一种基于RNA测序数据进行癌症溯源的方法与流程
本发明涉及生物信息领域,具体为一种基于rna测序数据进行癌症溯源的方法。
背景技术:
癌基因是细胞内一种控制细胞生长和分化的基因,在结构异常或表达异常时将会引起细胞癌变。原癌基因是存在于生物正常细胞基因组中的癌基因,一般情况下不出现致癌活性,在发生突变或被异常激活后会变成具有致癌能力的癌基因,即细胞的原癌基因被不适当的激活后,会造成蛋白质产物的结构改变、原癌基因的过量表达或不能在适当的时刻关闭表达等。
rna是基因表达的直接产物,对rna的研究集中于对细胞中基因转录表达情况和调控规律的研究。区别于同一个体所有细胞中的基因在不发生突变的情况下是相同的,同一细胞在不同生长时期和生长环境下的rna情况不完全相同,这是由于基因的表达具有细胞和组织特异性,行使不同功能的细胞将表达不同的基因,即基因的差异性表达(differentialgeneexpression,dge),且dge相关分析已用于探索癌组织中差异性表达的基因。
研究表明cdh5、tek、calcrl等基因为非小细胞肺癌信号通路关键基因,在患者中的表达显著提高,并与患者预后相关;乳腺癌中表达增高的基因包括gata3、cd2、egfr等,表达降低的基因则有brca1、dbc2等;sfrp4基因在结直肠癌组织中有明显的高表达,而fap基因的表达下降则可以抑制结直肠癌中肿瘤的生长。一些类似以上在不同癌症中具有显著差异性表达的基因已作为一种癌症标志物,用于癌症的诊断、肿瘤阶段划分和预后的预测等。原发灶不明性转移性肿瘤是一类经活检验证但找不到原发部位的转移性恶性肿瘤,患有此种癌症的病人占所有癌症病人的0.5%~0.7%,由于病灶较小、部位隐匿等原因而不易发现。对于另一些原发性肿瘤,虽然被诊断为转移性实体瘤,但通过传统的方法临床上也很难找到原发病灶,因此耽误最佳治疗时期。因此在临床上寻找原发病灶极其重要,临床上多用循环血液中检查到的癌细胞来寻找原发病灶。
经美国fda认证的基于rt-pcr技术的cancertypeid是寻找病灶进行肿瘤溯源的产品之一,该产品使用遗传算法挑选目标基因,并使用k-近邻算法进行肿瘤的溯源。美国约翰-霍普金斯大学癌症中心开发的delfi癌症溯源方法通过cfdna独特的片段化模式可对7种不同类型的癌症进行检测溯源,为癌症早期筛查提供了原理和验证方法。
随着精准健康领域的发展,临床上需要快速、简便且准确的进行早期癌症溯源以提高患者生存率和预后等。以上产品和技术虽然已达到较高的准确率,但是操作相对繁琐,且可溯源的癌症种类有限,具有应用方面的局限性。
目前尚缺少一种方法或分析平台来解决上述问题,因此,设计一种基于rna测序数据进行癌症溯源的方法,可溯源的癌症种类包括胃癌、结直肠癌、肺癌等18种肿瘤,在保证准确率的同时简化了操作步骤,具有现实意义和良好的应用前景。
技术实现要素:
针对上述背景技术中的不足,本发明提供一种基于rna测序数据进行癌症溯源的方法,即结合基因表达差异性分析和机器学习模型,使用从肿瘤基因组计划(thecancergenomeatlas,tcga)公共数据库中获取的18种肿瘤样本的rna测序数据进行分析和模型的训练,从而得到一种癌症溯源的预测模型,基于rna测序数据的使用和分析囊括了多个差异性表达基因,包括表达量低的基因,从而保证了方法的准确性,同时模型使用操作简便时效性高。
为实现上述目的,本发明提供如下技术方案:
一种基于rna测序数据进行癌症溯源的方法,其特征在于,包括如下步骤:
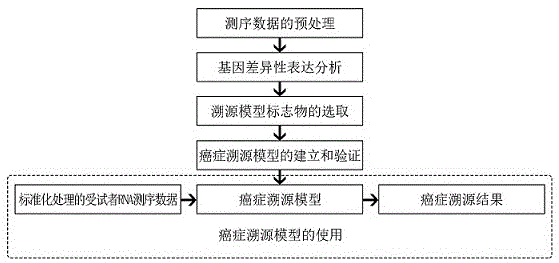
步骤1)测序数据的预处理,将从tcga数据库中获取的18种癌症的原始数据进行预处理,并将每一种癌症的所有样本数据整理成为一个基因表达矩阵;
