单细胞遗传结构变异的综合检测
1.本发明提供了一种通过整合测序读段深度、读段链方向和单倍型定相(phase)的三层信息来检测单细胞或单细胞群体的基因组内的结构变异(sv)的方法。本发明的方法可以检测缺失、重复、多倍体、易位、倒位和拷贝数中性的杂合性缺失(cnn-loh)等。本发明的方法可以充分地对基因组进行综合地核型分析,以及可以被应用于研究和临床方法。例如,本发明的方法可以用于分析患者的细胞样本以进行诊断或辅助诊断,在生殖医学中检测胚胎异常,或者在基于细胞疗法的治疗方法中质量控制基因工程细胞,例如在过继性t细胞疗法等中。本发明的方法可以进一步被应用于研究中来解释细胞模型(细胞系)、患者样本的核型,或进一步揭示导致基因组内任何sv产生的遗传和机械途径。
2.描述
3.结构变异(sv)(其中重整缺失、重复、倒位或易位的dna片段多达百万碱基)是与许多疾病相关的遗传变异的主要来源。最近的方法和技术的进步使得能够对不同人群中的sv进行编目。除了这些种系变体外,越来越清楚的是,人类组织表现出丰富的体细胞变异,特别是sv,一种动态的、高比率出现的变异类别,导致广泛的遗传异质性。细胞群中的体细胞sv分析能够有助于研究遗传嵌合和异常克隆扩增,允许谱系追踪,以及在癌症背景下能够有助于改进疾病分类和管理。然而,sv发现仍然具有挑战性,重复区域中出现的易位、倒位、复杂sv类别、细胞倍性改变以及sv通常会逃过在遗传异质性背景下的检测。
4.体细胞结构变异在健康和疾病中起着关键作用
10,2
。例如,癌症在单个肿瘤细胞中表现出染色体数量和细胞遗传学结构的巨大差异
79
。癌症中的sv显示出动态的形成模式,并且可以在基因组不稳定期间出现间断爆发
4,5
,从而导致肿瘤内的异质性。它们代表了几种癌症类型中基因组驱动因素改变的主要类别
2,1
,包括拷贝数改变(cna)和拷贝平衡sv,其通过引起基因破坏、基因缺失或扩增、基因融合、增强子劫持(hijacking)和重组拓扑结构域(tad)产生严重后果
2,5
。最近的研究也在正常组织(包括大脑、皮肤和血液)中检测到体细胞/合子后sv1,这些变体可以通过组织功能下降和/或促进疾病进程(包括癌症和白血病形成)影响健康。事实上,老年供体的血液中的合子后cna与白血病、实体瘤以及包括2型糖尿病和冠心病在内的常见疾病有关。合子后sv也出现在早期发育过程中,由此产生的嵌合体能够导致遗传疾病,对遗传咨询和测试产生影响
56
。由于其动态性质,体细胞sv能够深深地影响疾病进程。在前列腺癌患者中,影响雄激素受体位点的不同sv类别能够逐渐导致治疗耐药。此外,导致复杂sv(即染色体碎裂(chromothripsis))的间断爆发与whim综合征(一种先天性免疫疾病)的自发治愈有关。涉及体细胞sv的疾病种类繁多,其流行和动态发生需要有效的检测方法。单细胞分析原则上应该是此目的的理想方法,因为其能够使sv检测在低变异等位基因频率(vaf)下至单个细胞
15
。然而,目前扩展到数百或数千个细胞的单细胞方法面向cna
16-18
。其他sv类别(包括易位、倒位和复杂sv类别)通常可以逃过检测,尽管它们与多种疾病过程相关。
5.无论是在种系还是体细胞中出现,sv代表一种特别难以识别的变异类型。由于它们的尺寸往往远远超过dna序列的读段长度,目前的检测方法部分依赖于间接推断(包括成
对末端、读段深度、以及剪切或分割读段的解释)。这些方法需要广泛的序列覆盖度用于可靠的sv调用(使用批量测序时约为20倍或更高)
17
,这限制了它们在异质的环境中用于sv检测的实用性——除了读段深度分析,其能够被用于具有相对低的vaf(通常≥10%vaf)的变体,但仅限于cna
10
。相比之下,单细胞分析能够检测下至单个细胞的sv,并有助于剖析sv共现(co-occurrence)和细胞类型特异性sv的模式
17
。然而,尽管cna已经是单细胞中的常规分析,并且可扩展的
16
和商业的应用(例如,10x genomics公司的“the chromium single cell cnv solution”)正在变得可行,但在单细胞中检测另外的sv类别(如平衡sv和复杂sv)面临着重大挑战:当前可用的sv检测方法要求识别穿过sv断点的读段(或读段匹配(read pair))
55
;由于这种方法的高覆盖度需求和低且不均匀的覆盖度水平(包括单细胞中的局部等位基因缺失)
17
,这仍然是一个挑战。由于需要跨断点读取,一旦sv断点存在于重复区域(其在基因组中是大量的且sv显示富集),这些检测方法就会失效。此外,用于增加dna可获得量的全基因组扩增(wga)能够产生类似sv的读取嵌合体
19
,从而导致调用伪迹(artefact)。尽管最近的研究表明,结合充分的序列覆盖度,嵌合体过滤是可行的
19,20
,但在数百(或数千)个单细胞中sv发现将需要巨大的测序成本,因此尚未进行。此外,大多数当前的方法没有表明给定变体位于哪个单倍型,与单倍型识别(aware)单细胞分析相比,这可能导致调用能力降低
57
。
6.本领域已知的是单细胞/单链基因组测序(strand-seq)
67,21
,一种基于在复制过程中用核苷类似物(brdu)标记新生(即,非模板)dna链,然后去除非模板链,并且随后对剩余链进行短读段测序的技术
67,21
。strand-seq先前被证明能够成功地映射姐妹染色单体交换
21,71
、错误定向的基因组重叠
21
和遗传(种系)倒位
37
。最近进一步证明,strand-seq能够进行全染色体长度单倍型分析
322,72
并指导从头基因组组装。
7.因此,本发明的目的是提供一种手段和方法来促进基因组和染色体内复杂遗传变异、复杂结构变异的综合检测,以及量化细胞染色体稳定性。
8.发明简述
9.一般地,通过简要描述,本发明的主要方面可以描述如下:
10.在第一方面,本发明涉及一种通过单细胞三通道处理(sctrip)分析至少一个目标染色体区域的测序数据的方法,包括提供至少一个单细胞的至少一个目标染色体区域的链特异性序列数据,其中链特异性序列数据包括多个通过至少一个单细胞的目标染色体区域的测序获得的链特异性序列读段,将序列读段或者序列读段的每个片段化部分(如果序列读段被平均地片段化)与参考组件(assembly)比对,然后在任何给定的选定窗口中分配三层序列信息中的至少两层:(i)总序列读段或其部分的数量(也称为“读段深度”);(ii)正向(或watson)序列读段或其部分的数量,以及反向(或crick)序列读段或其部分的数量;(iii)分配有特异性单倍型身份(如h1和/或h2)的序列读段或其部分的数量。
11.在第二方面,本发明涉及一种检测目标染色体区域中结构变异(sv)的方法,该方法包含执行根据第一方面的方法并进一步包括以下步骤:通过对按位置顺序排列和比对的序列读段的序列数据内的多个(至少两个)窗口执行步骤(d)以及在多个窗口内识别子区域来识别结构变异(sv),该子区域包括具有通道(i)至(iii)中的任何一个、或所有、或任何组合的异常的/变化的/改变的信息分布的一个或多个窗口。
12.在第三方面,本发明涉及一种对单细胞或多个单细胞的群体进行核型分析的方
法,该方法包括,
13.(a)提供至少一个单细胞或单细胞群体中的每一个细胞的至少一个目标染色体区域(优选完整基因组)的链特异性序列数据,(b)执行第一或第二方面的方法,
14.(c)检测所述单细胞或单细胞群体的目标染色体区域内的sv,以及
15.(d)根据所有检测的sv获得计算机模拟(in-silico)核型。
16.在第四方面,本发明涉及一种诊断对象中疾病的方法,该方法包括,提供对象的一个或多个细胞的链特异性序列数据,执行根据第一或第二方面的方法,检测一个或多个细胞内的任何sv,以及将检测的sv与参考状态进行比较,其中对象的样本中的一个或多个sv的数量、类型或位置的改变表明存在病况,例如疾病(例如癌症)。
17.在第五方面,本发明涉及一种用于评估单细胞或单细胞群体内的染色体稳定性的方法,该方法包括执行根据上述方面中任何一个的方法,其中所述单细胞或单细胞群体中sv的总数量增加或sv的任何一种类型或多种类型的数量增加表明染色体不稳定。
18.在第六方面,本发明涉及一种计算机可读介质,其包括存储在其上的计算机可读指令,当其在计算机上运行时,指示计算机执行根据本发明的任何方面或实施方案的方法。
19.发明详述
20.在下文中将描述本发明的元素。这些元素与特定实施方案一起列出,然而,应当理解,它们可以以任何方式和任何数量组合以创造其他的实施方案。各种描述的实施例和优选实施方案不应被解释为将本发明限制为仅明确描述的实施方案。该描述应当被理解为支持并涵盖将两个或更多个明确描述的实施方案组合或将一个或多个明确描述的实施方案与任意数量的所公开和/或优选元素组合的实施方案。此外,除非上下文另有指示,否则应当认为本技术中的描述中公开了本技术中所有描述的元素的任何排列和组合。
21.在第一方面,本发明涉及一种通过单细胞三通道处理(sctrip)分析至少一个目标染色体区域的测序数据的方法,包括提供至少一个单细胞的至少一个目标染色体区域的链特异性序列数据,其中链特异性序列数据包括多个通过对至少一个单细胞的目标染色体区域的测序获得的链特异性序列读段,将序列读段或者序列读段的每个部分(如果序列读段被平均地片段化)与参考比对,然后在任何给定的选定窗口中分配三层信息中的至少两层:(i)总序列读段或其部分的数量(也称为“读段深度”);(ii)正向(或watson)序列读段或其部分的数量,以及反向(或crick)序列读段或其部分的数量;(iii)分配有特异性单倍型身份(例如,h1或h2)的序列读段或其部分的数量。
22.更具体地,本发明的第一方面涉及以下方法步骤,其可以以技术上可能或合理的任何顺序进行:
23.(a)提供至少一个单细胞的至少一个目标染色体区域的链特异性序列数据,其中链特异性序列数据包括多个通过至少一个单细胞的目标染色体区域的测序获得的链特异性序列读段;
