一种细菌菌株的分析方法、分析装置和存储介质与流程
本公开涉及微生物信息的分析方法和分析装置,更具体地,涉及利用计算机实现的细菌菌株的分析方法、分析装置和存储介质。
背景技术:
对菌群的生理功能研究已经进入菌株时代。越来越多的研究发现,同一菌种下的不同类的菌株在功能和生理作用上存在着巨大的差异,但是对这些菌株的基因组研究却严重滞后。传统的菌株基因组分析需要先从样品中对细菌进行分离和培养,然后再对菌落进行测序,以了解菌株的基因组序列。其中,感兴趣的菌株的分离培养是一个漫长的试错过程,也是传统的菌株基因组分析的瓶颈所在。
随着高通量测序技术的普及和宏基因组分析技术的出现,研究者可以直接从样品测序数据中通过生物信息分析工具提取菌群组成、基因家族以及功能信息。这一过程不依赖于实验培养,从而大大加速了微生物组研究的进程。早先对宏基因组菌株分析主要采用非参(denovo)的方案。但是受限于组装中的各种错误,宏基因组的菌株分析准确度较差。
基于参考数据库(有参)的基因组的菌株分析方法逐渐兴起。一类菌株分析方法基于单核苷酸多态性(snp),从数据中寻找保守基因片段上的突变位点,并以此确定菌种的菌株组成。这类方法已经越来越多地在宏基因组分析中使用,应用于环境病原菌溯源、粪菌移植(fmt)菌株追踪等场景。但是,这类菌株分析方法无法反映菌株的全体基因信息,例如,保守基因片段上的这些保守基因有时对于菌种中的菌株的特异性功能没有体现。
另一类菌株分析方法以panphlan为代表的另一类方法则使用菌种的泛基因组(pan-genome),根据该菌种的泛基因组上基因的存在与缺失来判断菌株的基因组成。相比于第一类方法,panphlan提供了菌株的基因组成,因而让研究者可以从基因的功能层面对菌株进行深入的解读分析。但是panphlan只提取了处于优势的单类菌株的基因组成。由于样品中同一菌种下面往往多种菌株同时存在,且菌株组成根据时间或者采样条件而变化,相应地,菌株比例的变动可能导致优势菌株的动态更迭。有时,优势菌株仅仅具有低位优势,在正常扰动下也会变化。panphlan并不能够反映多类菌株的组成状况,也无法探知优势菌株变化是由于正常扰动引起还是因为菌株微生态环境发生剧烈变化引起,在菌株分析中具有相当多的局限。
技术实现要素:
提供了本公开以解决现有技术中存在的上述问题。
需要一种细菌菌株的分析方法、分析装置和存储介质,其能够从泛基因组层面进行菌株的分析,准确地得到各个样品中多个菌种的各类菌株的组成比例情况以及其中每类菌株的基因家族包含情况,从而能够对各个样品中多个菌种下各类菌株的功能情况进行全面分析。
根据本公开的第一方面,提供了一种细菌菌株的分析方法。所述分析方法可以包括获取目标菌种的泛基因组数据(所述泛基因组数据可以包括目标菌种的多类菌株的参考基因序列),以及获取多个样品的测序数据,每个样品包括多个菌种。该分析方法可以包括经由至少一个处理器,将每个样品的测序数据与所述目标菌种的泛基因组数据进行比对,以得到相对于泛基因组中各个参考基因的比对计数,并将相似基因的比对计数合并为基因家族的比对计数,以确定第一矩阵。所述第一矩阵的各个元素可以表征各个基因家族在各个样品中的比对计数相关信息。该分析方法可以进一步包括经由所述至少一个处理器,将所述第一矩阵分解为第二矩阵和第三矩阵。其中,所述第二矩阵的各个元素可以表征各个基因家族在各类菌株中的存在相关信息,所述第三矩阵的各个元素可以表征各类菌株在各个样品中的相对丰度相关信息。
