预先训练完成的位点拷贝数变异识别模型中,得到模型输出参数,其中模型输出的序列与模型的输入序列的长度一致,但序列中的每个snp位点通过模型输出参数来进行表征。
其中模型输出参数包括但不限于每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置。可选地,模型的输出参数包括父本的染色体个数m、母本的染色体个数p、总共细胞数n、发生突变的细胞个数fc、均一的父本染色体个数m*以及均一的母本染色体个数p*,其中:
m*=((mfc+(1-fc))*n)/n(3)
p*=((pfc+(1-fc))*n)/n(4)
其中,rpj=log((m*+p*)/2),该参数满足正太分布;orpj=log(p*/m*)orlog(m*/p*),该参数满足卡方分布。
具体地,参阅图4所示,图4为一个实施例中的拷贝数变异预测方法的框架图,其中位点拷贝数变异识别模型为seq2seq模型,seq2seq是一个encoder–decoder结构的网络,它的输入是一个序列,输出也是一个序列,encoder中将一个可变长度的信号序列变为固定长度的向量表达,decoder将这个固定长度的向量变成可变长度的目标的信号序列。从染色体起始位置滑窗,每一个窗口固定长度,将其中的序列作为一个输入数据,直到滑到染色体结束为止;这里主要将序列作为输入因为每一个位点的信息确定了周围数据的状态,因为二代测序会由于附近数据影响很大。
s110:将每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比,染色体拷贝数变异识别模型是根据长短期记忆网络训练模型得到的。
具体地,染色体拷贝数变异识别模型是根据长短期记忆网络模型训练得到的,其输入是位点拷贝数变异识别模型的输出,具体地,参阅图5所示,图5为一个实施例中的染色体拷贝数变异识别模型的示意图,终端将分割好的decode数据作为染色体拷贝数变异识别模型的输入进行预测,导出的模型输出结果包括该区域占整体样本该区域的含量,相当于具有这种拷贝数变异的亚克隆事件的百分比,并且给出整个区域的拷贝数状态。上一步因为是每一个位点给出,数据存在偏差,通过这一步数据得到校准,最后输出数据的长度与样本的具有拷贝数区域一致。
上述拷贝数变异预测方法,首先将待测样本以及对照样本对应的基因序列文件进行分析得到单核苷酸多态性位点,并计算对应的模型输入参数,将所述模型输入参数输入至预先训练完成的位点拷贝数变异识别模型中,得到每个所述单核苷酸多态性位点对应的拷贝数状态以及分割点位置,这样将原始数据进行碎片化,识别是是每一个片段的拷贝数,且根据结果进行重新编码,再通过根据长短期记忆网络训练模型得到的染色体拷贝数变异识别模型进行处理得到所述待测样本对应的拷贝数变异状态以及对应每一所述拷贝数变异状态的百分比,充分利用了长短期记忆网络的优势,保证不会像马尔可夫序列因为序列连续性有限、模式单一造成的误差,提高了拷贝数变异预测的准确性。
在其中一个实施例中,将每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比之后,还包括:将待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比输入至肿瘤预测模型中,得到待测样本对应的肿瘤含量以及倍型,肿瘤预测模型是根据长短期记忆网络训练模型得到的。
具体地,肿瘤含量(fc)=肿瘤细胞个数(nt)/所有细胞个数(tn),克隆事件(i)含量(fcc_i)=第i个亚克隆肿瘤细胞个数(nct_i)/所有细胞(tn)。
请参见图6所示,图6为一个实施例中的肿瘤预测模型的结构示意图,在该实施例中,肿瘤预测模型是根据长短期记忆网络训练模型得到的,这样可以解决长序列训练过程中的梯度消失和梯度爆炸的问题,在通过lstm模型,将样本整体的拷贝状态及亚克隆事件百分比作为输入,预测整个样本的肿瘤含量及样本整体的倍型进行预测。
其中可以首先获取病理学家通过阅读病理图染色得到的肿瘤含量和倍型,这样将病理学家给出的肿瘤含量和倍型以及对应的样本进行模型训练得到肿瘤预测模型。