24.(b)将每个序列读段或其部分与至少一个目标染色体区域的参考序列比对,以使所述序列读段或其部分沿着至少一个目标染色体区域的参考序列按位置顺序排列;
25.(c)将染色体单倍型身份(h1/h2)沿着至少一个目标染色体区域分配至来自(b)的每个比对的序列读段或其部分;和
26.(d)将下列序列信息通道中的任意两个分配至按位置顺序排列且比对的序列读段
或其部分的至少一个预定序列窗口:
27.(i)在至少一个预定序列窗口中比对的总序列读段或其部分的数量;
28.(ii)在至少一个预定序列窗口中比对的正向序列读段或其部分的数量,以及反向序列读段或其部分的数量;
29.(iii)在至少一个预定序列窗口中比对的分配至第一(h1)单倍型身份的序列读段或其部分的数量;和/或分配至第二(h2)单倍型身份的序列读段或其部分的数量。
30.本发明优选地应用本文描述的方法以对候选细胞、组织或对象进行核型分析,作为诊断或质量控制目的的实施例。例如,在本发明第一方面的一个实施方式中,任选地或另外地,涉及一种对感兴趣的至少一个单细胞的基因组进行核型分析的方法,包括:a)从至少一个单细胞的基因组的随机位置获得多个(优选非重叠的)链特异性序列;b)将所述测试链特异性序列映射至基因组参考支架以获得映射的链特异性序列的测试分布;c)将(i)映射的序列读段的数量,(ii)映射的正链读段的数量和反链读段的数量,优选其比例,以及(iii)单倍型身份(h1/h2)(优选h1的数量和h2单倍型身份读段或其部分的数量)分配至参考支架内的预定序列窗口以获得映射的序列的三层测试分布;d)识别预期分布之间统计学上显著的改变,其中这种改变表明至少一个单细胞的基因组中的核型异常;或者e)将三层测试分布与从参考细胞(例如健康细胞)获得的参考分布比较,其中如果存在显著差异,则所述差异表明至少一个单细胞与参考细胞之间的核型差异。
31.发明人开发了一种技术,将三种有价值的信息整合至经测序的目标染色体区域(例如完整的染色体或基因组),这三种信息由读段深度、模板链识别(复制后源自母细胞的正链或反链)和单倍型定相或单倍型组成,单倍型定相或单倍型表明来自所有二倍体生物中存在的父本或母本染色体的序列的身份。发明人惊奇地发现,当分析经测序的单细胞的数据并将数据与三层信息的遗传预期分布比较时,通过仅分析单细胞的序列数据,或者通过观察相同遗传起源的多个细胞的分离模式,能够容易地识别染色体区域内许多以前难以检测到的结构变异。对于后一种方法,群体内完整或部分染色体的异常分离或分布能够被用于识别经测序的基因组内的多倍体或易位。
32.本发明的方法利用strand-seq对单细胞中的体细胞变异进行单倍型识别检测。经检测的变异类别包括缺失、重复、倒位、易位、复杂sv类别、拷贝数中性的杂合性丢失(cnn-loh)和细胞倍性改变。本发明的方法利用模板链的有丝分裂分离模式(即染色单体分离模式),其反映以前未考虑过的用于检测细胞群体中sv的“遗传信号”。本发明通过在每个单细胞中分析三个正交数据层(或“通道”)(读段深度,链方向和单倍型定相)来利用该信息,通过根据本发明的本文称为“三通道处理”的新方法(图1),整合产生一组鉴别sv诊断足迹(diagnostic footprint)。令人惊讶的是,本发明的方法不需要穿过sv断点的读段匹配,这使得该方法与单细胞序列的情况一样适用于具有低序列覆盖度的可扩展低通量测序策略,并且能够检测重复序列两侧的sv。本文中的实施例通过分析细胞系和原发性白血病展示实用性,揭示先前未解决或未完全解决的变异类别以及重复相关的和间断平衡的类sv形成,并解析通过单细胞sv图谱确定的亚克隆。本发明将通过对单细胞中对各种sv类别进行可扩展、经济高效的分析,从而打开一系列研究机会。
33.本发明上下文中使用的以下术语应被详细定义,这些定义通常包括本文描述的发明的特别优选的实施方案。对于此类实施方案或某些术语的优选的定义,上述关于实施方
案和方面的组合的描述同样适用。
34.术语“序列数据”应指通过对多核苷酸测序获得的数据,其中该序列数据包含多个序列读段,且每个序列读段源自对模板多核苷酸链的测序。在本发明的优选实施方案中,模板多核苷酸链为正向或反向(w或c)链。
35.如本文所用,术语“序列读段(read)”是指从获得自生物细胞或病毒的核酸分子获得或读取的核苷酸序列。序列读段能够通过本领域已知的各种方法获得。通常,序列读段是从测试样本获得或富集的核酸片段扩增(例如,聚合酶链式反应,如桥式扩增)后获得的。序列读段的长度可以因所使用的测序方法而变化。能够用于本发明上下文的序列读段的优选长度为50至500个核苷酸,优选约100至200个核苷酸。
36.可用于本发明上下文的测序方法选自技术人员已知的任何方法。然而,目前所谓的“下一代测序”方法是优选的,包括目前由例如illumina、life technologies和roche使用的所谓的并行合成测序或连接测序平台,或基于电子检测的方法(如thermofisher商业化的ion torrent技术)等。测序方法还可以包括所谓的“第三代测序(tgs)”技术,如纳米孔测序方法。其他方法包括“单分子实时(smrt)”测序(例如通过pacific biosciences),以及能够获得超过1kb的序列读段的所谓的“长读段测序”。这些都提供通常称为长读段序列数据(即序列读段》1000个碱基对)。
37.在本发明的上下文中,特别优选提供目标染色体区域(例如测试细胞的)的序列作为链特异性序列读段或其部分。该序列读段或其部分保留例如测序读段的染色体区域的模板链的链特异性信息,并且在母细胞有丝分裂后由经测序的单细胞遗传。如本文将进一步解释的,此类模板链可以是正向的或反向的,或者通常也称为watson或crick。任何允许保留链身份信息的方法应包含在本发明的方法中,并适用于本发明的方法,因为关键的只是链特异性信息,而不是如何获得链身份信息的方法。在测序过程中保持链身份的一种方法是通过链特异性测序或“strand-seq”。falconer et al.2012nature methods.9(11):1107
–
1112详细描述了该方法,通过引用将其全部并入本文。特别是该出版物的方法部分通过引用并入本文。简而言之,strand-seq涉及在细胞的一个合成阶段(s期)使用brdu核苷酸,使得在有丝分裂之前,每个染色体的新生成的姐妹染色单体中有一条被并入的brdu核苷酸标记的链和不含brdu的另一条链(模板链)。有丝分裂后,子细胞被处理,使得brdu链被切割,因此在pcr过程中只能扩增无brdu标记的链。使用特异性衔接子(adapter),原始模板链信息被保留在扩增片段中,以便在测序后只能确定模板链的链身份。将这样获得的序列读段与参考基因组支架比对,然后指示读段方向以及从哪个链(watson或crick)获得读段。
38.术语“核型”是指给定物种或测试样本的单个细胞或细胞系的基因组特征;例如,由染色体的数量和形态来定义。通常,核型被呈现为来自显微照片或计算机生成的图像的前期或中期(或以其他方式浓缩)染色体的系统化阵列。替代地,间期染色体可以以间期细胞核释放的组蛋白缺失的dna纤维被检测。在一个实施方案中,本发明的核型分析方法特别适用于检测拷贝数中性的sv。本发明的方法还可以被用于确定测试细胞或测试基因组中的拷贝数多态性(或也被称为“拷贝数变异”)。由于基于序列的核型分析方法可以在原核细胞上进行,因此染色体的存在对于本发明的方法不是必需的。
39.如本文所用,术语“结构变异”、“sv”、“染色体改变”或“染色体异常”可互换使用,并指对象染色体或核型的结构与正常(即“非异常”)同源染色体或核型之间的偏差。当提及
染色体或核型时,术语“正常”或“非异常”是指在特定物种和性别的健康个体中发现的主要核型或带型(banding pattern)。通过本发明的方法检测的sv优选是大型或中型sv(200kb或更大)。
40.sv在本质上可以是数值的或结构的,包括非整倍体、多倍体、倒位、平衡或不平衡易位、缺失、重复、倒位重复等。sv可能与病理状况的存在相关(例如,唐氏综合征中的21三体,猫叫综合征(cri-du-chat syndrome)中的染色体5p缺失,以及导致形态异常和精神障碍的各种不平衡染色体重排,以及增殖性疾病,尤其是癌症)或者与发展成病理状况的倾向相关。出于本发明的目的,染色体异常也指基因组异常,其中测试生物(例如原核细胞)可能没有传统定义的染色体。
41.此外,染色体异常包括fish的任何种类的遗传异常,包括使用光学显微镜、传统染色法在传统核型上通常不可见的遗传异常。本发明的一个优点是,由于三层信息的整合,可以检测到以前无法通过光学方法或者甚至测序方法检测到的染色体异常(例如,涉及4mb、600kb、200kb、40kb或更小的异常)。
42.出于本发明的目的,术语“拷贝数变异(cnv)”是指基因组dna的结构变异的一种形式,导致细胞出现异常,或对于某些基因而言dna的一个或多个片段的拷贝数出现正常变异。cnv对应于基因组中相对大的区域,这些区域在某些染色体上缺失(少于正常数量)或重复(多于正常数量)。相应地,术语“拷贝数中性的”应表示不会导致细胞具有异常的序列元件(如基因)的拷贝数的变异。
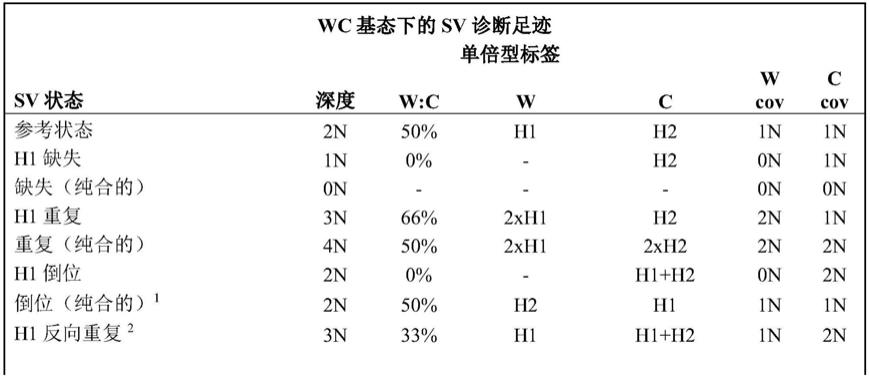
43.在本发明的上下文中,术语“诊断足迹”应指对sv是特异性的或至少指示性的本发明的三层信息的模式。因此,诊断足迹的特征在于对特定实验预期数据分布的改变。表明sv的特定模式将根据分析的数据而变化。例如,二倍体细胞可以被测序以包含每个染色体的ww、cc或wc链分布。根据链分布,相同的sv可以具有不同的诊断足迹。例如,本文表1中提供了此类足迹或模式。
44.在本发明的上下文中,术语“目标染色体区域”应指任何生物或病毒的一个或多个完整或部分染色体的dna序列,其是本发明上下文中的查询对象。目标染色体区域可以仅指单个染色体部分的一段序列,或指任何染色体的父本和母本区域。在一些实施方案中,作为根据本发明的查询对象的目标染色体区域是单细胞或多个单细胞的整个染色体或整个基因组。