根据本公开的第二方面,提供了一种细菌菌株的分析装置。所述分析装置可以包括接口和至少一个处理器。接口可以配置为:接收多个样品的测序数据,每个样品包括多个菌种。所述至少一个处理器可以配置为执行根据本公开各个实施例所述的细菌菌株的分析方法。
根据本公开的第三方面,提供了一种细菌菌株的分析装置。所述分析装置可以包括第一获取单元、第二获取单元、比对单元、合并单元和分解单元。第一获取单元可以配置为获取目标菌种的泛基因组数据(所述泛基因组数据可以包括目标菌种的多类菌株的参考基因序列)。第二获取单元可以配置为获取多个样品的测序数据,每个样品包括多个菌种。比对单元可以配置为将每个样品的测序数据与所述目标菌种的泛基因组数据进行比对,以得到各个样品相对于泛基因组中各个参考基因的比对计数。合并单元可以配置为将相似基因的比对计数合并为基因家族的比对计数,以确定第一矩阵,所述第一矩阵的各个元素表征各个基因家族在各个样品中的比对计数相关信息。分解单元可以配置为将所述第一矩阵分解为第二矩阵和第三矩阵。其中,所述第二矩阵的各个元素可以表征各个基因家族在各类菌株中的存在相关信息,所述第三矩阵的各个元素可以表征各类菌株在各个样品中的相对丰度相关信息。
根据本公开的第四方面,提供了一种非易失性计算机可读存储介质,其上存储有可执行指令。所述可执行指令由至少一个处理器执行时,实现根据本公开各个实施例所述的细菌菌株的分析方法。
利用根据本公开的各个实施例的细菌菌株的分析方法、分析装置和存储介质,其能够从泛基因组层面进行菌株的分析,准确地得到各个样品中多个菌种的各类菌株的整体组成情况以及其中每类菌株的基因家族包含情况,从而能够对各个样品中多个菌种下各类菌株的功能情况进行全面分析。
附图说明
在不一定按比例绘制的附图中,相同的附图标记可以在不同的视图中描述相似的部件。具有字母后缀或不同字母后缀的相同附图标记可以表示相似部件的不同实例。附图大体上通过举例而不是限制的方式示出各种实施例,并且与说明书以及权利要求书一起用于对所公开的实施例进行说明。这样的实施例是例证性的,而并非旨在作为本装置或方法的穷尽或排他实施例。
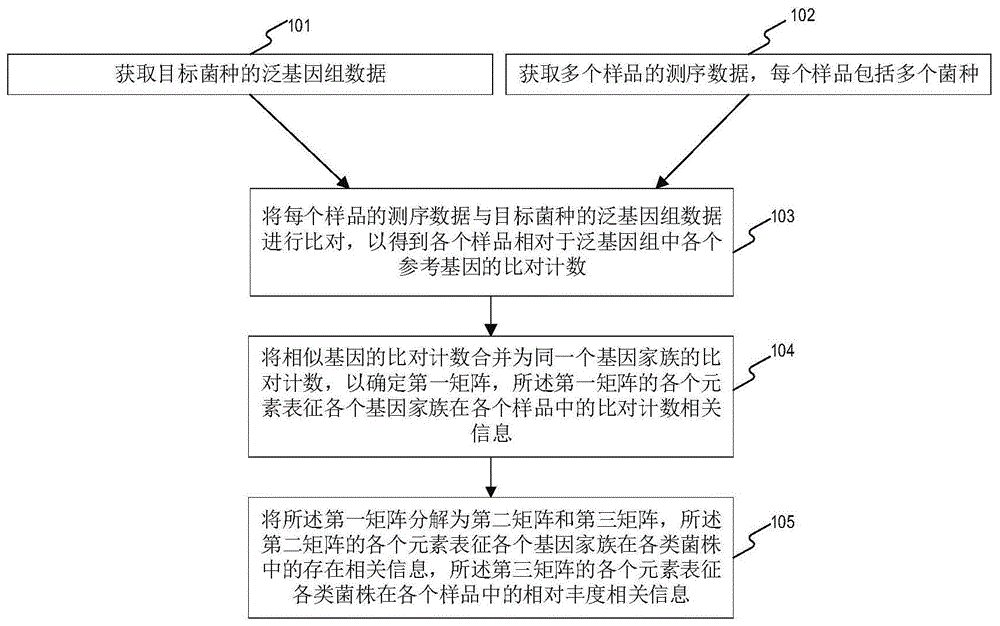
图1示出根据本公开实施例的细菌菌株的分析方法的过程的流程图;
图2示出根据本公开实施例的细菌菌株的分析方法中将第一矩阵分解为第二矩阵和第三矩阵的子流程的流程图;
图3示出根据本公开实施例的细菌菌株的分析方法中初始化子流程的流程图;
图4示出根据本公开实施例的细菌菌株的分析装置的配置图示;
图5示出根据本公开实施例的细菌菌株的分析装置的框图;
图6(a)示出预设10个样品的预设的5类菌株的参考菌株组成与利用根据本公开实施例的细菌菌株的分析方法对预设10个样品分析所得的菌株组成的对比图;
图6(b)示出利用根据本公开实施例的细菌菌株的分析方法对预设10个样品分析所得的5类菌株包含情况的精确率-召回率曲线的图示;
图7(a)-图7(f)分别示出将根据本公开实施例的细菌菌株的分析方法应用于粪菌移植(fmt)数据集来分别评估4名供体与各自的受体fmt前后各时间点的普拉梭菌(faecalibacteriumprausnitzii,也称为f.prausnitzii)的菌株的组成变化的图示;以及
图8示出图7(a)-图7(f)中所示的普拉梭菌的三类菌株的基因分别对应的通路功能。
具体实施方式