上述实施例中,由于影响cnv的因素有很多,比如每一次测序组织中,各种细胞(正常细胞,癌症细胞)比例不均衡,所以正常时是二倍体,癌症组织是非二倍体,混合后就容易判别不出来,更复杂情况是,有多种癌症细胞,所以不好判断,而采用lstm模型进行训练得到肿瘤预测模型其会选择性删除之前信息,这就可以保证不会像马尔可夫序列因为序列连续性有限、模式单一造成的误差。
在其中一个实施例中,根据预设输入参数计算逻辑,计算所得到的单核苷酸多态性位点对应的模型输入参数,包括:统计待测样本的基因序列与标准基因组相同的第一序列以及不同的第二序列;读取第一序列中的目标位点的观测值、第二序列中的目标位点的观测值,以及待测样本中的基因序列中的目标位点的观测值;统计对照样本的基因序列与标准基因组相同的第三序列和不同的第四序列;读取第三序列中的目标位点的观测值、第四序列中的目标位点的观测值,以及对照样本中的基因序列中的目标位点的观测值;根据目标位点前后预设长度区域内的点位计算得到目标点位对应的聚合酶链式反应亲和度;根据待测样本中的基因序列中目标位点的观测值、对照样本中的基因序列中目标位点的观测值计算得到染色体扩增参考指标;根据第二序列中的目标位点的观测值、待测样本中的基因序列中目标位点的观测值、第四序列中的目标位点的观测值以及对照样本中的基因序列中目标位点的观测值计算得到染色体缺失参考指标。
具体地,标准基因组可以是指人类标准基因组,例如hg19。终端通过gatk软件可以获取到单核苷酸多态性位点,即将待测样本中的基因序列分别与标准基因组进行比对,这样就可以得到待测样本与标准基因组不一致的基因序列的数量mt*、待测样本与标准基因组一致的基因序列的数量pt*以及待测样本总的基因序列的数量dt*,其中一个基因序列是指一条read,即150bp的碱基,t标识序列的编号。将对照样本中的基因序列分别与标准基因组进行比对,这样就可以得到对照样本与标准基因组不一致的基因序列的数量mn*、对照样本与标准基因组一致的基因序列的数量pn*以及对照样本总的基因序列的数量dn*,其中一个基因序列是指一条read,即150bp的碱基,n标识序列的标号。这样终端可以读取到所得到的上述各个序列组中各个序列的对应点位j的观测值,也就是二代测序的值。
终端然后根据上述参数计算得到聚合酶链式反应亲和度gcp_j、染色体扩增参考指标rpj和染色体缺失参考指标orpj,具体可以参见上文中的公式进行计算,这样终端计算得到了各个snp位点对应的模型输入参数。
上述实施例中,终端在获取到snp位点后,则根据二代测序的结果计算各个snp位点对应的模型输入参数,为后续模型处理奠定基础。
在其中一个实施例中,将模型输入参数输入至预先训练完成的位点拷贝数变异识别模型中,得到每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置,包括:将模型输入参数作为待测样本对应的基因序列的输入矩阵;获取待测样本对应的基因序列的预设数量的点位作为初始序列,并将初始序列进行逆序拼接得到输入序列;将输入序列输入至预先训练完成的位点拷贝数变异识别模型中,得到每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置。
具体地,参见图7和图8所示,图7为一个实施例中的滑窗模型的结构示意图;图8为一个实施例中的位点拷贝数变异识别模型的结构示意图,其中,输入序列是每个基因上左右snp位点形成的,且考虑到方向性,终端构建滑窗模型,即将目标snp位点接下来的k-1个位点作为附近信息,合并成一个序列,作为输入序列,此外,考虑到方向性,终端将每一个输入序列进行逆序拼接,这样保证数据从两个方向上都是一致的,结合图2,这样(2*k)*9作为seq2seq的encode-decode过程的输入文件,其中9是指上文中每个snp位点存在9个模型输入参数,这样将输入文件输入至位点拷贝数变异识别模型中,通过对每个snp位点的模型输入参数的学习以确定每个snp位点对应的模型输出参数,即encoder将一个可变长度的信号序列变为固定长度的向量表达,decoder将这个固定长度的向量变成可变长度的目标的信号序列,其中目标的信号序列与输入序列的长度一致,但是序列中每个snp位点是通过父本染色体个数、母本染色体个数、总细胞数以及发生突变的细胞个数来进行表征。