45.在本发明的上下文中,术语“单细胞”应指通过例如链特异性测序获得的单个细胞,生成单细胞文库。本发明上下文中的单细胞文库描述了通过对所述单细胞的基因组进行测序而获得的多个序列读段。此外,本发明在一些方面和实施方案中涉及多个单细胞或大量单细胞,在这种情况下,指的是对包含在多个单细胞中的每个单细胞生成的多个单独且独立的序列库。在本发明的一个优选实施方案中,细胞系的多达96个单细胞被单独测序。此类实施方案是优选的,因为此类分析能够在多孔板(例如96孔板或384孔板)中进行。
46.术语“至少一个目标染色体区域的参考序列”是指目标的完全测序参考的数据库版本。通常,这样的参考是一个完整的染色体序列。在某些情况下,参考序列也被表示为“参考支架”或“参考基因组支架”或“参考元件”或类似表达。例如,对于人类序列,基因组参考联盟经常发布和更新人类基因组,以及其他基因组(如小鼠、斑马鱼和鸡基因组)的参考序列(https://www.ncbi.nlm.nih.gov/grc)。
47.在本发明的上下文中,术语“参考状态”应指用作与样本数据集进行比较的参考的序列数据的状态或分布,例如为了识别异常。这种参考状态可以是用作参考的一组真实的序列数据,或者可以是对于某个潜在的取样染色体区域预期的数据状态。通常,本发明上下文中的参考状态应涉及一条染色体或一组染色体(基因组)内的序列分布,其预期用于非异常单细胞或细胞群。例如,通常的二倍体人类基因组的参考状态是大多数人类共有的体细胞中人类染色体的分布。然而,在某些方面和实施方案中,参考状态还可以包含异常的染色体结构或非整倍性——根据本发明的参考状态是基于所分析的样本和用本发明的方法回答的问题来确定的。作为仅仅说明性实施例,使用本发明方法分析的样本可以单独源自被筛选其他sv的21三体。最重要的是,本发明上下文中的术语“参考状态”不应与“参考序列”混淆,后者如上文所定义并指用于比对序列读段的序列的元件。
48.在本发明上下文所公开的序列中,术语“比对”或“对齐”应表示链特异性序列至参考支架的映射,例如本文所描述的与相应链特异性序列匹配的参考基因组或参考染色体。将序列读段及其部分与相应的参考支架比对是本领域众所周知的。这些方法可以包括bowtie(genome biol,2009;10(3):r25)或burrows wheeler alignment(bwa)(bioinformatics,2009jul 15;25(14):1754-60.doi:10.1093/bioinformatics/btp324)。将所有序列读段或其部分与参考染色体支架比对导致序列信息沿参考(例如至少一个目标染色体区域)的两条链按位置顺序排列。
49.如本文所用,术语“定相”是指确定两个或更多个核酸序列(通常包括序列变异区域)是否位于相同的核酸模板上(例如染色体或染色体片段)的过程。定相可以指在单一测序读段中解析两个或更多个单核苷酸变体或多态性(snp)。优选地,定相可以指在大基因组区域上解析序列数据,或解析整个基因组序列。
50.在两个或更多个多态性位点的序列的上下文中使用的术语“定相的”是指在这些多态性位点存在的序列是否来自单个染色体是已知的。
51.在单个染色体的上下文中使用的术语“定相的核酸序列”是指单个染色体的核酸序列,其中核酸序列是从单个染色体的测序获得。在单个染色体片段的上下文中使用的术语“定相的核酸序列”是指单个染色体片段的核酸序列,其中核酸序列是从单个染色体片段的测序获得。
52.术语“单倍型”是短语“单倍型基因型”的缩写,目前被认为是指存在于单个母本染色体或父本染色体上的一组核苷酸序列多态性或等位基因,通常作为一个单元遗传。替代地,单倍型可以指在单一染色体上一起连接或存在的一组单核苷酸多态性(snp)。术语单倍型可以用来指在单一染色体上一起连接或存在的至少两个等位基因或snp。
53.术语“单倍型身份(identity)”是指感兴趣序列中观察到的单倍型与参考序列(如染色体)的已知单倍型的对应关系。例如,单倍型身份可以对应于二倍体生物的母本或父本单倍型序列的身份。在本发明的上下文中,对于每个目标染色体区域,单倍型身份“h1”或“h2”可以被分配对应于在文库或实验中观察到的所有序列的观察到的单倍型分布。在一些优选实施方案中,h1是在一条链上测序的单倍型,h2是在互补链上测序的单倍型。
[0054]“聚合酶链式反应”或“pcr”是指通过dna的互补链的同时引物延伸,对特定dna序列进行体外扩增的反应。换句话说,pcr是一种用于对两侧有引物结合位点的目标核酸进行多个拷贝或复制的反应,该反应包含以下步骤中的一个或多个的重复:(i)使目标核酸变
性,(ii)退火使引物至引物结合位点,以及(iii)在三磷酸核苷的存在下通过核酸聚合酶延伸引物。通常,在热循环仪中,反应在对每个步骤最优的不同温度下循环。特定温度、每个步骤的持续时间和步骤之间的变化比例取决于本领域普通技术人员熟知的许多因素。
[0055]
术语“互补”是指多核苷酸彼此形成碱基对的能力。碱基对通常由反向平行的多核苷酸链中核苷酸单元之间的氢键形成。互补多核苷酸链能够以watson-crick方式(例如,a与t、a与u、c与g)或允许形成双链体的任何其他方式进行碱基配对。术语“互补”也被用于表示各自的互补dna链。例如,watson链的互补链指crick链,反之亦然。
[0056]
术语“多核苷酸”或“核酸”指任何长度的核苷酸聚合物,包括但不限于dna、rna或dna/rna杂交体的单链或双链分子,包括规则和不规则交替的脱氧核糖部分和核糖部分的多核苷酸链(即,其中交替的核苷酸单元在糖部分的2
′
位置具有-oh,然后-h、然后-oh、然后-h,以此类推),以及这类多核苷酸的修饰,其中包括各种实体或部分在任何位置对核苷酸单元的取代或连接,以及天然存在或非天然存在的骨架(backbone)。多核苷酸可以在聚合后进一步被修饰,例如通过与标记组分连接。核酸的“片段”或“区段”是该核酸的一小部分。优选地,本发明中使用或分析的多核苷酸是dna分子,例如真核生物的染色体或基因组。
[0057]“纯合”状态指当相同的等位基因位于同源染色体的相应位点时存在的遗传状态。相反,“杂合”状态指当不同的等位基因位于同源染色体的相应位点时存在的遗传状态。
[0058]“基因”指包含至少一个开放阅读框的多核苷酸,该开放阅读框能够在转录和翻译后编码特定的蛋白质。
[0059]“对象”、“个体”或“患者”在本文中可互换使用,指脊椎动物,例如哺乳动物,例如人类。
[0060]
如本文所用,术语“扩增”指使用目标核酸作为模板生成目标核酸的一个或多个拷贝。
[0061]
如本文所用,术语“基因组”指通常以核酸(dna或rna)编码的个体的遗传信息,包括基因和非编码序列。基因组可以指构成生物的一组染色体(单倍体基因组)或生物的两组染色体(二倍体基因组)的核酸,取决于其被使用的上下文。
[0062]
如本文所用,“目标染色体对”指相同类型的一对染色体,其中该对染色体的一个成员是母系遗传的(从母亲那里继承的),并且该对染色体的另一个成员是父系遗传的(从父亲那里继承的)。例如,目标染色体对指一对1号染色体、2号染色体、3号染色体,并且包括多达21号染色体、22号染色体和x染色体。可以通过本文公开的方法同时分析一个或多个目标染色体对,以确定目标染色体对的母系和父系遗传染色体的序列。
[0063]
如本文所用,目标染色体对的“单拷贝”或“单个拷贝”是指单个物理的(physical)dna分子,或者是染色体本身,或者包装成染色体形式(借助于染色体蛋白质(例如组蛋白))。在正常的二倍体人类细胞中,有46条染色单体,23条染色单体来自母亲且23条染色单体来自父亲。目标染色体的单拷贝也称为染色体类型的单拷贝。在本文所述的方法中,一种或多种染色体类型的单拷贝通常被分离到单独的容器中。
[0064]
如本文所用,“染色体类型”指存在于细胞中的特定染色体。在女性的正常二倍体人类细胞中,有22种常染色体和一种性染色体(x染色体)。在男性的正常二倍体人类细胞中,有22种常染色体和两种性染色体(x和y染色体)。
[0065]
如本文所用,术语“多态性位点”或“多态性”是指染色体内的局部区域,其中核苷
酸序列与群体中至少一个个体中的参考序列不同。序列变异可以是一个或多个碱基的取代、插入或缺失。改变染色体或较大核酸分子的结构的多态性是如本文其他地方所述的sv。
[0066]
如本文所用,术语“单核苷酸多态性或snp”指由特定位置的单个碱基的取代引起序列变异的多态性位点。snp是指群体中确定的基因组位置处的核苷酸变异。编码区内的snp,其中两种形式导致相同的蛋白质序列,被称为同义的;如果产生不同的蛋白质,它们是非同义的。例如,snp可以对基因剪接、转录因子结合或非编码rna的序列产生影响,和/或可以表明生物的单倍型。
[0067]
如本文所用,术语“杂交”指一种或多种用于与单链或双链核酸共同定位互补单链核酸和/或共同定位互补的非传统分子的过程,例如通过链分离(例如,通过变性)和重新退火。在说明性实施方案中,互补核酸分子(任选地寡核苷酸)可以与单链或双链dna杂交。本领域已知的杂交方法,包括但不限于用于低严格杂交和高严格杂交的条件(sambrook and russell.(2001)molecular cloning:alaboratory manual 3rd edition.cold spring harbor laboratory press;sambrook,fritsch,maniatis.molecular cloning:alaboratory manual 3rd edition)。可以控制杂交的严格性(例如,通过洗涤条件)以要求探针和目标序列之间达到100%的互补性(高严格性),或者允许探针和目标序列之间存在一些错配(低严格性)。基于目标和探针确定适当杂交和洗涤条件因素是本领域已知的。在说明性实施方案中,在68℃使用0.2
