基于16S微生物扩增测序数据的分析方法、装置及设备与流程
本发明涉及微生物分析技术领域,尤其涉及一种基于16s微生物扩增测序数据的分析方法、装置、计算机设备及存储介质。
背景技术:
16srdna位于原核细胞核糖体小亚基上,包括10个保守区域(conservedregions)和9个高变区域(hypervariableregions),其中保守区在细菌间差异不大,高变区具有属或种的特异性,随亲缘关系不同而有一定的差异。因此,16srdna可以做作为揭示生物物种的特征核酸序列,被认为是最适于细菌系统发育和分类鉴定的指标。
16s(即16srdna)微生物测序技术的实施主要分两部分,一是实验样本建库测序,二是测序数据的生物信息分析。目前实验样本建库测序主要依靠illumina平台完成,而对实验数据的分析主要依靠测序数据的生物信息分析,目前生物信息分析技术尚未有完整的分析方法,即使有,但分析流程过于简单,导致不能很多地进行差异分析。
技术实现要素:
基于此,有必要针对上述问题,提出一种基于16s微生物扩增测序数据的分析方法、装置、计算机设备及存储介质,该分析方法具有完整的分析流程,有利于准确地识别差异基因。
一种基于16s微生物扩增测序数据的分析方法,包括:
获取对16s微生物进行测序得到的原始序列数据,对所述原始序列数据进行过滤得到有效序列数据;
确定所述有效序列数据中正向序列和反向序列的质量值;
根据所述质量值对所述正向序列和反向序列进行过滤,得到过滤后的正向序列和反向序列;
将所述正向序列和所述反向序列进行匹配合并,输出合并得到的otus序列;
将所述otus序列与微生物数据库进行比对注释,确定所述otus序列对应的物种注释和丰度信息;
根据所述otus序列对应的物种注释和丰度信息进行聚类分组,得到多个组;
基于得到的多个组进行组间差异分析。
一种基于16s微生物扩增测序数据的分析装置,包括:
第一过滤模块,用于获取对16s微生物进行测序得到的原始序列数据,对所述原始序列数据进行过滤得到有效序列数据;
质量确定模块,用于确定所述有效序列数据中正向序列和反向序列的质量值;
第二过滤模块,用于根据所述质量值对所述正向序列和反向序列进行过滤,得到过滤后的正向序列和反向序列;
合并模块,用于将所述正向序列和所述反向序列进行匹配合并,输出合并得到的otus序列;
比对模块,用于将所述otus序列与微生物数据库进行比对注释,确定所述otus序列对应的物种注释和丰度信息;
聚类模块,用于根据所述otus序列对应的物种注释和丰度信息进行聚类分组,得到多个组;
分析模块,用于基于得到的多个组进行组间的差异分析。
一种计算机设备,包括存储器和处理器,所述存储器存储有计算机程序,所述计算机程序被所述处理器执行时,使得所述处理器执行以下步骤:
获取对16s微生物进行测序得到的原始序列数据,对所述原始序列数据进行过滤得到有效序列数据;
确定所述有效序列数据中正向序列和反向序列的质量值;
根据所述质量值对所述正向序列和反向序列进行过滤,得到过滤后的正向序列和反向序列;
将所述正向序列和所述反向序列进行匹配合并,输出合并得到的otus序列;
将所述otus序列与微生物数据库进行比对注释,确定所述otus序列对应的物种注释和丰度信息;
根据所述otus序列对应的物种注释和丰度信息进行聚类分组,得到多个组;
基于得到的多个组进行组间差异分析。
一种计算机可读存储介质,存储有计算机程序,所述计算机程序被处理器执行时,使得所述处理器执行以下步骤:
获取对16s微生物进行测序得到的原始序列数据,对所述原始序列数据进行过滤得到有效序列数据;
确定所述有效序列数据中正向序列和反向序列的质量值;
根据所述质量值对所述正向序列和反向序列进行过滤,得到过滤后的正向序列和反向序列;
将所述正向序列和所述反向序列进行匹配合并,输出合并得到的otus序列;