为使本领域技术人员更好的理解本公开的技术方案,下面结合附图和具体实施方式对本公开作详细说明。下面结合附图和具体实施例对本公开的实施例作进一步详细描述,但不作为对本公开的限定。在本公开中使用的“第一”、“第二”和“第三”的措辞,仅仅旨在区分相应的特征,并不代表需要这样的排序,也未必仅表示单数形式。在本公开中使用的“所述泛基因组数据”应采取“包括目标菌种的多类菌株的参考基因序列”的宽泛解释,也就是说,可以理解为包括任何数量的目标菌种的任何类菌株的任何参考基因序列。
图1示出了根据本公开实施例的细菌菌株的分析方法的过程的流程图。如图1所示,该过程可以始于步骤101和步骤102。在步骤101,可以获取目标菌种的泛基因组数据,所述泛基因组数据可包括目标菌种的多类菌株的参考基因序列。在一些实施例中,可以经由网络连接到泛基因组数据库,以便从中取得目标菌种的泛基因组数据,下载到本地以供调用。在一些实施例中,也可以获取目标菌种的各类菌株的基因组序列,并基于此来生成目标菌种的泛基因组数据以供使用。在步骤102可以获取多个样品的测序数据(例如但不限于fastq格式的dna测序下机数据等),每个样品可包括多个菌种,每个菌种可能具有多类菌株。
在步骤103,经由至少一个处理器,将每个样品的测序数据与所述目标菌种的泛基因组数据进行比对,以得到各个样品相对于泛基因组中各个参考基因的比对计数。通过使用目标菌种的泛基因组数据(尤其是泛基因组中各个参考基因的基因序列)作为比对参考,可以准确地反映各个样品的综合基因信息,从泛基因组层面进行样品级别的分析。
在步骤104,可以将相似基因的比对计数合并为基因家族的比对计数,以确定第一矩阵。所述第一矩阵的各个元素表征各个基因家族在各个样品中的比对计数相关信息。为了描述方便,在本公开中也将第一矩阵简称为d矩阵。基因家族是由一个共同的祖先基因经过重复和突变产生的、具有结构和功能相似性序列的一组相关基因;在本公开中认为相似基因属于同个基因家族。可以根据基因名称、基因注释、序列相似性等中的至少一种来选出相似基因。可以认为相似基因归属于同一个基因家族。通过将各组相似基因的比对计数进行合并,可以得到各个样品中基因家族级别的比对计数相关信息,相较合并之前的基因级别的比对计数相关信息,显著降低计算矩阵的维度并且增强了比对计数相关信息的分布规律的鲁棒性,从而为步骤105的分解稳定性奠定基础。
在步骤105,可以经由所述至少一个处理器,将d矩阵(即所述第一矩阵)分解为第二矩阵和第三矩阵。所述第二矩阵的各个元素表征各个基因家族在各类菌株中的存在相关信息,所述第三矩阵的各个元素表征各类菌株在各个样品中的相对丰度相关信息。为了描述方便,在本公开中也将第二矩阵简称为p矩阵而将第三矩阵简称为s矩阵。p矩阵也可以称为菌株基因家族谱,s矩阵也可以称为菌株丰度谱。发明人创造性地发现,通过在步骤104确定各个元素表征各个基因家族在各个样品中的比对计数相关信息的d矩阵,可以采用各种方式来迭代得到稳定的p矩阵和s矩阵。通过分解得到的p矩阵和s矩阵,可以准确得到每个基因家族在每类菌株中的存在或缺失状况,并且可以准确得到每类菌株在每个样品中的相对丰度,从而能够对各个样品中多个菌种下各类菌株的功能情况进行全面分析。
在一些实施例中,所述d矩阵的行可以对应各个基因家族而列可以对应各个样品,相应地,所述p矩阵的行可以对应各个基因家族而列可以对应各类菌株,并且所述s矩阵的行可以对应各类菌株而列可以对应各个样品。当然,d矩阵、p矩阵和s矩阵的行和列的设置并不限于此,例如,可以分别对其中各个矩阵进行转置等。