×
ssc/0.1%sds进行10分钟的第一次洗涤之后,对于高严格性洗涤,在68℃使用0.2
×
ssc/0.1%sds进行两次15分钟的另外的洗涤;对于中严格性洗涤,在42℃使用0.2
×
ssc/0.1%sds进行两次15分钟的另外的洗涤。对于低严格性洗涤,在室温使用0.2
×
ssc/0.1%sds进行两次15分钟的另外的洗涤。
[0068]
如本文所用,术语“等位基因”指通过其特定核苷酸序列区别于其他形式的遗传位点、基因组区域或整个染色体的特定形式。
[0069]
如本文所用,术语“位点”指染色体或dna分子上与基因或物理或表型特征相对应的位置。
[0070]
如本文所用,术语“样本”涉及材料或材料混合物,通常(尽管不一定)是液体形式,其含有一种或多种感兴趣的分析物,在本发明的上下文中,是含有细胞材料或至少一种或多种细胞的基因组材料的样本。如本文所用,术语“染色体样本”涉及包含来自对象的染色体的材料或材料混合物。类似地,术语“基因组样本”涉及包含来自对象或细胞的基因组材料的材料或材料混合物。
[0071]
在本发明上下文中,关于信息的术语“分配”指任何种类的信息被连接至特定序列实体(例如参考支架的预定或预选窗口)或序列读段。优选地,根据本文公开的三个通道(i)至(iii),将观察的或映射的读段或读段的部分的数量分配为信息。
[0072]“序列窗口”指支架序列的一部分,其中一个或多个序列读段或其部分可在比对过程中被映射。序列窗口的尺寸取决于序列数据的覆盖度来选择,或者取决于本发明方法的应用任意选择。在本发明的上下文中,序列窗口的尺寸可以为1至50kb,或者优选地为1至10kb,或者最优选地为约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或2 0kb。本发明的窗口也可以更大,例如50kb、100kb、200kb或500kb。根据本文给出的实施例的示例窗口约为50kb。
[0073]
在本发明的上下文中,术语“三层信息”是指三个独立信息通道的整合,该信息通
道可以源自与序列读段信息的单倍型定相结合的链特异性测序。
[0074]
出于本发明的目的,术语“覆盖度”指代表重构序列中给定核苷酸的读段的平均数量。它可以根据原始基因组的长度(g)、读段数量(n)和平均读段长度(l)计算为n
×
l/g。例如,一个假设的基因组有2,000个碱基对,由平均长度为500个核苷酸的8个读段重构而成,其冗余度约为2倍(2x)。该参数还可以估计其他数量,例如由读段覆盖的基因组百分比(有时也称为覆盖度)。相比于30x,其通常是通过测序1000多个细胞的基因组材料获得的覆盖度,本发明的优点之一是在单细胞的目标序列内稳定识别sv,其测序覆盖度仅为0.01x。在本发明的上下文中,优选地序列读段具有目标染色体区域的0.001x至100x的总覆盖度,优选地约0.01x至0.05x。
[0075]
术语“生殖细胞系”指生物的细胞,其可以追溯它们的最终细胞谱系至生物的雄性或雌性生殖细胞。其他被称为“体细胞”的细胞是不直接产生配子或生殖系细胞的细胞。根据应用,生殖系细胞和体细胞均可在本发明的一些实施方案中使用。
[0076]
如本文所用,术语“染色体不稳定性”(cin)和“基因组不稳定性”以及类似表达涉及染色体结构和数量异常的数量或程度,即整个染色体或部分染色体的缺失或重复,例如导致非整倍体(染色体数目不正确)。高cin通常与增殖性疾病(如癌症)相关或在增殖性疾病中检测到。
[0077]
在本发明的上下文中,术语“读段深度”或“深度”指映射到预定或预选序列窗口的读段的数量。
[0078]
在本发明上下文中,术语“诊断特征”或“诊断足迹”或类似表达指与参考状态相比根据本发明分析的测序数据中的预期差异或预示sv或其他改变原因。本文表1提供了二倍体基因组中sv的实施例。然而,本领域技术人员应当理解,遗传的遗传模式将能够根据潜在情况确定任何其他诊断特征或诊断足迹。
[0079]
当在本发明的上下文中使用术语“基态”时,表示单细胞或单细胞群体内亲本模板链的分布。因此,在本发明的优选的实施方案中,基态应表示单细胞是否包含任意数量的w或c模板链。在二倍体情况下,作为非限制性实施例,基态可以是ww、cc、wc或cw(也参见表1)。
[0080]
在一些实施方案中,提供链特异性序列数据以开始本发明的方法。在其他实施方案中,同样优选地,该方法可以包括准备步骤,以制备或过滤序列数据,或者甚至通过对包含目标染色体区域的遗传物质的样本进行链测序来获得序列数据。
[0081]
在一个实施方案中,链特异性序列数据可以已经包括映射至参考支架的序列读段或其部分。在其他实施方案中,使用本领域已知的标准比对工具将序列读段或其部分映射或比对至相应的参考支架。
[0082]
优选地,作为sctrip特定方法的第一步,跨每个单独细胞或实验的读段被分配至给定宽度的窗口(“分箱(binned)”)。在一些实施方案中,根据数据或应用的覆盖度和特定条件选择窗口的宽度。本文其他地方描述了窗口的优选长度。在一些优选的实施方案中,基于其起始位置将映射读段分配至窗口;但是,也可以使用其他参考位置。此外,在一些实施方案中,链状态被分配至所述窗口中的每一个,其指示染色体区域的模板链分布或w和c读段的相对丰度。在二倍体数据集中,链状态表示为ww(watson-watson)、cc(crick-crick)或wc(watson-crick)。在优选的实施方案中,可以使用隐藏markov模型(hmm)执行链状态分
配。
[0083]
任选地,本发明可以包括根据本文实施例1中使用的特定方法的质量控制和数据标准化的各个步骤。
[0084]
在本发明的优选的实施方案中,本文公开的方法整合所有三个信息通道,例如深度、方向和单倍型定相。因此,优选地,在步骤(d)中,所有三个序列信息通道(i)至(iii)被分配至至少一个预定序列窗口。
[0085]
在一些实施方案中,链特异性序列数据包括源自至少一个目标染色体区域的至少两条单独的链中的一条的序列读段,优选地,链特异性序列数据包括源自至少两条单独的链中的另一条的进一步序列读段,例如,其中一条链来自父本染色体,另一条链来自母本染色体(但可以进一步包括源自另外的链的序列读段,如在三倍体的情况下等)。因此,在本发明的方法的一些实施方案中,在步骤(b)中每个序列读段或其部分与正向或反向方向比对,其保留链特异性序列信息。
[0086]
在本发明的一些实施方案中,该方法可以包括在序列数据中识别链状态和/或检测姐妹染色单体交换(sce)。在一些实施方案中优选地在链状态检测步骤期间同时检测sce。使用strand-seq,单细胞内的每个染色体同源物在w或c链上测序(在二倍体情况下导致观察到ww、wc或cc链模式)。链状态检测和sv发现通过检测sce事件(每个二倍体细胞基因组通常有6个sce)来改进,其可以沿着染色体改变同源物的链状态。
[0087]
在一些实施方案中,本发明的方法可以包含片段化至少一个目标染色体区域的步骤,其中片段化基于序列信息(i)至(iii)的通道各自单独或一起执行。原则上,片段化旨在识别沿目标染色体区域的信息分布中的断点,从而确定候选sv的边界。由于本发明还使用链特异性序列数据,因此在片段化期间也可以检测拷贝数中性的候选sv的断点。在分析单细胞群体序列数据的实施方案中,优选地同时对所有细胞片段化。在一些实施方案中,这样的片段也称为子区域。
[0088]
本发明包括对序列单倍型定相的步骤。在一些优选的实施方案中,单倍型定相将wc区域分类为wc或cw状态,其中第一个位置指h1,第二位置个指h2。这样的步骤是优选的,因为该区别随后在sv识别期间被用于以单倍型识别方式预测sv,这是本发明的优点。为了执行该区别,在一些实施方案中,使用至少几十个snv的整个染色体单倍型;这些可以从外部数据源获得,或者替代地作为本发明方法的一个步骤在链特异性序列数据中直接被识别。给定杂合snv列表,作为非限制性实施例,本发明的工作流程可包括strandphaser算法(porubsky,d.et al.dense and accurate whole-chromosome haplotyping of individual genomes.nat.commun.8,1293(2017))以生成染色体规模的单倍型(详情参见实施例中的方法部分)。在本发明的一些实施方案中,步骤(c)涉及通过分配单核苷酸多态性(snp)沿着至少一个目标染色体区域染色体单倍型身份(h1/h2)被分配至任何给定读段,优选地其中此类snp不具有疾病关联。在一些情况下,这种分配在本文中被称为“单倍型标记”序列读段。在本发明的一些实施方案中,基于来自含有一个或多个snp或与一个或多个snp重叠的相同链的一些读段,将源自单链(w或c)的所有读段的单倍型身份被分配为单倍型身份(h1/h2)。该实施方案允许不包含任何snp或不与任何snp重叠的单倍型定相读段。
[0089]
特别优选地,以“链识别(aware)方式”进行单倍型定相。在本发明的上下文中,该实施方案将需要任何给定序列读段的分配的单倍型身份与相同序列读段具有的方向的信
息相关联。因此,在本发明的优选的实施方案中,用于每个序列读段或其部分的通道(ii)和(iii)的信息是关联的。
[0090]
在某些情况下(可能是优选地),测序数据包括多个非重叠的和/或重叠的序列读段。然而,特别是在单细胞分析的情况下(其通常需要低的测序覆盖度),通常是伪迹的读段重复(例如通过pcr)被移除。因此,在优选的实施方案中,链特异性序列数据不包括重叠的序列读段。
[0091]
如本文所述,本发明的方法可用于检测各种sv。因此,优选地,第一方面的方法可以包括步骤(e),其通过对按位置顺序排列和比对的序列读段的序列数据内的多个(至少两个)窗口执行步骤(d)以及在多个窗口内识别子区域来识别结构变异(sv),子区域包括具有通道(i)至(iii)中的任何一个、或所有、或任何组合的异常的/变化的/改变的信息分布的一个或多个窗口。本发明中的异常的/变化的/改变的分布优选为任何本文公开的表明一个或多个sv的诊断足迹。根据本发明的此类诊断足迹描述如下:
[0092]
本发明的整合数据中sv检测的诊断足迹考虑了三个数据层——读段深度、读段方向和定相。在一些实施方案中,除了单细胞外,还可以分析单细胞群体以增加实现类似的sv可能性的两个不同sv类别之间的检测和/或区分,,例如单倍型标签或“单倍型标记”(含有杂合snp的定相读段)也可以优选地被考虑用于分类。在没有单倍型标签的实施方案中,本发明的方法考虑总片段覆盖度(这里表示为倍性水平;例如n=2表示二倍体或与参考相同的拷贝数)和watson读段的分数(简称为w.frac',用w/(w+c)计算)。为本发明开发的sv发现特征取决于目标染色体区域的潜在链状态,以及sv是否是纯合的还是杂合的——即,例如它们在wc、cw、ww或cc染色体区域以及纯合与杂合重复中是不同的。表1显示了在杂合的和纯合的sv背景下以及对有丝分裂链分离的不同模式的sv诊断足迹的概述,这些足迹是本发明的优选的实施方案:
[0093]
表1:根据本发明的诊断足迹
[0094]
[0095][0096]1不能与wc染色体中的参考状态区分*(但能够解析cc和ww染色体,以及因此当评估细胞群体中的亚克隆sv时)
[0097]2不能与wc染色体中的杂合子重复区分*(但能够解析cc和ww染色体,以及因此当评估细胞群体中的亚克隆sv时)
[0098]3不能在ww或cc染色体中定相*(但能够解析wc染色体,以及因此当评估细胞群体中的亚克隆sv时)
[0099]
如已经解释过的片段化,所述子区域或片段可由至少一个但优选地两个断点定
义,其中这些断点表示与参考状态相比和/或与序列数据中的所述通道信息的整体分布相比通道(i)至(iii)的信息中的任何一个、或任何组合、或全部的变化。
[0100]
在一些实施方案中,所述染色体区域的所述参考状态是通道的信息的状态,其预期为所述染色体区域的信息的非异常分布和/或预定状态。
[0101]
在一些实施方案中,目标二倍体染色体区域中的参考状态是在二倍体目标染色体区域包含源自第一亲本目标染色体区域的第一模板链和源自第二亲本目标染色体区域的第二模板链的情况下;所述参考状态为:
[0102]
如果第一亲本目标染色体区域为watson(w),第二个亲本目标染色体区域为crick(c)——wc参考状态:
[0103]
通道(i):总读段的数量对应约2x目标染色体区域(2n)的存在;
[0104]
通道(ii):每个w链和c链的读段的数量对应约1x目标染色体区域(1n)的存在;
[0105]
通道(iii):h1身份的w读段的数量对应1x,以及h2身份的c读段的数量对应1x;或者
[0106]
如果第一亲本目标染色体区域为c,第二亲本目标染色体区域为w——cw参考状态:
[0107]
通道(i):总读段的数量对应约2x目标染色体区域(2n)的存在;
[0108]
通道(ii):每个w链和c链的读段的数量对应约1x目标染色体区域(1n)的存在;
[0109]
通道(iii):h2身份的w读段的数量对应1x,以及h1身份的c读段的数量对应1x;或者
[0110]
如果第一和第二亲本目标染色体区域为w——ww参考状态:
[0111]
通道(i):总读段的数量对应约2x目标染色体区域(2n)的存在;
[0112]
通道(ii):w链的读段的数量对应约2x目标染色体区域(2n)的存在,以及其中仅存在剩余的(0n)读段;
[0113]
通道(iii):h1身份的w读段的数量对应1x,以及h2身份的w读段的数量对应1x,以及其中仅存在对应0n的剩余的读段;或者
[0114]
如果第一和第二亲本目标染色体区域为c——cc参考状态:
[0115]
通道(i):总读段的数量对应约2x目标染色体区域(2n)的存在;
[0116]
通道(ii):c链的读段的数量对应约2x目标染色体区域(2n)的存在,以及其中仅存在对应0n的剩余的w读段;
[0117]
通道(iii):h1身份的c读段的数量对应1x,以及h2身份的c读段的数量对应1x,以及其中仅存在对应0n的剩余的w读段;
[0118]
其中,如果参考状态存在变化,检测到sv,并且任选地,其中根据表1中所示的变化对sv进行分类。
[0119]
特别优选地,表1中提到的任何sv是基于指示的诊断足迹检测的,此类sv将根据细胞的相应基态显示。
[0120]
在一些实施方案中,sv是改变的倍性状态,其中序列数据包括不同染色体的多个目标染色体区域,并且其中改变的倍性状态通过一条染色体的候选多倍性染色体区域与其他染色体的一个或多个其他染色体区域之间的通道(i)至(iii)的信息中的任何一个、全部或任何组合的整体分布的差异来识别。优选地,本发明的方法涉及确定w和c链在单细胞群
体中的分布,并由此得到每个目标染色体区域(优选地是目标染色体)的倍性状态。
[0121]
实施例部分提供了非整倍体鉴定的详细描述。检测单细胞的目标染色体区域的倍性状态是基于这样一个事实,即在通过strand-seq测序的二倍体细胞中,复制的染色体随机且独立地有丝分裂分离至产生的子细胞。这意味着所有常染色体的大约50%将显示一种特征模式,其中一个同源物在正链(这里是watson的w)上被测序,另一个同源物在负链(crick的c)上被测序——以下称为wc模式。其余常染色体分别仅在c链(约25%;cc模式)或仅在w链(约25%;ww模式)上被测序(图2)。二项分布(参见实施例部分)可以被用于计算不同细胞倍性状态下常染色体链模式的预期频率。例如,在三倍体细胞中,ccc模式(常染色体的所有读段映射到c链)和www模式(常染色体的所有读段映射到w链)分别出现在所有常染色体的12.5%中。cww型和ccw型分别出现在所有常染色体的37.5%中。相比之下,四倍体和单倍体将产生各自可识别的链模式(表2)。这些不同的链状态模式(即w和c读段的相对丰度)和/或给定染色体区域的链遗传模式的预期频率可以被用于识别样本中的非整倍体。与现有方法不同的是,这些诊断足迹不需要另外的数据(例如在给定细胞中检测另外的体细胞变异)来进行倍性分配,因此,它们在检测细胞中潜在的致病性倍性改变方面更为有效和适用。
[0122]
表2中示出了几种细胞倍性状态的诊断足迹特征。二项分布可以被用于计算不同倍性状态下常染色体链模式的预期频率。w,基因组的watson链。c,crick链。
[0123]
表2:非整倍体的诊断链模式(足迹)
[0124][0125]
检测细胞倍性的方法优选地是至少使用链特异性序列数据和读段深度的方法。更优选地,单倍型定相也被整合。
[0126]
在其他实施方案中,倍性的检测涉及包含源自单细胞群体的数据以允许检测w和/或c链的分布的链特异性序列数据。包括的单细胞数据越多,通过本发明的方法可以检测到的非整倍体就越复杂。
[0127]
在一些实施方案中,使用至少两个或更多个单细胞的至少一个目标染色体区域的链特异性序列数据执行本发明的方法,优选地10个或更多个,更优选地50个或更多个,最优选地90个或更多个或350个或更多个;并且优选地,其中多个单细胞源自相同或同一的来源,例如相同个体和/或相同组织或样本类型。这种单细胞群体或多个单细胞优选地具有相同的起源,并且预期共享所述多倍体和/或易位。如果群体内链方向的分布与预期模式不同,则优选地检测多倍体或易位。在一些实施方案中,如果每个染色体的序列的正链或反链的分布与预期的二倍体染色体(常染色体)分离的整体分布不同,例如50%wc、25%ww和25%cc,则检测到多倍体。
[0128]
在本发明的上下文中,细胞或单细胞可以是包括多核苷酸基因组或其部分的任何生物细胞或细胞样结构。因此,细胞可以是病毒、原核细胞或真核细胞,例如动物或植物细胞,其中动物细胞优选地是哺乳动物细胞,例如小鼠、大鼠或人类细胞。任何细胞类型或任何组织来源的任何细胞均可用于本发明。优选地,从患者的细胞样本中获得至少一个单细胞,其中所述单细胞是与疾病相关的细胞或者是所述患者的健康细胞,优选地其中所述方法被用于与疾病和/或健康细胞相关的多个单细胞。
[0129]
本发明的方法特别适用于诊断疾病或对象发生疾病的概率,以及最终对疾病进行分期或监测,甚至估计疾病严重程度。有许多遗传性疾病与任何类型的sv有关。因此,本发明的一些优选的实施方案还包括进一步的步骤(f),基于目标染色体区域内检测的sv的身份、位置或数量诊断疾病。下文提供了诊断应用的详细信息。在一些实施方案中,可以将所述目标染色体区域的检测的sv与所述染色体区域的已知参考状态(例如健康细胞的染色体区域的已知状态)进行比较。此外,为了检测sv可能的病理影响,本发明可以包括检测目标染色体区域内sv影响的基因或遗传元件。由于本发明识别了每个检测的sv的染色体位置,因此进一步识别受sv影响的遗传元件(优选地是基因)可以是优选的实施方案,例如,如果其开放阅读框被sv的断点、或者被拷贝数改变、或者被基因区域中任何调节元件的损伤所破坏。
[0130]
在一些优选的实施方案中,根据本文公开的发明的任何方法为体外方法和/或计算机模拟方法。
[0131]
在一些进一步的实施方案中,如本文其他地方所述的,该方法使用多个单细胞文库来执行。在这样的实施方案中,该方法可以进一步包括计算在给定位置sv出现的概率的步骤,例如通过使用所分析的单细胞群体的通道(i)至(iii)中任何一个、任何组合,或所有的bayesian网络。
[0132]
使用sctrip核型分析
[0133]
对基因组进行核型分析在临床实践和研究中是一种有价值的方法。诊断患者、与疾病相关的组织、或生殖医学中的胚胎细胞中的遗传异常。在研究中,核型分析允许研究这类sv、进化事件和表型的遗传模式。传统的核型分析通常使用劳动密集型方法(如giemsa染色(g-banding)),对淋巴细胞和羊水细胞进行。因为染色体在光学显微镜上是可见的,所以解析详细突变(仅涉及染色体的一小部分)的能力是有限的。虽然有更详细的核型分析技术,如fish(荧光原位杂交),但它们依赖于特异性探针,并且在整个染色体组(即完整基因组)上进行fish在经济上或技术上都不可行。