将所述otus序列与微生物数据库进行比对注释,确定所述otus序列对应的物种注释和丰度信息;
根据所述otus序列对应的物种注释和丰度信息进行聚类分组,得到多个组;
基于得到的多个组进行组间差异分析。
上述基于16s微生物扩增测序数据的分析方法、装置、计算机设备及存储介质,首先,获取对16s微生物进行测序得到的原始序列数据,对所述原始序列数据进行过滤得到有效序列数据;确定所述有效序列数据中正向序列和反向序列的质量值;根据所述质量值对所述正向序列和反向序列进行过滤,得到过滤后的正向序列和反向序列;将所述正向序列和所述反向序列进行匹配合并,输出合并得到的otus序列;将所述otus序列与微生物数据库进行比对注释,确定所述otus序列对应的物种注释和丰度信息;根据所述otus序列对应的物种注释和丰度信息进行聚类分组,得到多个组;基于得到的多个组进行组间差异分析。通过上述完整的分析处理流程,可以实现对微生物的聚类分组,以及基于聚类分组实现了组间差异分析,提高了组间差异分析的准确度,而且基于聚类分组有利于进行进一步的分析。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
其中:
图1为一个实施例中基于16s微生物扩增测序数据的分析方法的流程图;
图2为一个实施例中基于16s微生物扩增测序数据的分析装置的结构框图;
图3为另一个实施例中基于16s微生物扩增测序数据的分析装置的结构框图;
图4为一个实施例中计算机设备的内部结构图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
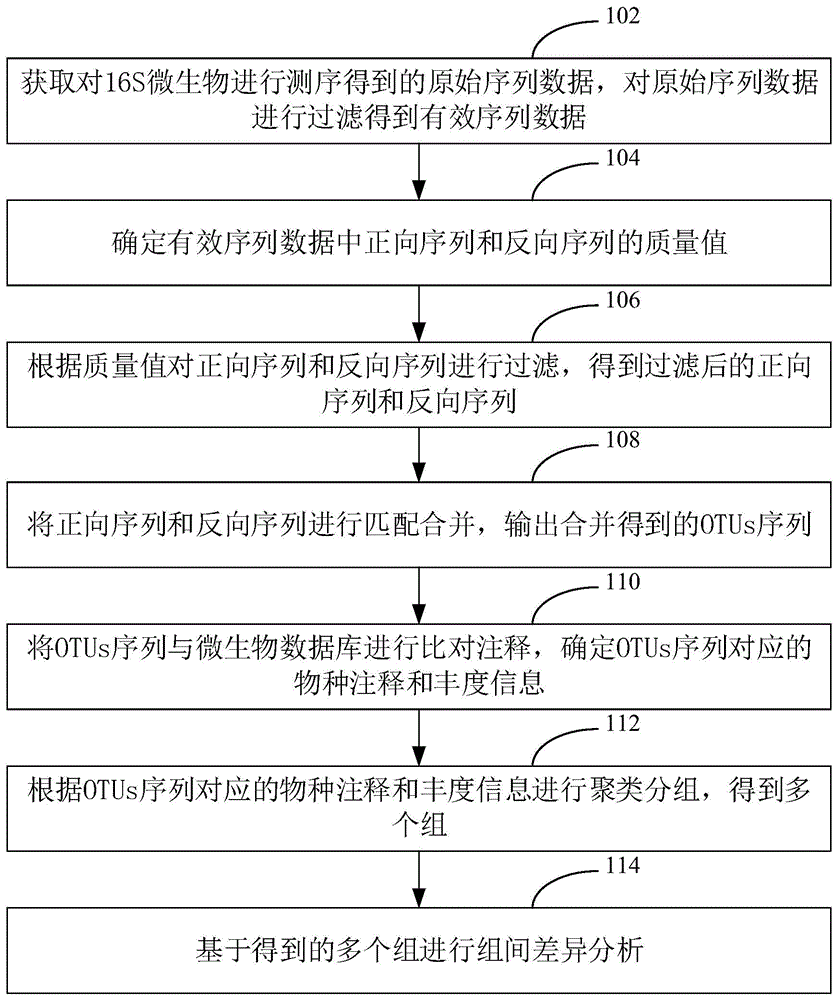
如图1所示,提出了一种基于16s微生物扩增测序数据的分析方法,该基于16s微生物扩增测序数据的分析方法可以应用于终端,本实施例以应用于终端举例说明。该基于16s微生物扩增测序数据的分析方法具体包括以下步骤:
步骤102,获取对16s微生物进行测序得到的原始序列数据,对原始序列数据进行过滤得到有效序列数据。
其中,16s是指16srdna。原始序列数据是指对16s微生物进行测序直接得到的序列数据。有效序列数据是指过滤掉干扰数据之后得到的序列数据。
具体地,16srdna中包括9个可变区和10个保守区,保守区序列反映了物种间的亲缘关系,而可变区序列则能反映物种间的差异。所以为了研究物种间的差异,我们通常选择16srdna某个或某几个变异区域,选择通用引物对环境样本微生物进行pcr扩增,然后对pcr产物进行高通量测序。在一个实施例中,选择v1-v2或v3-v4区域进行pcr扩增,然后进行测序得到原始序列数据。
由于测序得到的原始序列数据中肯定包含有干扰数据,所以需要对原始序列数据进行过滤,过滤是实现后续准确地进行差异分析的前提。
步骤104,确定有效序列数据中正向序列和反向序列的质量值。
其中,有效序列数据中包括正向序列和反向序列,其中,正向序列是指正向进行测序得到的序列,反向序列是指反向测序得到的序列。为了过滤掉质量不合格的正向序列或反向序列,首先计算得到正向序列和反向序列的质量值。质量值可以采用dada2进行分析得到。
步骤106,根据质量值对正向序列和反向序列进行过滤,得到过滤后的正向序列和反向序列。
其中,根据计算得到质量值,将不合格的正向序列和反向序列过滤掉,通过过滤到质量不合格的正向序列和反向序列,有利于提高后续组间差异分析的准确度。
步骤108,将正向序列和反向序列进行匹配合并,输出合并得到的otus序列。
其中,将正向序列和反向序列合并在一起以获得完整的序列,该完整的序列即为otus序列。otus(operationaltaxonomicunits)序列是指可操作分类单元。具体地,将过滤后的正向序列和过滤后的反向序列进行互补序列比对,当两者之间的重合度高于预设值时,则输出两者的合并序列,即得到了otus序列。