在一些实施例中,可以将p矩阵的各个元素二元化处理,使得所述存在相关信息为表示是否存在的二元信息,例如但不限于,用1表示存在而用0表示缺失。如此,可以使得p矩阵稀疏化,也就是将d矩阵的分解转换为稀疏化分解,从而降低运算负荷并加快分析速度。
图2示出根据本公开实施例的细菌菌株的分析方法中将d矩阵分解为p矩阵和s的子流程的流程图。如图2所示,在得到d矩阵之后,可以进行s矩阵的初始化处理(步骤202),以生成初始s矩阵。可以根据d矩阵和初始p矩阵的信息,生成初始s矩阵,例如可以通过将所述d矩阵与初始p矩阵的逆矩阵相乘,来得到初始s矩阵,也可以经由回归(例如但不限于线性回归)来得到初始s矩阵。具体说来,可以通过以下优化方程(1)通过线性回归来求解得到初始s矩阵。
其中,dij表示d矩阵的第i行第j列的元素,pik表示p矩阵的第i行第k列的元素,skj表示s矩阵的第k行第j列的元素。本发明人发现,d矩阵相对于上述定义的p矩阵和s矩阵具有良好的线性可分解性,通过计算负担较小的线性回归,可以利用公式(1)定义的优化方程来稳定分解出初始s矩阵。
初始s矩阵(下文中表示为sini)可以代入,以实现p矩阵和s矩阵的交替迭代求解(参见步骤203和204),直到收敛(参见步骤209),或者达到预设的迭代次数为止。虽然在图2中先后示出了步骤203(“估算p矩阵”)和步骤204(“估算s矩阵”),但须知并不限于此,也可以调换估算的次序,只要p矩阵和s矩阵是交替迭代估算的即可。
在一些实施例中,在每个迭代步,可以采用如下方式来求解p矩阵。为了便于描述,将每个迭代步求解得到的p矩阵和s矩阵分别表示为p和s,在每个迭代步中使用的分别与p和s相关的中间矩阵用上角标区别标识出来。
可以根据公式(2)来确定第四矩阵p′:
p′=ds′-1公式(2)
其中,s′表示当前得到的s矩阵。以图2中所示的p矩阵和s矩阵的交替迭代求解次序为例,第n个(n为大于1的自然数)迭代步使用的当前得到的s矩阵s′即第n-1个迭代步最后得到的s矩阵,第1个迭代步使用的当前得到的s矩阵s′即初始s矩阵sini。
第四矩阵p′可以作为p计算的中间矩阵,区别在于,p′的各个元素为连续数据,每列的元素可表征各个基因家族相对于对应类的菌株的量化分布信息。可以确定每列与各个基因家族相关联的概率密度函数。所得到的概率密度函数具有低位分布峰(即靠近0的峰)和非低位分布峰(通常对应于该类菌株中实际存在的基因家族)。发明人发现,低位分布峰反映的是噪声信息,可以从第四矩阵p′的各列中去除与对应概率密度函数的低位分布峰对应的元素,从而有效地滤除噪声。所谓噪声信息对应的基因家族实际上在菌株中并不该存在,而是因为误差或者扰动错误地引入;通过针对性且高效地滤除噪声,可以显著降低d矩阵分解的失败率。接着,可以将滤除噪声后的第四矩阵p′中的各个元素与第一阈值进行比较,例如对超过第一阈值的元素赋予非零值(比如1)而对其他元素赋予零值,从而得到二元化的p矩阵。通过基于第一阈值的二元化处理,不仅便于提高后续s的迭代求解的效率,也有利于改善p的鲁棒性,进一步提高d矩阵分解的稳定性。
对于第n个迭代步来说,在得到p之后,可以基于d矩阵和当前得到的p矩阵(即p)通过回归方法来求解所述s矩阵。所述回归方法包括但不限于线性回归。
下面结合图3对步骤202即初始化处理步骤的实例进行进一步说明。