[0134]
因此,在另一方面本发明的目的通过对单细胞、或者多个单细胞的群体、或者获得此类细胞的对象进行核型分析的方法得以解决,该方法包括,
[0135]
(a)提供至少一个单细胞或单细胞群体中每个细胞的至少一个目标染色体区域(优选完整基因组)的链特异性序列数据,
[0136]
(b)执行本文其他地方所述的sctrip方法,
[0137]
(c)在所述单细胞或单细胞群体的目标染色体区域内检测一个或多个sv,以及
[0138]
(d)根据从sctrip方法的输出中检测的所有sv获得计算机模拟核型;例如,核型可以通过sv的位置、概率和/或类型在分析的基因组的示意图上可视化。这种表现可以对应于染色体处于其中期或前中期状态中的分析的基因组。图中提供了此类计算机核型的实施
lupski综合征)、17p11.2(微重复17p11.2)、17q21.31(微缺失17q21.31)、19q13.11(微缺失19q13.11)、22q11.2(远端微缺失22q11.2)、xq28(微重复xq28)、1p32.1-p31.1(微缺失和重复1p32-p31)、7q32.2-q34(微缺失7q33)和6q22.33-q23.3(微缺失6q22.33)。
[0147]
许多癌症疾病与染色体异常有关。因此,如果患者样本与参考相比显示cin异常或增加,则一般可诊断为癌症。本发明上下文中分析、预测、诊断或监测的癌症选自以下非限制性癌症列表:
[0148]
听神经瘤;腺癌;肾上腺癌;肛门癌;血管肉瘤(如淋巴管肉瘤、淋巴管内皮肉瘤、血管肉瘤);阑尾癌;良性单克隆丙球蛋白病;胆管肿瘤(如胆管癌);膀胱癌;乳腺癌(如乳房的腺癌、乳房的乳头状癌、乳腺瘤、乳房的髓样癌);脑癌(如脑膜瘤、胶质母细胞瘤、神经胶质瘤(如星形细胞瘤、少突胶质细胞瘤)、成神经管细胞瘤);支气管癌瘤;类癌瘤;宫颈癌(如宫颈腺癌);绒毛膜癌;脊索瘤;颅咽管瘤;结肠直肠癌(如结肠癌、直肠癌、结直肠腺癌);结缔组织癌;上皮癌;室管膜瘤;内皮肉瘤(如kaposi肉瘤、多发性特发性出血性肉瘤);子宫内膜癌(如子宫癌、子宫肉瘤);食管癌(如食管腺癌、barrett腺癌);尤因氏肉瘤(ewing's sarcoma);眼癌(如眼内黑色素瘤、视网膜母细胞瘤);常见嗜酸性粒细胞增多症(familiar hypereosinophilia);胆囊癌;胃癌(如胃腺癌);胃肠道间质瘤(gist);生殖细胞癌;头颈癌(如头颈部鳞状细胞癌、口腔癌(如口腔鳞状细胞癌)、咽喉癌(如喉癌、咽癌、鼻咽癌、口咽癌);造血系统癌(如白血病,如急性淋巴细胞白血病(all)(如b细胞all、t细胞all)、急性髓细胞白血病(aml)(如b细胞aml、t细胞aml)、慢性髓细胞白血病(cml)(如b细胞cml、t细胞cml)和慢性淋巴细胞白血病(cll)(如b细胞cll、t细胞cll));淋巴瘤,如霍奇金(hodgkin)淋巴瘤(hl)(如b细胞hl、t细胞hl)和非霍奇金淋巴瘤(nhl)(如b细胞nhl,如弥漫性大细胞淋巴瘤(dlcl)(如弥漫性大b细胞淋巴瘤)、滤泡性淋巴瘤、慢性淋巴细胞白血病/小淋巴细胞淋巴瘤(cll/sll)、套细胞淋巴瘤(mcl)、边缘区b细胞淋巴瘤(如粘膜相关淋巴组织(malt)淋巴瘤、淋巴结边缘区b细胞淋巴瘤、脾边缘区b细胞淋巴瘤)、原发性纵隔b细胞淋巴瘤、burkitt淋巴瘤、淋巴浆细胞淋巴瘤(即waldenstrom巨球蛋白血症)、毛细胞白血病(hcl)、免疫母细胞大细胞淋巴瘤、前体b淋巴细胞淋巴瘤和原发性中枢神经系统(cns)淋巴瘤;以及t细胞nhl,如前体t淋巴细胞淋巴瘤/白血病、外周t细胞淋巴瘤(ptcl)(如皮肤t细胞淋巴瘤(ctcl)(如真菌病、sezary综合征)、血管免疫母细胞性t细胞淋巴瘤、结外自然杀伤性t细胞淋巴瘤、肠病型t细胞淋巴瘤、皮下脂膜炎样t细胞淋巴瘤和间变性大细胞淋巴瘤),如上所述的一种或多种白血病/淋巴瘤的混合物和多发性骨髓瘤(mm))、重链疾病(例如,α链疾病、γ链疾病、μ链疾病);血管母细胞瘤;下咽癌;炎性肌纤维母细胞瘤;免疫细胞性淀粉样变;肾癌(如肾母细胞瘤又称wilms瘤、肾细胞癌);肝癌(如肝细胞癌(hcc)、恶性肝癌);肺癌(例如,支气管肺癌、小细胞肺癌(sclc)、非小细胞肺癌(nsclc)、肺腺癌);平滑肌肉瘤(lms);肥大细胞增多症(如系统性肥大细胞增多症);肌肉癌;骨髓增生异常综合征(mds);间皮瘤;骨髓增生性疾病(mpd)(如真性红细胞增多症(pv)、原发性血小板增多症(et)、原因不明性髓样化生(amm)又称骨髓纤维化(mf)、慢性特发性骨髓纤维化、慢性粒细胞白血病(cml)、慢性中性粒细胞白血病(cnl)、高嗜酸性粒细胞综合征(hes));成神经细胞瘤;神经纤维瘤(如神经纤维瘤病(nf)1型或2型,神经鞘瘤病);神经内分泌癌(如胃肠胰神经内分泌肿瘤(gep-net)、类癌瘤);骨肉瘤(如骨癌);卵巢癌(如囊腺癌、卵巢胚胎癌、卵巢腺癌);乳头状腺癌;胰腺癌(如胰腺腺癌、导管内乳头状粘液瘤(ipmn)、胰岛细胞瘤);阴茎
癌(如阴茎和阴囊的paget病);松果体瘤;原始神经外胚层肿瘤(pnt);浆细胞瘤;副肿瘤综合征;上皮内肿瘤;前列腺癌(如前列腺腺癌);直肠癌;横纹肌肉瘤;唾液腺癌;皮肤癌(如鳞状细胞癌(scc)、角化棘皮瘤(ka)、黑色素瘤、基底细胞癌(bcc));小肠癌(如阑尾癌);软组织肉瘤(如恶性纤维组织细胞瘤(mfh)、脂肪肉瘤、恶性周围神经鞘瘤(mpnst)、软骨肉瘤、纤维肉瘤、粘液肉瘤);皮脂腺癌;小肠癌;汗腺癌;滑膜瘤;睾丸癌(如精原细胞瘤、睾丸胚胎癌);甲状腺癌(如甲状腺乳头状癌、乳头状甲状腺癌(ptc)、甲状腺髓样癌);尿道癌;阴道癌;以及外阴癌(如外阴的paget病)。
[0149]
在优选的实施方案中,本发明用于诊断疾病的方法是纯体外或甚至计算机模拟执行的方法。
[0150]
在其他的实施方案中,本发明的诊断可以包括以下步骤中的任何一个或所有:获得待诊断对象的样本。此类样品可以是包含基因组材料的任何生物样品,优选对象的细胞样品。此类样本可以从任何来源获得以分析对象的总体基因组状态,或者可以从怀疑涉及病理的组织或细胞类型中具体获得。因此,除了本文提供的样本的一般定义之外,此类生物样本还可以包括任何生物组织、器官、器官系统或流体。此类样本包括但不限于痰、血液、血细胞(如白细胞)、羊水、血浆、精液、骨髓和组织或髓部(core)、细针或穿刺活检样本、尿液、腹膜液和胸膜液,或其细胞。生物样本还可以包括组织切片,例如为组织学目的而采集的冷冻切片。生物样本也可以被称为“患者样本”。
[0151]
诊断中包括的进一步步骤可能是用本发明的方法分离待分析的dna。此类获得dna、纯化和制备以用于测序用途的方法是本领域技术人员所熟知的。此外,本发明的诊断方法可以包括链特异性测序以获得链特异性序列数据。
[0152]
细胞质量控制
[0153]
另一方面,本发明提供了一种用于评估单细胞或单细胞群体内的染色体稳定性的方法,该方法包括执行根据前述权利要求中任一项所述的方法,其中所述单细胞或单细胞群体中sv的总数量增加或sv的任何一种类型或多种类型的数量增加表明染色体不稳定。
[0154]
如本文已经提到的,cin是许多疾病(特别是癌症)的一般指标。因此,用本发明的sctrip测试cin提供了一种应用,当细胞显示增加的cin时,轻松了解细胞群体是否是低质量的。该方法用于基因工程细胞或细胞群体的质量控制,其中不稳定性的增加表示质量损失。
[0155]
在涉及治疗目的的自体或异源或外源细胞的基因工程的基因编辑和自体t细胞疗法的时代,将工程化细胞应用于人类患者之前对其质量控制的需求增加。细胞的基因工程总是承担着将可能影响遗传稳定性的工程化细胞改变引入基因组的风险。在最坏的情况下,cin增加能够在给患者施用后导致癌症疾病的发展,这必须不惜一切代价避免。由于本发明提供了一种快速且便宜的方法来评估细胞群体的sv,因此其可以被用作再输注之前此类工程化细胞的质量控制程序。在一个实施方案中,该方法需要检测工程化细胞或细胞系样本中的sv,并将其与参考细胞或参考状态进行比较。观察的cin的增加将导致工程细胞的质量下降。此外,某些类型的有问题的sv的出现可能会导致丢弃工程化细胞。
[0156]
优选地,在这方面,所分析的单细胞或单细胞群体是遗传工程化细胞,例如通过基因编辑、病毒整合。优选的工程化细胞是免疫细胞,如嵌合抗原受体(car)-t细胞、t细胞受体(tcr)工程化细胞或抗体工程化细胞。然而,任何细胞或细胞系可以用本发明的方法进行
质量控制测试。这些应用包括干细胞研究,如控制诱导多能干细胞(ipsc)。因此,此类干细胞(优选ipsc)是根据本发明的各个方面和实施方案分析的优选的单细胞或细胞群体。
[0157]
在一些实施方案中,单细胞或单细胞群体被用于患者的细胞疗法,例如自体免疫细胞疗法。
[0158]
另一方面,本发明还涉及筛选影响染色体稳定性的候选化合物的方法。优选地该方法涉及将至少一个单细胞或细胞群体与候选化合物接触,然后执行本文之前所述的sctrip的任何方法,以便在处理的细胞中获得sv。该方法的另一步骤可以包括将经处理的细胞中检测的sv与参考、或与经处理前的细胞、或与平行的未经处理的细胞进行比较。
[0159]
筛选方法可以被应用于例如测试治疗性化合物对基因组稳定性的副作用。此类化合物可以是可能被怀疑对基因组稳定性有影响的任何化合物,且优选地选自多肽、肽、糖蛋白、拟肽、抗体或抗体样分子;核酸,例如dna或rna,例如反义dna或rna、核酶、rna或dna适配体、sirna、shrna等,包括其变体或衍生物,例如肽核酸(pna);靶向基因编辑构建体,例如crispr/cas9构建体,碳水化合物,例如多糖或低聚糖等,包括其变体或衍生物;脂质,例如脂肪酸等,包括其变体或衍生物;或者小有机分子,包括但不限于小分子配体、小细胞渗透分子和拟肽化合物。因此,术语候选化合物还应包括处理或改变细胞的任何方法,以测试此类方法在基因组稳定性方面的能力。然而,优选地是测试抗癌药物,如化疗药物。