步骤110,将otus序列与微生物数据库进行比对注释,确定otus序列对应的物种注释和丰度信息。
其中,将得到otus序列与微生物数据库进行比对注释,分别注释到门、纲、目、科、属水平,然后选取丰度排名靠前的物种。即与微生物数据库进行比对注释后,可以到otus序列对应的物种注释,以及丰度信息。otus和物种是映射关系,它们可能是一对一关系,也可能是多对一关系。
步骤112,根据otus序列对应的物种注释和丰度信息进行聚类分组,得到多个组。
其中,根据所有otus序列在门、纲、目、科、属水平的物种注释以及丰度信息,在物种和丰度两个层面进行聚类分组,得到多个组。
步骤114,基于得到的多个组进行组间差异分析。
其中,组间差异分析包括:组件差异显著物种的分析。可以利用样本间的物种丰度信息,评估组间物种丰度差异。对于组间的物种差异,可以采用lefse(ldaeffectsize)方法来进行分析。具体地,lefse用于发现高维生物标识和揭示基因组特征,主要包括基因,代谢和分类,用于区别两个或两个以上生物条件,能够在组与组之间寻找具有统计学差异的biomarker(生物标记),即组间差异显著的物种。
主要分为三个步骤:
1)首先在多组样本中采用的非参数因子kruskal-wallis(一种检验方法)秩和检验检测不同分组间丰度差异显著的物种;
2)然后在上一步中获得的显著差异物种,用成组的wilcoxon(一种检验方法)秩和检验来进行组间差异分析;
3)最后用线性判别分析(lda)对数据进行降维和评估差异显著的物种的影响力(即ldascore)。
上述基于16s微生物扩增测序数据的分析方法,首先,获取对16s微生物进行测序得到的原始序列数据,对所述原始序列数据进行过滤得到有效序列数据;确定所述有效序列数据中正向序列和反向序列的质量值;根据所述质量值对所述正向序列和反向序列进行过滤,得到过滤后的正向序列和反向序列;将所述正向序列和所述反向序列进行匹配合并,输出合并得到的otus序列;将所述otus序列与微生物数据库进行比对注释,确定所述otus序列对应的物种注释和丰度信息;根据所述otus序列对应的物种注释和丰度信息进行聚类分组,得到多个组;基于得到的多个组进行组间差异分析。通过上述完整的分析处理流程,可以实现对微生物的聚类分组,以及基于聚类分组实现了组间差异分析,提高了组间差异分析的准确度,而且基于聚类分组有利于进行进一步的分析。
在一个实施例中,所述对原始序列数据进行过滤得到有效序列数据包括:去除所述原始序列数据中包含的barcode序列和pcr引物扩增序列,得到目标序列数据;根据目标序列数据的质量值进行过滤,得到有效序列数据。
其中,原始序列数据中包括barcode序列和pcr引物扩增序列,barcode序列是一种标签序列,用于将数据拆分为不同样品数据。pcr引物扩增序列是采用pcr进行扩增得到的序列。两者对物种差异的研究没有用处,所以要将从原始数据中去掉,得到目标序列数据,然后再对目标序列数据进行质量过滤得到有效序列数据,具体地,过滤掉质量值小于10碱基所占比例大于50%的序列,然后去掉含有n的序列,含有n的序列是指识别到的未知序列。
在一个实施例中,所述根据所述质量值对所述正向序列和反向序列进行过滤,得到过滤后的正向序列和反向序列,包括:对所述正向序列和所述反向序列进行质量检测;当所述正向序列或反向序列中的一个或多个片段的质量不符合要求时,则对质量不符合要求的片段进行剪切过滤,得到剪切过滤后的正向序列和反向序列。
其中,在对正向序列和反向序列进行质量检测时,实际上是评估的碱基的质量,当某些碱基的质量不合格时,剪切掉相应的片段。
在一个实施例中,所述对所述正向序列和所述反向序列进行质量检测,包括:采用质量评估模型对过滤后的正向序列和反向序列进行质量检测,得到质检结果。