在一些实施例中,可以在步骤301,首先基于目标菌种的泛基因组数据生成初始p矩阵pini,并基于d矩阵和初始p矩阵pini,经由回归来求解得到初始s矩阵sini,例如根据公式(1)来优化回归求解。在一些实施例中,可以基于目标菌种的多类菌株的基因相关信息(其可以构成泛基因组数据的至少一部分)来生成初始p矩阵pini。在一些实施例中,可以对目标菌种的多类菌株的基因相关信息进行聚类(例如但不限于层次聚类),从各个聚类中选择代表性菌株(例如可以选择居中的菌株作为该代表性菌株),并将各个代表性菌株的基因相关信息转化为基因家族的存在信息列表,从而得到初始p矩阵pini。
在一些实施例中,基于d矩阵和初始p矩阵pini经由回归求解得到的s矩阵(为了描述方便也称为第五矩阵,以与迭代处理中的s矩阵和初始s矩阵本身相区别开)可以直接用作sini,所述第五矩阵的各个元素表征各类菌株在各个样品中的相对丰度相关信息(参见步骤302)。
在另一些实施例中,可以对步骤302得到的第五矩阵进行进一步处理。例如,可以在步骤303将所述第五矩阵的各个元素的代表值与第二阈值进行比较,来选出对应元素的代表值超出所述第二阈值的菌株类。注意,本公开中“各个元素的代表值”可以表示各个元素自身,也可以表示各个元素进行代表性处理所得到的值,例如平均值、中值、最小值等等。对应元素的代表值意味着对应类的菌株在全部样品中的代表性的相对丰度,通过选出前者超出第二阈值的菌株类,可以筛选出在全部样品中的代表性的相对丰度超过一定阈值的菌株类。如此,不仅可以滤除由于干扰或噪声引入的并非真实存在的菌株类的干扰信息(也可以称为“假阳性信息”),也可以滤除虽然真实存在但丰度低而生物学意义不大确定的菌株类的不确定信息。本发明人创造性地发现,通过滤除干扰信息和不确定信息两者,保留所述初始p矩阵中选出的菌株类对应的列来得到处理后的初始p矩阵(步骤304),基于d矩阵和处理后的初始p矩阵,经过回归来求解得到sini(步骤305),可以显著提高回归求解sini的稳定性和成功率。
在本公开的各个实施例中,用于求解初始s矩阵sini的各种方法都可以转用于迭代求解s矩阵,在此不赘述。
馈送到初始化步骤202的d矩阵可以预先进行标准化处理(参见步骤201)。具体说来,可以将多个样品的测序数据与所述目标菌种的泛基因组数据进行比对,确定每个样品相对于所述目标菌种的泛基因组数据中各个基因的比对计数,以得到第六矩阵。所述第六矩阵的各个元素可以表示各个基因在各个样品中的比对计数。
基于基因长度和样品测序深度,对所述第六矩阵的各个元素进行标准化处理。作为示例而非限制,可以根据公式(3)来对所述第六矩阵的各个元素进行标准化处理。
其中,readcount表示第六矩阵的各个元素即每个样品相对于所述目标菌种的泛基因组中各个参考基因的比对计数,genelength表示基因长度,librarysize表示样品测序深度,rpkm表示标准化处理后的各个元素。通过这样的标准化处理,可以消除基因长度和样品测序深度对比对计数的影响。注意,公式(3)仅仅作为示例,也可以采用其他方式,例如,在公式(3)的分母中采用使用基因长度和样品测序深度作为参数的其他表达式(例如但不限于多项式、对数处理等)来执行标准化处理,以便消除基因长度和样品测序深度的影响。
可以根据公式(4),对标准化处理后的第六矩阵中归属于同个基因家族的各个基因的元素求和,来得到所述d矩阵。
其中,genefamilyi表示d矩阵的第i行对应的基因家族,g表示归属于该基因家族的各个基因。