[0160]
此外,在一些实施方案和方面中本发明涉及以下特别优选的逐项列出的实施方案:
[0161]
项目1:一种通过单细胞三通道处理(sctrip)分析至少一个目标染色体区域的测序数据的方法,其包括提供至少一个单细胞的至少一个目标染色体区域的链特异性序列数据,其中链特异性序列数据包括多个通过至少一个单细胞的目标染色体区域的测序获得的链特异性序列读段,将序列读段或者序列读段的每个片段化部分(如果序列读段被平均地片段化)与参考组件比对,然后在任何给定的选定窗口中分配三层序列信息中的至少两层:(i)总序列读段或其部分的数量(也称为“读段深度”);(ii)正向(或watson)序列读段或其部分的数量,以及反向(或crick)序列读段或其部分的数量;(iii)分配有特异性单倍型身份(如h1和/或h2)的序列读段或其部分的数量。
[0162]
项目2:根据项目1所述的方法,其包括以下具体步骤:
[0163]
(a)提供至少一个单细胞的至少一个目标染色体区域的链特异性序列数据,其中链特异性序列数据包括多个通过至少一个单细胞的目标染色体区域的测序获得的链特异性序列读段;
[0164]
(b)将每个序列读段或其部分与至少一个目标染色体区域的参考序列比对,以使所述序列读段或其部分沿着所述至少一个目标染色体区域的参考序列按位置顺序排列;
[0165]
(c)将染色体单倍型身份(h1/h2)沿着至少一个目标染色体区域分配至来自(b)的每个比对的序列读段或其部分;和
[0166]
(d)将下列序列信息通道中的任意两个分配至按位置顺序排列且比对的序列读段或其部分的至少一个预定序列窗口:
[0167]
(i)在至少一个预定序列窗口中比对的总序列读段或其部分的数量;
[0168]
(ii)在至少一个预定序列窗口中比对的正向序列读段或其部分的数量,以及反向序列读段或其部分的数量;
[0169]
(iii)在至少一个预定序列窗口中比对的分配至第一(h1)单倍型身份的序列读段或其部分的数量;和/或分配至第二(h2)单倍型身份的序列读段或其部分的数量。
[0170]
项目3:根据项目1或项目2所述的方法,其中所有三个序列信息(i)至(iii)通道被分配。
[0171]
项目4:根据项目1至项目3中任一项所述的方法,其包括片段化至少一个目标染色体区域的步骤,其中片段化是基于序列信息通道(i)至(iii)各自单独或一起执行的。
[0172]
项目5:根据项目1至项目3中任一项所述的方法,其中提供的序列读段(如项目2中的步骤(a))独立于序列读段的读段长度被提供。
[0173]
项目6:根据项目1至项目5中任一项所述的方法,其中所述链特异性序列数据包括映射至所述至少一个目标染色体区域的至少两条单独的链中的一条的序列读段,优选地包括映射至所述至少两条单独的链中的另一条的进一步序列读段,例如其中一条链来自父本染色体,另一条链来自母本染色体(但可以进一步包括映射至单链(在单倍体的情况下),或另外的链(在三倍体的情况下)的序列读段等等)。
[0174]
项目7:根据项目1至项目6中任一项所述的方法,其中通过strand-seq(falconer et al.2012nature methods.9(11):1107
–
1112.)获得链特异性序列数据。
[0175]
项目8:根据上述项目中任一项所述的方法,其中测序数据包括多个非重叠的和/或重叠的序列读段。
[0176]
项目9:根据上述项目中任一项所述的方法,其中如项目2中的步骤(b)中,每个序列读段或其部分与正向或反向方向比对,以保持链特异性序列信息。
[0177]
项目10:根据上述项目中任一项所述的方法,其进一步包括以下步骤:
[0178]
(e)通过对按位置顺序排列和比对的序列读段的序列数据内的多个(至少两个)窗口执行步骤(d),以及在多个窗口内识别子区域,来识别结构变异(sv),该子区域包含具有通道(i)至(iii)中的任何一个、或所有、或任何组合的异常的/变化的/改变的信息分布的一个或多个窗口。
[0179]
项目11:根据项目10所述的方法,其中所述子区域由至少一个(优选两个)断点定义,并且其中所述断点表示与参考状态相比和/或与序列数据中的所述通道信息的整体分布相比通道(i)至(iii)的信息中的任何一个、或任何组合、或全部的变化。
[0180]
项目12:根据项目9或项目10所述的方法,其中所述染色体区域的所述参考状态是通道的信息的状态,其预期为所述染色体区域的信息的非异常分布和/或预定状态。
[0181]
项目13:根据项目12所述的方法,其中目标二倍体染色体区域中的所述参考状态是在二倍体目标染色体区域包括源自第一亲本目标染色体区域的第一模板链和源自第二亲本目标染色体区域的第二模板链的情况下;所述参考状态为:如果第一个亲本目标染色体区域为watson(w),第二个亲本目标染色体区域为crick(c)——wc参考状态:
[0182]
通道(i):总读段的数量对应约2x目标染色体区域(2n)的存在;
[0183]
通道(ii):每个w链和c链的读段的数量对应约1x目标染色体区域(1n)的存在;
[0184]
通道(iii):h1身份的w读段的数量对应1x,以及h2身份的c读段的数量对应1x;或者
[0185]
如果第一亲本目标染色体区域为c,第二亲本目标染色体区域为w——cw参考状态:
[0186]
通道(i):总读段的数量对应约2x目标染色体区域(2n)的存在;
[0187]
通道(ii):每个w链和c链的读段的数量对应约1x目标染色体区域(1n)的存在;
[0188]
通道(iii):h2身份的w读段的数量对应1x,以及h1身份的c读段的数量对应1x;或者
[0189]
如果第一和第二亲本目标染色体区域为w——ww参考状态:
[0190]
通道(i):总读段的数量对应约2x目标染色体区域(2n)的存在;
[0191]
通道(ii):w链的读段的数量对应约2x目标染色体区域(2n)的存在,以及其中仅存在剩余的(0n)读取;
[0192]
通道(iii):h1身份的w读段的数量对应1x,以及h2身份的w读段的数量对应1x,以及其中仅存在对应0n的剩余的读段;或者
[0193]
如果第一和第二亲本目标染色体区域为c——cc参考状态:
[0194]
通道(i):总读段的数量对应约2x目标染色体区域(2n)的存在;
[0195]
通道(ii):c链的读段的数量对应约2x目标染色体区域(2n)的存在,以及其中仅存在对应0n的剩余的w读段;
[0196]
通道(iii):h1身份的c读段的数量对应1x,以及h2身份的c读段的数量对应1x,以及其中仅存在对应0n的剩余的w读段;
[0197]
其中,如果参考状态存在变化,则检测到sv,并且任选地,其中根据表1中所示的变化对sv进行分类。
[0198]
项目14:根据项目10至项目12中任一项所述的方法,其中sv为易位,以及其中序列数据包括不同染色体的多个目标染色体区域,以及其中易位通过一条染色体的候选染色体区域与其他染色体的一个或多个其他染色体区域之间通道(i)至(iii)的信息中的任何一个、或全部或任何组合的整体分布的差异来识别。
[0199]
项目15:根据项目10至项目12中任一项所述的方法,其中sv为改变的倍性状态,以及其中序列数据包括不同染色体的多个目标染色体区域,以及其中改变的倍性状态通过一条染色体的候选多倍体染色体区域与其他染色体的一个或多个其他染色体区域之间的通道(i)至(iii)的信息中的任何一个、全部或任何组合的整体分布的差异来识别。
[0200]
项目16:根据上述项目中任何一项所述的方法,其中序列读段具有20至500个核苷酸的长度,以及其中在序列读段超过长度阈值(500个,优选1000个或更多个核苷酸)的情况下使用部分序列读段,以及这种长序列读段被计算机模拟片段化成具有优选的20至500(~150)个核苷酸长度的序列读段的较小部分,优选地其中数据集内所述序列读段或其部分具有整体相当的序列长度。
[0201]
项目17:根据上述项目中任一项所述的方法,其中所述序列读段具有目标染色体区域的0.001x至100x(优选约0.01x至0.05x)的整体覆盖度。
[0202]
项目18:根据上述项目中任一项所述的方法,其中在步骤(c)中,染色体单倍型身份(h1/h2)沿着至少一个目标染色体区域被分配,优选地同时保留链方向信息(即,链识别方式),以及优选地,通过分配单核苷酸多态性(snp)将所述单倍型分配至序列读段或其部分,优选地其中所述snp不具有疾病关联。
[0203]
项目19:根据上述项目中任一项所述的方法,其中所述单倍型身份被分配至包含snp的序列读段或其部分,以及通过与snp数据库比较或者替代地通过将等位基因与相同来
源的多个进一步测序的单细胞进行比较(例如,使用strandphaser
–
porubsky et al.2017)来识别snp的等位基因;以及任选地,其中通过以链身份推断所述单倍型身份并与具有相同链身份且包含该snp的其他序列读段或其部分进行比较,单倍型身份被分配至不包含snp的序列读段或其部分。
[0204]
项目20:根据上述项目中任一项所述的方法,其中所述方法用至少两个或更多个(优选地10个或更多个,更优选地50个或更多个,最优选地90个或更多个或350个或更多个)单细胞的至少一个目标染色体区域的链特异性序列数据执行;以及优选地,其中多个单细胞源自相同或同一来源,例如相同个体和/或相同组织或样本类型。
[0205]
项目21:根据上述项目中任一项所述的方法,其中所述目标染色体区域是一条或多条染色体,优选地是二倍体生物的一条或多条染色体。
[0206]
项目22:根据上述项目中任一项所述的方法,其中至少一个单细胞的至少一个目标染色体区域的链特异性序列数据包括覆盖所述单细胞的完整基因组的数据。