其中,在进行质量检测时,采用质量评估模型,质量评估模型用于评估正向序列和反向序列中各个片段的质量,当有不符合质量要求的片段时,则进行过滤掉。质量评估模型是采用机器学习进行训练得到的。采用训练好的质量评估模型对正向序列和反向序列进行质量检测,有利于提高质量检测的速度和准确度。
在一个实施例中,所述将所述正向序列和所述反向序列进行匹配合并,输出合并得到的otus序列,包括:计算所述正向序列和所述反向序列的重叠度,当所述正向序列和所述反向序列的重叠度符合预设要求时,根据对应的重叠区域将所述正向序列和所述反向序列进行合并输出得到otus序列。
其中,当正向序列和反向序列进行匹配的重叠度达到预设要求时(比如,重叠至少12个碱基),将正向序列和反向序列进行合并,得到otus序列。
在一个实施例中,所述基于得到的多个组进行组间差异分析,包括:根据每个组对应的物种注释和丰度信息确定不同组间的丰度差异显著的物种;基于组间的所述丰度差异显著的物种进行差异分析。
其中,首先确定出组与组之间丰度差异显著的物种,然后基于丰度差异显著的物种进行进一步的差异分析,得出组间差异的原因。
在一个实施例中,所述方法还包括:基于得到的多个组进行多样性分析,所述多样性分析包括:alpha多样性分析和beta多样性分析。
其中,alpha多样性是指一个特定环境或生态系统内的多样性,主要关注局域均匀生境下的物种数目,因此又称为生境内的多样性。alpha多样性常用的指数有四种:observedspecies,chao1,shannon,simpson。其中observedspecies指数是指样本中实际包含的otus数目,chao1指数是指估算样本中otus的数目,这两个指数均反映样本中otus(物种)数目的高低,这两个指数越高表明样本中的物种丰富度越高。shanon指数是用来描述otus出现的紊乱和不确定性,不确定性越高,多样性指数越高,simpson指数是指从样品中随机取两条序列,这两条序列属于不同的otus的概率,如果样本中只有一个otus,simpson指数为0,多样性最低。shannon和simpson指数不仅反映了样本中物种的数量(即丰富度)又反映了样本中各物种的丰度分布情况(即均匀度)。总之,样品中otus数目越多,otus丰度分布越均匀,多样性指数越高。
beta多样性指沿环境梯度不同生境群落之间物种组成的相异性或物种沿环境梯度的更替速率,也被称为生境间的多样性,主要是衡量群落之间的差别。beta多样性的意义在于:1)它可以反映生境变化的程度或指示生境被物种分割的程度;2)beta多样性的高低可以用来比较不同生境的多样性。总的来说,alpha多样性主要关注某一个群落中的物种多样性,而beta多样性主要关注不同群落之间的物种多样性差别。
如图2所示,在一个实施例中,提出了一种基于16s微生物扩增测序数据的分析装置,包括:
第一过滤模块202,用于获取对16s微生物进行测序得到的原始序列数据,对所述原始序列数据进行过滤得到有效序列数据;
质量确定模块204,用于确定所述有效序列数据中正向序列和反向序列的质量值;
第二过滤模块206,用于根据所述质量值对所述正向序列和反向序列进行过滤,得到过滤后的正向序列和反向序列;
合并模块208,用于将所述正向序列和所述反向序列进行匹配合并,输出合并得到的otus序列;
比对模块210,用于将所述otus序列与微生物数据库进行比对注释,确定所述otus序列对应的物种注释和丰度信息;
聚类模块212,用于根据所述otus序列对应的物种注释和丰度信息进行聚类分组,得到多个组;
分析模块214,用于基于得到的多个组进行组间的差异分析。
在一个实施例中,第一过滤模块202还用于去除所述原始序列数据中包含的barcode序列和pcr引物扩增序列,得到目标序列数据;根据目标序列数据的质量值进行过滤,得到有效序列数据。
在一个实施例中,第二过滤模块206还用于对所述正向序列和所述反向序列进行质量检测;当所述正向序列或反向序列中的一个或多个片段的质量不符合要求时,则对质量不符合要求的片段进行剪切过滤,得到剪切过滤后的正向序列和反向序列。