图4示出根据本公开实施例的细菌菌株的分析装置400的配置图示。如图4所示,该分析装置400可以包括第一获取单元401、第二获取单元402、比对单元403、合并单元404和分解单元405。第一获取单元401可以配置为获取目标菌种的泛基因组数据,例如可以获取本地的目标菌种的泛基因组数据,也可以经由网络下载并(例如从菌种的泛基因组数据库)获取目标菌种的泛基因组数据,其中,所述泛基因组数据包括目标菌种的多类菌株的参考基因序列。
第二获取单元402可以配置为获取多个样品的测序数据,每个样品包括多个菌种,其中每个菌种可以具有数类菌株。比对单元403可以配置为将多个样品的测序数据与所述目标菌种的泛基因组数据进行比对,以得到各个样品相对于泛基因组中各个参考基因的比对计数。合并单元404可以配置为将相似基因的比对计数合并为基因家族的比对计数,以确定d矩阵,所述d矩阵的各个元素表征各个基因家族在各个样品中的比对计数相关信息。分解单元405可以配置为:将所述d矩阵分解为p矩阵和s矩阵,所述p矩阵的各个元素可以表征各个基因家族在各类菌株中的存在相关信息,所述s矩阵的各个元素可以表征各类菌株在各个样品中的相对丰度相关信息。
在一些实施例中,分解单元404可以进一步包括初始化单元406和迭代求解单元407,以分别执行根据本公开各个实施例的初始化处理和迭代求解处理,根据本公开各个实施例的初始化处理和迭代求解处理的变型都可以结合与此,在此不赘述。
具体说来,初始化单元406可以配置为:基于目标菌种的泛基因组数据,生成初始p矩阵;并根据d矩阵和初始p矩阵的信息,经由回归(包括不限于一次或数次回归)来得到初始s矩阵。迭代求解单元407可以配置为迭代地求解p矩阵和s矩阵。具体说来,迭代求解单元407可以进一步配置为,在各个迭代步骤中:通过将所述d矩阵与当前得到的s矩阵的逆矩阵相乘,来确定第四矩阵;确定所述第四矩阵每列与各个基因家族相关联的概率密度函数;从所述第四矩阵的各列中去除与对应概率密度函数的低位分布峰对应的元素,以滤除噪声;将滤除噪声后的第四矩阵中的各个元素与第一阈值进行比较,以得到当前的p矩阵;基于所述d矩阵和当前得到的p矩阵通过回归方法来求解s矩阵,以便用于在下个迭代步骤中确定所述第四矩阵。
在一些实施例中,图4中的各个单元可以实现为软件模块,该软件模块的计算机可执行指令可由处理器执行来实现相应的计算处理。作为示例,该软件模块可以利用r语言在nextflow的框架下编写实现,但这仅仅作为示例,也可以利用c++、python等语言来编写,也可以利用其它框架来实现。
图5示出根据本公开实施例的细菌菌株的分析装置500的框图。如图5所示,所述分析装置500可以包括通信接口503和至少一个处理器504。通信接口503可以配置为:接收多个样品的测序数据,每个样品包括多个菌种。在一些实施例中,所述通信接口503还可以配置为经由网络来下载目标菌种的泛基因组数据,以供本地使用。所述泛基因组数据可以包括目标菌种的多类菌株的参考基因序列。或者,在一些实施例中,所述通信接口503还可以配置为接收目标菌种的菌株基因组序列,以便利用所述至少一个处理器504基于所接收的目标菌种的菌株基因组序列来生成目标菌种的泛基因组数据。所述至少一个处理器504可以进一步配置为执行根据本公开各个实施例的细菌菌株的分析方法。
分析装置500可以是专用计算机或通用计算机。例如,分析装置500可以是定制的计算机,以执行处理任务。如图5中所示,分析装置500可以包括通信接口503、至少一个处理器504、存储器505、储存器506和显示器507。在一些实施例中,分析装置500可以实现为用户终端或者服务器(例如但不限于云端服务器等)。