上述实施例中,通过位点拷贝数变异识别模型得到了每个位点的拷贝数状态以及是否具有分割位点的数据,便于后续通过染色体拷贝数变异识别模型的处理。
在其中一个实施例中,每个单核苷酸多态性位点对应的拷贝数状态包括父本染色体个数、母本染色体个数、细胞总数以及突变的细胞数;将每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比,包括:根据分割点位置对待测样本对应的基因序列进行分割;根据细胞总数、突变的细胞数、父本染色体个数和母本染色体个数计算得到均一的父本染色体个数、均一母本染色体个数;将分割后的基因序列、均一的父本染色体个数和均一母本染色体个数输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比。
具体地,结合图5所示,在位点拷贝数变异识别模型得到输出数据后,终端根据位点拷贝数变异识别模型的输出数据计算得到染色体拷贝数变异识别模型的输入数据,包括根据总细胞个数n、发生图片的细胞个数fc、父本染色体个数m和母本染色体个数p计算得到均一的父本染色体个数、均一母本染色体个数:
其中均一的父本染色体个数为:m*=((mfc+(1-fc))*n)/n
均一母本染色体个数为:p*=((pfc+(1-fc))*n)/n
这样,终端将得到的均一的父本染色体个数、均一母本染色体个数以及分割后的基因序列输入至染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比。
上述实施例中,通过每一个snp位点的拷贝数状况结果及是否具有分割位点的数据得到染色体层面上的拷贝数变异状态以及对应每一拷贝数变异状态的百分比,且通过长短期记忆网络进行训练,可以保证预测的准确性。
应该理解的是,虽然图1的流程图中的各个步骤按照箭头的指示依次显示,但是这些步骤并不是必然按照箭头指示的顺序依次执行。除非本文中有明确的说明,这些步骤的执行并没有严格的顺序限制,这些步骤可以以其它的顺序执行。而且,图1中的至少一部分步骤可以包括多个步骤或者多个阶段,这些步骤或者阶段并不必然是在同一时刻执行完成,而是可以在不同的时刻执行,这些步骤或者阶段的执行顺序也不必然是依次进行,而是可以与其它步骤或者其它步骤中的步骤或者阶段的至少一部分轮流或者交替地执行。
在一个实施例中,如图9所示,提供了一种拷贝数变异预测装置,包括:接收模块100、单核苷酸多态性位点提取模块200、入参计算模块300、位点拷贝数变异预测模块400和染色体拷贝数变异预测模块500,其中:
接收模块100,用于接收输入的待测样本以及对照样本对应的基因序列文件;
单核苷酸多态性位点提取模块200,用于对基因序列文件进行分析得到单核苷酸多态性位点;
入参计算模块300,用于根据预设输入参数计算逻辑,计算所得到的单核苷酸多态性位点对应的模型输入参数;
位点拷贝数变异预测模块400,用于将模型输入参数输入至预先训练完成的位点拷贝数变异识别模型中,得到每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置;
染色体拷贝数变异预测模块500,用于将每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比,染色体拷贝数变异识别模型是根据长短期记忆网络训练模型得到的。
在其中一个实施例中,上述装置还可以包括:
肿瘤含量预测模块,用于将待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比输入至肿瘤预测模型中,得到待测样本对应的肿瘤含量以及倍型,肿瘤预测模型是根据长短期记忆网络训练模型得到的。
在其中一个实施例中,上述的单核苷酸多态性位点提取模块200用于从待测样本和对照样本对应的基因序列文件中的提取基因序列,并将所提取的基因序列分别与标准基因组进行比对,得到单核苷酸多态性位点。
在其中一个实施例中,上述的入参计算模块300可以包括:
第一统计单元,用于统计待测样本的基因序列与标准基因组相同的第一序列以及不同的第二序列;
第一读取单元,用于读取第一序列中的目标位点的观测值、第二序列中的目标位点的观测值,以及待测样本中的基因序列中目标位点的观测值;