在一个实施例中,第二过滤模块206还用于采用质量评估模型对过滤后的正向序列和反向序列进行质量检测,得到质检结果。
在一个实施例中,合并模块还用于计算所述正向序列和所述反向序列的重叠度,当所述正向序列和所述反向序列的重叠度符合预设要求时,根据对应的重叠区域将所述正向序列和所述反向序列进行合并输出得到otus序列。
在一个实施例中,分析模块还用于根据每个组对应的物种注释和丰度信息确定不同组间的丰度差异显著的物种;基于组间的所述丰度差异显著的物种进行差异分析。
如图3所示,在一个实施例中,上述装置还包括:
多样性模块216,用于基于得到的多个组进行多样性分析,所述多样性分析包括:alpha多样性分析和beta多样性分析。
图4示出了一个实施例中计算机设备的内部结构图。该计算机设备具体可以是终端,也可以是服务器。如图4所示,该计算机设备包括通过系统总线连接的处理器、存储器和网络接口。其中,存储器包括非易失性存储介质和内存储器。该计算机设备的非易失性存储介质存储有操作系统,还可存储有计算机程序,该计算机程序被处理器执行时,可使得处理器实现上述的基于16s微生物扩增测序数据的分析方法。该内存储器中也可储存有计算机程序,该计算机程序被处理器执行时,可使得处理器执行上述的基于16s微生物扩增测序数据的分析方法。本领域技术人员可以理解,图4中示出的结构,仅仅是与本申请方案相关的部分结构的框图,并不构成对本申请方案所应用于其上的计算机设备的限定,具体的计算机设备可以包括比图中所示更多或更少的部件,或者组合某些部件,或者具有不同的部件布置。
在一个实施例中,提出了一种计算机设备,包括存储器和处理器,所述存储器存储有计算机程序,所述计算机程序被所述处理器执行时,使得所述处理器执行以下步骤:获取对16s微生物进行测序得到的原始序列数据,对所述原始序列数据进行过滤得到有效序列数据;确定所述有效序列数据中正向序列和反向序列的质量值;根据所述质量值对所述正向序列和反向序列进行过滤,得到过滤后的正向序列和反向序列;将所述正向序列和所述反向序列进行匹配合并,输出合并得到的otus序列;将所述otus序列与微生物数据库进行比对注释,确定所述otus序列对应的物种注释和丰度信息;根据所述otus序列对应的物种注释和丰度信息进行聚类分组,得到多个组;基于得到的多个组进行组间差异分析。
在一个实施例中,所述对原始序列数据进行过滤得到有效序列数据包括:去除所述原始序列数据中包含的barcode序列和pcr引物扩增序列,得到目标序列数据;根据目标序列数据的质量值进行过滤,得到有效序列数据。
在一个实施例中,所述根据所述质量值对所述正向序列和反向序列进行过滤,得到过滤后的正向序列和反向序列,包括:对所述正向序列和所述反向序列进行质量检测;当所述正向序列或反向序列中的一个或多个片段的质量不符合要求时,则对质量不符合要求的片段进行剪切过滤,得到剪切过滤后的正向序列和反向序列。
在一个实施例中,所述对所述正向序列和所述反向序列进行质量检测,包括:采用质量评估模型对过滤后的正向序列和反向序列进行质量检测,得到质检结果。
在一个实施例中,所述将所述正向序列和所述反向序列进行匹配合并,输出合并得到的otus序列,包括:计算所述正向序列和所述反向序列的重叠度,当所述正向序列和所述反向序列的重叠度符合预设要求时,根据对应的重叠区域将所述正向序列和所述反向序列进行合并输出得到otus序列。
在一个实施例中,所述基于得到的多个组进行组间差异分析,包括:根据每个组对应的物种注释和丰度信息确定不同组间的丰度差异显著的物种;基于组间的所述丰度差异显著的物种进行差异分析。
在一个实施例中,所述处理器执行所述计算机程序时,还用于执行以下步骤:基于得到的多个组进行多样性分析,所述多样性分析包括:alpha多样性分析和beta多样性分析。