在一些实施例中,通信接口503可以包括网络适配器、电缆连接器、串行连接器,usb连接器、并行连接器、高速数据传输适配器(诸如光纤、usb3.0、雷电接口等)、无线网络适配器(诸如wifi适配器)、电信(3g、4g/lte、5g等)适配器等。分析装置500可以通过通信接口503连接到其他构件,例如但不限于基因测序装置、泛基因组数据库等。相应地,通信接口503可以配置为获取来自基因测序装置的测序下机数据,或者获取来自泛基因组数据库的目标菌种的泛基因组数据等。
处理器504可以是包括一个以上通用处理设备的处理设备,诸如微处理器、中央处理单元(cpu)、图形处理单元(gpu)等。更具体地,该处理器可以是复杂指令集计算(cisc)微处理器、精简指令集计算(risc)微处理器、超长指令字(vliw)微处理器、运行其他指令集的处理器或运行指令集的组合的处理器。该处理器还可以是一个以上专用处理设备,诸如专用集成电路(asic)、现场可编程门阵列(fpga)、数字信号处理器(dsp)、片上系统(soc)等。处理器504可以通信地耦合到存储器505并且被配置为执行存储在其上的计算机可执行指令,以执行诸如在本公开的各个实施例中描述的细菌菌株的分析方法的处理。
存储器505/储存器506可以是非暂时性计算机可读的介质,诸如只读存储器(rom)、随机存取存储器(ram)、相变随机存取存储器(pram)、静态随机存取存储器(sram)、动态随机存取存储器(dram)、电可擦除可编程只读存储器(eeprom)、其他类型的随机存取存储器(ram)、闪存盘或其他形式的闪存、缓存、寄存器、静态存储器、光盘只读存储器(cd-rom)、数字通用光盘(dvd)或其他光学存储器、盒式磁带或其他磁存储设备,或被用于储存能够被计算机设备访问的信息或指令的任何其他可能的非暂时性的介质等。
在一些实施例中,计算机可执行指令可以储存在储存器506上,以便加载到存储器505由处理器504执行,从而实现根据本公开各个实施例的细菌菌株的分析方法的处理。
在一些实施例中,显示器507可以包括液晶显示器(lcd)、发光二极管显示器(led)、等离子显示器或任何其他类型的显示器,并提供显示器上呈现的图形用户界面(gui)用于用户输入和图像/数据/图表显示。所述显示器可以包括许多不同类型的材料(诸如塑料或玻璃),并且可以是触敏的以从用户接收命令。例如,显示器可以包括基本上刚性的触敏材料(诸如gorilla玻璃tm)或基本上柔韧的(诸如willow玻璃tm)的触敏材料。
通过模拟生成预设了10个样品。具体说来,可以选择5类经过测序的普拉梭菌(faecalibacteriumprausnitzii)菌株,在ncbi数据库中的id分别为gcf_000162015、gcf_002549755、gcf_002549975、gcf_003287415和gcf_003433905(分别称为菌株1、菌株2、菌株3、菌株4和菌株5),从每类菌株的全基因组测序数据中随机抽取数据,并按照预先设置比例混合成10个样品,即样品1-样品10。
每个预设样品的参考菌株组成是已知的,可以用作地面真值(图6(a)中示出为“参考菌株组成”),以与利用根据本公开实施例的细菌菌株的分析方法对预设10个样品分析所得的菌株组成(即s矩阵,图6(a)中示出为“预测菌株组成”)进行比较。如图6(a)所示,根据本公开实施例的细菌菌株的分析方法对预设10个样品分析所得的菌株组成与地面真值吻合度高。