第二统计单元,用于统计对照样本的基因序列与标准基因组相同的第三序列和不同的第四序列;
第二读取单元,用于读取第三序列中的目标位点的观测值、第四序列中的目标位点的观测值,以及对照样本中的基因序列中目标位点的观测值;
聚合酶链式反应亲和度计算单元,用于根据目标位点前后预设长度区域内的点位计算得到目标点位对应的聚合酶链式反应亲和度;
染色体扩增参考指标计算单元,用于根据待测样本中的基因序列中目标位点的观测值、对照样本中的基因序列中目标位点的观测值计算得到染色体扩增参考指标;
染色体缺失参考指标计算单元,用于根据第二序列中的目标位点的观测值、待测样本中的基因序列中目标位点的观测值、第四序列中的目标位点的观测值以及对照样本中的基因序列中目标位点的观测值计算得到染色体缺失参考指标。
在其中一个实施例中,上述位点拷贝数变异预测模块400可以包括:
输入矩阵计算单元,用于将模型输入参数作为待测样本对应的基因序列的输入矩阵;
逆序拼接单元,用于获取待测样本对应的基因序列的预设数量的点位作为初始序列,并将初始序列进行逆序拼接得到输入序列;
第一模型处理单元,用于将输入序列输入至预先训练完成的位点拷贝数变异识别模型中,得到每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置。
在其中一个实施例中,每个单核苷酸多态性位点对应的拷贝数状态包括父本染色体个数、母本染色体个数、细胞总数以及突变的细胞数;上述的染色体拷贝数变异预测模块500可以包括:
分割单元,用于根据分割点位置对待测样本对应的基因序列进行分割;
参数计算单元,用于根据细胞总数、突变的细胞数、父本染色体个数和母本染色体个数计算得到均一的父本染色体个数、均一母本染色体个数;
第二模型处理单元,用于将分割后的基因序列、均一的父本染色体个数和均一母本染色体个数输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比。
关于拷贝数变异预测装置的具体限定可以参见上文中对于拷贝数变异预测方法的限定,在此不再赘述。上述拷贝数变异预测装置中的各个模块可全部或部分通过软件、硬件及其组合来实现。上述各模块可以硬件形式内嵌于或独立于计算机设备中的处理器中,也可以以软件形式存储于计算机设备中的存储器中,以便于处理器调用执行以上各个模块对应的操作。
在一个实施例中,提供了一种计算机设备,该计算机设备可以是终端,其内部结构图可以如图10所示。该计算机设备包括通过系统总线连接的处理器、存储器、通信接口、显示屏和输入装置。其中,该计算机设备的处理器用于提供计算和控制能力。该计算机设备的存储器包括非易失性存储介质、内存储器。该非易失性存储介质存储有操作系统和计算机程序。该内存储器为非易失性存储介质中的操作系统和计算机程序的运行提供环境。该计算机设备的通信接口用于与外部的终端进行有线或无线方式的通信,无线方式可通过wifi、运营商网络、nfc(近场通信)或其他技术实现。该计算机程序被处理器执行时以实现一种拷贝数变异预测方法。该计算机设备的显示屏可以是液晶显示屏或者电子墨水显示屏,该计算机设备的输入装置可以是显示屏上覆盖的触摸层,也可以是计算机设备外壳上设置的按键、轨迹球或触控板,还可以是外接的键盘、触控板或鼠标等。
本领域技术人员可以理解,图10中示出的结构,仅仅是与本申请方案相关的部分结构的框图,并不构成对本申请方案所应用于其上的计算机设备的限定,具体的计算机设备可以包括比图中所示更多或更少的部件,或者组合某些部件,或者具有不同的部件布置。
在一个实施例中,提供了一种计算机设备,包括存储器和处理器,存储器中存储有计算机程序,该处理器执行计算机程序时实现以下步骤:接收输入的待测样本以及对照样本对应的基因序列文件;对基因序列文件进行分析得到单核苷酸多态性位点;根据预设输入参数计算逻辑,计算所得到的单核苷酸多态性位点对应的模型输入参数;将模型输入参数输入至预先训练完成的位点拷贝数变异识别模型中,得到每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置;将每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比,染色体拷贝数变异识别模型是根据长短期记忆网络训练模型得到的。