在一个实施例中,提出了一种计算机可读存储介质,存储有计算机程序,所述计算机程序被处理器执行时,使得所述处理器执行以下步骤:获取对16s微生物进行测序得到的原始序列数据,对所述原始序列数据进行过滤得到有效序列数据;确定所述有效序列数据中正向序列和反向序列的质量值;根据所述质量值对所述正向序列和反向序列进行过滤,得到过滤后的正向序列和反向序列;将所述正向序列和所述反向序列进行匹配合并,输出合并得到的otus序列;将所述otus序列与微生物数据库进行比对注释,确定所述otus序列对应的物种注释和丰度信息;根据所述otus序列对应的物种注释和丰度信息进行聚类分组,得到多个组;基于得到的多个组进行组间差异分析。
在一个实施例中,所述对原始序列数据进行过滤得到有效序列数据包括:去除所述原始序列数据中包含的barcode序列和pcr引物扩增序列,得到目标序列数据;根据目标序列数据的质量值进行过滤,得到有效序列数据。
在一个实施例中,所述根据所述质量值对所述正向序列和反向序列进行过滤,得到过滤后的正向序列和反向序列,包括:对所述正向序列和所述反向序列进行质量检测;当所述正向序列或反向序列中的一个或多个片段的质量不符合要求时,则对质量不符合要求的片段进行剪切过滤,得到剪切过滤后的正向序列和反向序列。
在一个实施例中,所述对所述正向序列和所述反向序列进行质量检测,包括:采用质量评估模型对过滤后的正向序列和反向序列进行质量检测,得到质检结果。
在一个实施例中,所述将所述正向序列和所述反向序列进行匹配合并,输出合并得到的otus序列,包括:计算所述正向序列和所述反向序列的重叠度,当所述正向序列和所述反向序列的重叠度符合预设要求时,根据对应的重叠区域将所述正向序列和所述反向序列进行合并输出得到otus序列。
在一个实施例中,所述基于得到的多个组进行组间差异分析,包括:根据每个组对应的物种注释和丰度信息确定不同组间的丰度差异显著的物种;基于组间的所述丰度差异显著的物种进行差异分析。
在一个实施例中,所述处理器执行所述计算机程序时,还用于执行以下步骤:基于得到的多个组进行多样性分析,所述多样性分析包括:alpha多样性分析和beta多样性分析。
本领域普通技术人员可以理解实现上述实施例方法中的全部或部分流程,是可以通过计算机程序来指令相关的硬件来完成,所述的程序可存储于一非易失性计算机可读取存储介质中,该程序在执行时,可包括如上述各方法的实施例的流程。其中,本申请所提供的各实施例中所使用的对存储器、存储、数据库或其它介质的任何引用,均可包括非易失性和/或易失性存储器。非易失性存储器可包括只读存储器(rom)、可编程rom(prom)、电可编程rom(eprom)、电可擦除可编程rom(eeprom)或闪存。易失性存储器可包括随机存取存储器(ram)或者外部高速缓冲存储器。作为说明而非局限,ram以多种形式可得,诸如静态ram(sram)、动态ram(dram)、同步dram(sdram)、双数据率sdram(ddrsdram)、增强型sdram(esdram)、同步链路(synchlink)dram(sldram)、存储器总线(rambus)直接ram(rdram)、直接存储器总线动态ram(drdram)、以及存储器总线动态ram(rdram)等。
以上实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本申请的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本申请专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本申请构思的前提下,还可以做出若干变形和改进,这些都属于本申请的保护范围。因此,本申请专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!