图6(b)示出利用根据本公开实施例的细菌菌株的分析方法对预设10个样品分析所得的5类菌株包含情况(例如但不限于p矩阵的打分排序)的精确率-召回率曲线的图示,可以看到精确率-召回率曲线下面积超过0.99,证实了其与5类菌株的包含情况高度一致。
发明人将根据本公开实施例的细菌菌株的分析方法应用于粪菌移植(fmt)数据集,来评估健康供体与受体fmt前后各时间点的普拉梭菌的菌株的组成变化。具体说来,该fmt数据集包括来自4名健康的供体和19名受体的多个时间点(例如可由横坐标轴上的fmt5、fmt6、fmt39、fmt41……等来标识出)的样品。图7(a)示出第1供体(图中示出为“供体mgh01d”)及其受体的菌株组成情况,图7(b)示出第2供体(图中示出为“供体mgh06d”)及其受体的菌株组成情况,图7(c)示出第3供体(图中示出为“供体mgh07d”)及其受体的菌株组成情况,图7(d)-图7(f)示出第4供体(图中示出为“供体mgh03d”)及其受体的菌株组成情况。
从图7(a)-图7(f)可以看到,这4名供体的样品中主要包含3类菌株(即菌株1、菌株2和菌株3,分别用黑色、深灰色和浅灰色表示);一些受体继承了来自供体的部分或者全部菌株,但是受体的菌株的组成比例不同于供体的菌株组成比例。根据本公开实施例的细菌菌株的分析方法可以方便且精确地跟踪健康供体与受体fmt前后的各时间点的普拉梭菌的菌株的组成变化。
图8示出了反映了这三个菌株的基因对应的通路功能。深色代表菌株有该通路功能,浅色代表该通路功能缺失。可以看出,这三类菌株在物质代谢上面存在差异,这可能也会影响到各自在宿主肠道环境中的适应性和生存能力。通过方便且精确地跟踪健康供体与受体fmt前后的各时间点的普拉梭菌的菌株的组成变化,结合各个菌株的基因对应的通路功能,有利于受体对供体的菌株组成比例的继承效果在代谢和通路功能层面的综合分析。
此外,尽管已经在本文中描述了示例性实施例,其范围包括任何和所有基于本公开的具有等同元件、修改、省略、组合(例如,各种实施例交叉的方案)、改编或改变的实施例。权利要求书中的元件将被基于权利要求中采用的语言宽泛地解释,并不限于在本说明书中或本申请的实施期间所描述的示例,其示例将被解释为非排他性的。因此,本说明书和示例旨在仅被认为是示例,真正的范围和精神由以下权利要求以及其等同物的全部范围所指示。
在本公开中的各个步骤的顺序仅仅是示例性的,而非限制性的。在不影响本公开的实现的情况下(不破坏所需的步骤之间的逻辑关系的情况下),可以对步骤的执行顺序进行调整,调整后得到的各种实施例依然落在本公开的范围内。
以上描述旨在是说明性的而不是限制性的。例如,上述示例(或其一个或更多方案)可以彼此组合使用。例如本领域普通技术人员在阅读上述描述时可以使用其它实施例。另外,在上述具体实施方式中,各种特征可以被分组在一起以简单化本公开。这不应解释为一种不要求保护的公开的特征对于任一权利要求是必要的意图。相反,本发明的主题可以少于特定的公开的实施例的全部特征。从而,以下权利要求书作为示例或实施例在此并入具体实施方式中,其中每个权利要求独立地作为单独的实施例,并且考虑这些实施例可以以各种组合或排列彼此组合。本发明的范围应参照所附权利要求以及这些权利要求赋权的等同形式的全部范围来确定。
- 还没有人留言评论。精彩留言会获得点赞!