步骤2)基因差异性表达分析,使用步骤1)中的表达矩阵和样本条件数据,对每一种癌症进行基因差异性表达分析,根据分析结果筛选出每个癌症中有显著性差异表达的基因数据;
步骤3)溯源模型标志物的选取,对步骤2)中分析出的每种癌症的差异性表达基因数据进行交集处理,并从每种癌症的差异性表达基因数据中去除交集中的基因,剩余基因即为每种癌症差异性表达的标志基因;
步骤4)癌症溯源模型的建立和验证,以步骤3)所得的差异性表达标志基因作为特征,以癌症种类作为标签输入随机森林模型,利用多棵决策树的集成学习策略对样本进行训练预测,并进行十次十倍交叉验证,最终得到癌症溯源模型;
步骤5)癌症溯源模型的使用,对受试者的rna测序数据进行标准化处理,获得模型输入的标准文件,并将标准文件输入所述癌症溯源模型中,模型即可输出该样本的癌症溯源结果。
优选的,所述的一种基于rna测序数据进行癌症溯源的方法,其特征在于:所述癌症种类包括前列腺癌、乳腺癌、宫颈癌、子宫内膜癌、胃癌、肺鳞癌、肺腺癌、结肠癌、肝细胞肝癌、多形成性胶质细胞瘤、肾透明细胞癌、肾乳头状细胞癌、头颈鳞状细胞癌、胰腺癌、直肠腺癌、甲状腺癌、食管癌和急性髓细胞白血病,总计18种癌症。
优选的,所述的一种基于rna测序数据进行癌症溯源的方法,其特征在于:所述步骤1)测序数据的预处理包括过滤掉该癌症种类下所有样本表达量小于5的基因表达信息,填补遗漏信息,进行标准化处理并注释基因信息。
优选的,所述的一种基于rna测序数据进行癌症溯源的方法,其特征在于:所述填补遗漏信息具体为使用k-近邻算法参考邻近样本的基因表达值来补充缺失值。
优选的,所述的一种基于rna测序数据进行癌症溯源的方法,其特征在于:所述注释基因信息包括将探针名称和基因名称一一对应起来,并做基因功能、坐标等注释。
优选的,所述的一种基于rna测序数据进行癌症溯源的方法,其特征在于:所述步骤2)基因差异性表达分析,使用依赖于r软件的deseq2软件进行基因差异性表达分析。
优选的,所述的一种基于rna测序数据进行癌症溯源的方法,其特征在于:所述步骤2)基因差异性表达分析中样本条件数据具体为记录该样本来源为癌症组织或者癌旁组织的样本分类条件数据。
与现有技术相比,本发明具备以下有益效果:
1.本方法使用的溯源模型标志物为rna测序数据的分析结果,相比于靶基因测序,基因覆盖范围更广泛和完整,很大程度避免了遗漏标志物基因的可能;
2.本方法使用随机森林机器学习模型对癌症进行溯源预测,预测和计算更加精准和简单;
3.本方法可对18种常见癌症进行溯源,癌症覆盖率高,很大程度避免了临床误诊的情况;
4.模型使用时操作简便,仅需输入样本的rna测序数据标准化文件即可对样本癌症种类进行溯源预测。
附图说明
图1为本发明方法的一种示例性流程图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,但本发明的保护范围不受具体的实施方式所限制,以权利要求书为准,显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例都属于本发明保护的范围。
请参阅图1,为本发明方法的一种示例性流程图,本实施例提供一种基于rna测序数据进行癌症溯源的方法,该方法是一个结合基因表达差异性分析和机器学习模型的方法,使用从肿瘤基因组计划(thecancergenomeatlas,tcga)公共数据库中获取的18种肿瘤样本的rna测序数据进行分析和模型的训练,从而得到一种癌症溯源的预测模型,基于rna测序数据的使用和分析囊括了多个差异性表达基因,包括表达量低的基因,从而保证了方法的准确性,同时模型使用操作简便时效性高,该方法的特征在于,包括如下步骤:
步骤1)测序数据的预处理,将从tcga数据库中获取的18种癌症的原始数据进行预处理,并将每一种癌症的所有样本数据整理成为一个基因表达矩阵,该表达矩阵是以基因名称和基因表达量(readcount)为轴建立的;
步骤2)基因差异性表达分析,使用步骤1)中的表达矩阵和样本条件数据,对每一种癌症进行基因差异性表达分析,根据分析结果筛选出每个癌症中有显著性差异表达的基因数据,筛选差异性表达基因的标准为差异倍数|log(foldchange)|>1,且差异显著性p<0.05;
步骤3)溯源模型标志物的选取,对步骤2)中分析出的每种癌症的差异性表达基因数据进行交集处理,并从每种癌症的差异性表达基因数据中去除交集中的基因,剩余基因即为每种癌症差异性表达的标志基因,剔除交集中基因的原因是防止这些在多个癌症中表达的基因对后续癌症溯源模型的训练造成影响,因此仅挑选出在每种癌症中特异性表达且有显著表达差异的基因作为溯源模型的标志物;
步骤4)癌症溯源模型的建立和验证,以步骤3)所得的差异性表达标志基因作为特征,以癌症种类作为标签输入随机森林模型,利用多棵决策树的集成学习策略对样本进行训练预测,并进行十次十倍交叉验证,可以避免一次交叉验证所导致的误差,最终得到癌症溯源模型;
步骤5)癌症溯源模型的使用,对受试者的rna测序数据进行标准化处理,包括基因名称注释和对应表达量的提取,同时过滤掉表达量小于5的基因,获得模型输入的标准文件,并将标准文件输入所述癌症溯源模型中,模型即可输出该样本的癌症溯源结果。
其中,所述一种基于rna测序数据进行癌症溯源的方法,其特征在于:所述癌症种类包括前列腺癌、乳腺癌、宫颈癌、子宫内膜癌、胃癌、肺鳞癌、肺腺癌、结肠癌、肝细胞肝癌、多形成性胶质细胞瘤、肾透明细胞癌、肾乳头状细胞癌、头颈鳞状细胞癌、胰腺癌、直肠腺癌、甲状腺癌、食管癌和急性髓细胞白血病,总计18种癌症,其中急性髓细胞白血病为血液瘤,其他癌症为实体瘤。
其中,所述的一种基于rna测序数据进行癌症溯源的方法,其特征在于:所述步骤1)测序数据的预处理包括过滤掉该癌症种类下所有样本表达量小于5的基因表达信息,填补遗漏信息,进行标准化处理并注释基因信息。过滤表达量过低的基因,一方面是为了减少后续基因差异性表达分析的计算量,另一方面也是防止由于测序原因造成基因表达量低的情况,避免对后续分析造成影响。
其中,所述的一种基于rna测序数据进行癌症溯源的方法,其特征在于:所述填补遗漏信息具体为使用k-近邻算法参考邻近样本的基因表达值来补充缺失值。
其中,所述的一种基于rna测序数据进行癌症溯源的方法,其特征在于:所述注释基因信息包括将探针名称和基因名称一一对应起来,并做基因功能、坐标等注释。
其中,所述的一种基于rna测序数据进行癌症溯源的方法,其特征在于:所述步骤2)基因差异性表达分析,使用依赖于r软件的deseq2软件进行基因差异性表达分析,deseq2软件是常用的基因表达数据分析软件,用于分析不同组的基因表达数据中差异性表达的基因,该软件的使用方法较其他软件更加简洁的同时,也兼顾了分析效率和分析准确率。
其中,所述的一种基于rna测序数据进行癌症溯源的方法,其特征在于:所述步骤2)基因差异性表达分析中样本条件数据具体为记录该样本来源为癌症组织或者癌旁组织的样本分类条件数据。
需要说明的是,在本文中,诸如第一和第二等之类的关系术语仅仅用来将一个实体或者操作与另一个实体或操作区分开来,而不一定要求或者暗示这些实体或操作之间存在任何这种实际的关系或者顺序。而且,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。
以上所述仅为本发明的优选实例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!