在一个实施例中,处理器执行计算机程序时所实现的将每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比之后,还包括:将待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比输入至肿瘤预测模型中,得到待测样本对应的肿瘤含量以及倍型,肿瘤预测模型是根据长短期记忆网络训练模型得到的。
在一个实施例中,处理器执行计算机程序时所实现的对基因序列文件进行分析得到单核苷酸多态性位点,包括:从待测样本和对照样本对应的基因序列文件中的提取基因序列,并将所提取的基因序列分别与标准基因组进行比对,得到单核苷酸多态性位点。
在一个实施例中,处理器执行计算机程序时所实现的根据预设输入参数计算逻辑,计算所得到的单核苷酸多态性位点对应的模型输入参数,包括:统计待测样本的基因序列与标准基因组相同的第一序列以及不同的第二序列;读取第一序列中的目标位点的观测值、第二序列中的目标位点的观测值,以及待测样本中的基因序列中目标位点的观测值;统计对照样本的基因序列与标准基因组相同的第三序列和不同的第四序列;读取第三序列中的目标位点的观测值、第四序列中的目标位点的观测值,以及对照样本中的基因序列中目标位点的观测值;根据目标位点前后预设长度区域内的点位计算得到目标点位对应的聚合酶链式反应亲和度;根据待测样本中的基因序列中目标位点的观测值、对照样本中的基因序列中目标位点的观测值计算得到染色体扩增参考指标;根据第二序列中的目标位点的观测值、待测样本中的基因序列中目标位点的观测值、第四序列中的目标位点的观测值以及对照样本中的基因序列中目标位点的观测值计算得到染色体缺失参考指标。
在一个实施例中,处理器执行计算机程序时所实现的将模型输入参数输入至预先训练完成的位点拷贝数变异识别模型中,得到每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置,包括:将模型输入参数作为待测样本对应的基因序列的输入矩阵;获取待测样本对应的基因序列的预设数量的点位作为初始序列,并将初始序列进行逆序拼接得到输入序列;将输入序列输入至预先训练完成的位点拷贝数变异识别模型中,得到每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置。
在一个实施例中,处理器执行计算机程序时所涉及的每个单核苷酸多态性位点对应的拷贝数状态包括父本染色体个数、母本染色体个数、细胞总数以及突变的细胞数;处理器执行计算机程序时所实现的将每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比,包括:根据分割点位置对待测样本对应的基因序列进行分割;根据细胞总数、突变的细胞数、父本染色体个数和母本染色体个数计算得到均一的父本染色体个数、均一母本染色体个数;将分割后的基因序列、均一的父本染色体个数和均一母本染色体个数输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比。
在一个实施例中,提供了一种计算机可读存储介质,其上存储有计算机程序,计算机程序被处理器执行时实现以下步骤:接收输入的待测样本以及对照样本对应的基因序列文件;对基因序列文件进行分析得到单核苷酸多态性位点;根据预设输入参数计算逻辑,计算所得到的单核苷酸多态性位点对应的模型输入参数;将模型输入参数输入至预先训练完成的位点拷贝数变异识别模型中,得到每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置;将每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比,染色体拷贝数变异识别模型是根据长短期记忆网络训练模型得到的。
在一个实施例中,计算机程序被处理器执行时所实现的将每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比之后,还包括:将待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比输入至肿瘤预测模型中,得到待测样本对应的肿瘤含量以及倍型,肿瘤预测模型是根据长短期记忆网络训练模型得到的。
在一个实施例中,计算机程序被处理器执行时所实现的对基因序列文件进行分析得到单核苷酸多态性位点,包括:从待测样本和对照样本对应的基因序列文件中的提取基因序列,并将所提取的基因序列分别与标准基因组进行比对,得到单核苷酸多态性位点。
在一个实施例中,计算机程序被处理器执行时所实现的根据预设输入参数计算逻辑,计算所得到的单核苷酸多态性位点对应的模型输入参数,包括:统计待测样本的基因序列与标准基因组相同的第一序列以及不同的第二序列;读取第一序列中的目标位点的观测值、第二序列中的目标位点的观测值,以及待测样本中的基因序列中目标位点的观测值;统计对照样本的基因序列与标准基因组相同的第三序列和不同的第四序列;读取第三序列中的目标位点的观测值、第四序列中的目标位点的观测值,以及对照样本中的基因序列中目标位点的观测值;根据目标位点前后预设长度区域内的点位计算得到目标点位对应的聚合酶链式反应亲和度;根据待测样本中的基因序列中目标位点的观测值、对照样本中的基因序列中目标位点的观测值计算得到染色体扩增参考指标;根据第二序列中的目标位点的观测值、待测样本中的基因序列中目标位点的观测值、第四序列中的目标位点的观测值以及对照样本中的基因序列中目标位点的观测值计算得到染色体缺失参考指标。
在一个实施例中,计算机程序被处理器执行时所实现的将模型输入参数输入至预先训练完成的位点拷贝数变异识别模型中,得到每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置,包括:将模型输入参数作为待测样本对应的基因序列的输入矩阵;获取待测样本对应的基因序列的预设数量的点位作为初始序列,并将初始序列进行逆序拼接得到输入序列;将输入序列输入至预先训练完成的位点拷贝数变异识别模型中,得到每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置。
在一个实施例中,计算机程序被处理器执行时所涉及的每个单核苷酸多态性位点对应的拷贝数状态包括父本染色体个数、母本染色体个数、细胞总数以及突变的细胞数;计算机程序被处理器执行时所实现的将每个单核苷酸多态性位点对应的拷贝数状态以及分割点位置输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比,包括:根据分割点位置对待测样本对应的基因序列进行分割;根据细胞总数、突变的细胞数、父本染色体个数和母本染色体个数计算得到均一的父本染色体个数、均一母本染色体个数;将分割后的基因序列、均一的父本染色体个数和均一母本染色体个数输入至预先训练完成的染色体拷贝数变异识别模型中,得到待测样本对应的拷贝数变异状态以及对应每一拷贝数变异状态的百分比。
本领域普通技术人员可以理解实现上述实施例方法中的全部或部分流程,是可以通过计算机程序来指令相关的硬件来完成,所述的计算机程序可存储于一非易失性计算机可读取存储介质中,该计算机程序在执行时,可包括如上述各方法的实施例的流程。其中,本申请所提供的各实施例中所使用的对存储器、存储、数据库或其它介质的任何引用,均可包括非易失性和易失性存储器中的至少一种。非易失性存储器可包括只读存储器(read-onlymemory,rom)、磁带、软盘、闪存或光存储器等。易失性存储器可包括随机存取存储器(randomaccessmemory,ram)或外部高速缓冲存储器。作为说明而非局限,ram可以是多种形式,比如静态随机存取存储器(staticrandomaccessmemory,sram)或动态随机存取存储器(dynamicrandomaccessmemory,dram)等。
以上实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本申请的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本申请构思的前提下,还可以做出若干变形和改进,这些都属于本申请的保护范围。因此,本申请专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!