[0207]
项目23:根据上述项目中任一项所述的方法,其中所述细胞为原核细胞、真核细胞(例如动物细胞或植物细胞),优选地其中所述动物细胞为哺乳动物细胞,例如小鼠、大鼠或人类细胞。
[0208]
项目24:根据上述项目中任一项所述的方法,其中从患者的细胞样本中获得至少一个单细胞的至少一个目标染色体区域的链特异性序列数据,以及其中所述单细胞是与疾病相关的细胞或者是所述患者的健康细胞,优选地其中所述方法被用于与疾病和/或健康细胞相关的多个单细胞。
[0209]
项目25:根据上述项目中任一项所述的方法,用于检测优选的二倍体细胞基因组内的多倍体状态和/或平衡或不平衡易位,其中所述方法包括覆盖单细胞的受影响染色体区域(例如染色体)的链特异性序列数据,以及其中上述项目中任一项的方法用相同来源和/或预期共享所述多倍体和/或易位的多个单细胞进行;以及其中,如果单细胞群体内的链方向的分布与预期模式不同,则检测到多倍体或易位。
[0210]
项目26:根据项目26所述的方法,其中,如果每个染色体的测序的正链或反链的分布与预期的二倍体染色体(常染色体)分离的整体分布不同,例如50%wc、25%ww和25%cc,则检测到多倍体。
[0211]
项目27:根据项目26所述的方法,其中如果任何给定目标染色体区域(例如染色体)内的任何给定子区域的正向或反向读段的分布与给定染色体的另一个子区域独立分离,如多个单细胞内其分布所证明的,则检测到易位。
[0212]
项目28:根据上述项目中任何一项所述的方法,其中所述方法包括基于目标染色体区域内检测的sv的身份、位置或数量诊断疾病的进一步的步骤(f)。
[0213]
项目29:根据项目28所述的方法,其中所述目标染色体区域的检测的sv与所述染色体区域的已知参考状态(例如健康细胞的染色体区域的已知状态)进行比较。
[0214]
项目30:根据上述项目中任一项所述的方法,其中所述方法进一步包括检测目标染色体区域内sv影响的基因或遗传元件。
[0215]
项目31:根据上述项目中任一项所述的方法,其为体外方法或计算机模拟方法。
[0216]
项目32:根据上述项目中任何一项所述的方法,其中所述方法进一步包括计算在给定位置sv出现的概率的步骤,例如通过使用所有通道(i)至(iii)的bayesian网络。
[0217]
项目33:一种检测目标染色体区域中结构变异(sv)的方法,该方法包括,执行如项目9所述所述的方法,以及参考项目9时执行如项目10至项目32所述的方法。
[0218]
项目34:一种对单细胞或多个单细胞的群体进行核型分析的方法,该方法包括:
[0219]
(a)提供至少一个单细胞或单细胞群体中的每一个细胞的至少一个目标染色体区域(优选完整基因组)的链特异性序列数据,
[0220]
(b)执行项目1至项目31的方法,
[0221]
(c)检测所述单细胞或单细胞群体的目标染色体区域内的sv,以及
[0222]
(d)根据所有检测的sv获得计算机模拟核型。
[0223]
项目35:一种诊断对象中疾病的方法,该方法包括,提供对象的一个或多个细胞的链特异性序列数据,执行项目33所述的方法,检测一个或多个细胞内的任何sv,以及将检测的sv与参考状态进行比较,其中对象的样本中的一个或多个sv的数量、类型或位置的改变表明存在病况,例如疾病,例如癌症。
[0224]
项目36:一种用于评估单细胞或单细胞群体内的染色体不稳定性(cin)的方法,该方法包括执行根据上述项目中任一项所述的方法,其中所述单细胞或单细胞群体中sv的总数量增加或sv的任何一种类型或多种类型的数量增加,表明染色体不稳定。
[0225]
项目37:根据项目36所述的方法,用于细胞或细胞群体的质量控制,其中不稳定性的增加表示质量损失,优选地在所述细胞或细胞群体的(遗传)改变之后。
[0226]
项目38:根据项目36或项目37所述的方法,其中所述单细胞或单细胞群体是基因工程化的,优选地例如通过重编程、基因编辑或病毒整合。
[0227]
项目39:根据项目36至项目38中任一项所述的方法,其中所述单细胞或单细胞群体被用于患者的细胞疗法,例如自体免疫细胞疗法。
[0228]
项目40:一种计算机可读介质,其包括存储在其上的计算机可读指令,当其在计算机上运行时,执行项目1至项目33项中任一项所述的方法。
[0229]
项目41:一种对感兴趣的至少一个单细胞的基因组进行核型分析的方法,包括:a)从至少一个单细胞的基因组的随机位置获得多个(优选非重叠)链特异性序列;b)将所述测试链特异性序列映射至基因组参考支架以获得映射的链特异性序列的测试分布;c)将(i)映射的序列读段的数量,(ii)映射的正链读段的数量和反链读段的数量,优选其比例,以及(iii)单倍型身份(h1/h2)分配至参考支架内的预定序列窗口以获得映射的序列的三层测试分布;d)识别预期分布之间统计上显著的改变,其中这种改变表明至少一个单细胞的基因组中的核型异常;或者e)将三层测试分布与从参考细胞(例如健康细胞)获得的参考分布比较,其中如果存在显著差异,则所述差异表明至少一个单细胞与参考细胞之间的核型差异。
[0230]
在最后一个方面,本发明还涉及一种计算机可读介质,其包括存储在其上的计算机可读指令,当其在计算机上运行时,执行根据本文公开的发明的方法,优选sctrip。
[0231]
上述实施方案能够以多种方式中的任何一种实施。
[0232]
例如,可以使用硬件、软件或其组合来实施实施方案。当在软件中实施时,软件代码可以在任何合适的处理器或处理器集合上执行,无论是在单个计算机中提供还是分布在多个计算机之间。应当理解,执行上述功能的任何组件或组件集合通常可以被视为控制上述功能的一个或多个控制器。一个或多个控制器可以通过多种方式实现,例如使用专用硬
件,或者使用微码或软件编程以执行上述功能的通用硬件(例如,一个或多个处理器)。
[0233]
在这方面,应当理解,一种实施方式包括至少一种计算机可读存储介质(即,至少一种有形的、非暂时的计算机可读介质),例如计算机存储器(例如,硬盘驱动器、闪存、处理器工作存储器等)、软盘、光盘、磁带、或其他有形的、非暂时的计算机可读介质,其用计算机程序(即,多条指令)编码,当在一个或多个处理器上执行时,执行上述功能。计算机可读存储介质可以是可传输的,使得存储在其上的程序可以被加载到任何计算机资源上以实施本文讨论的技术。此外,应当理解,对在执行时执行上述功能的计算机程序的引用不限于在主机上运行的应用程序。相反,本文使用的术语“计算机程序”在一般意义上指能够被用于编程一个或多个处理器以实施上述技术的任何类型的计算机代码(例如,软件或微码)。
[0234]
本文中使用的术语“本发明的”、“根据本发明”、“根据本发明的”等在本文中的使用意在指本文中所描述和/或所要保护的本发明的所有方面和实施方案。
[0235]
如本文所用,术语“包括”将被解释为涵盖“包含”和“由
……
组成”,这两种含义都是特别意图的,因此单独公开根据本发明的实施方案。在本文中使用时,“和/或”应被视为具有或不具有彼此的两个指定特征或组件中的每一个的特定公开。例如,“a和/或b”将被视为(i)a、(ii)b和(iii)a和b中每一个的具体公开,就如同它们每一个在本文中单独列出一样。在本发明的上下文中,术语“大约”和“约”表示本领域技术人员将理解为仍能确保所讨论特征的技术效果的精度范围。该术语通常表示与所示数值相差
±
20%、
±
15%、
±
10%,例如
±
5%。如普通技术人员所理解的,对于给定技术效果,数值的这种特定将偏差将取决于技术效果的性质。例如,自然或生物技术效通常比人为或工程技术效应具有更大的偏差。当提及单数名词时使用不定冠词或定冠词时,除非另有说明,否则例如“一”、“一个”或“该”包括该名词的复数形式。
[0236]
应当理解,将本发明的教导应用于特定问题或环境,以及包括本发明的变体或其附加特征(例如进一步的方面和实施方案),将在根据本文包含的教导下具有本领域普通技术的人员的能力范围内。
[0237]
除非上下文另有指示,否则以上阐述的特征的描述和定义不限于本发明的任何特定方面或实施方案,并且等同地适用于所描述的所有方面和实施方案。
[0238]
本文引用的所有参考文献、专利和出版物均通过引用整体并入本文。
[0239]
附图的简要说明
[0240]
附图显示出:
[0241]
图1显示出(a)strand-seq测序方案的概述。strand-seq涉及将brdu并入分裂细胞中,然后通过切割去除含有brdu的链,并对剩余的链进行短读段测序
21
。strand-seq文库保留了链方向和染色体同源物(单倍型)身份。虚线:链(brdu)标签。w,watson链(橙色);c,crick(绿色);h,单倍型。(b)描述有丝分裂期间模板链共分离模式如何显示单细胞中sv的方案。del,缺失;inv,倒位;tr,易位。衍生染色体的片段在dna复制过程中共享相同的模板链。h1/h2,染色体的单倍型1和2;h1/h2,另一条染色体的单倍型1和2。(c)sctrip计算方法利用三个数据层:读段深度、链比例和染色体长度单倍型定相。红色棒糖(lollipop):基于重叠snp分配至h1的读段;蓝色棒糖:分配至h2的读段。单倍型定相以链识别的方式进行评估,核型模式图(ideogram)的左侧棒糖显示定相w读段,右侧显示为定相c读段。与之前的sv检测方法相比,sctrip不依赖于不整合的或分开的读段,可扩展检测在单细胞中被认为是
不可行的。图d-f描绘了染色体的诊断足迹,其中两个单倍型在不同的链上(“wc/cw染色体”)被标记。我们的框架还检测并评分cc和ww染色体上的等效足迹(见表1)。(d)del,结合未改变的读段方向检测为影响单个单倍型的读段深度损失。dup,检测为读段方向不变的单倍型特异性深度增加。(e)平衡inv,识别为深度不变的单倍型定相读段方向“翻转”。invdup,其特征是检测到一种单倍型的反向读段与同一单倍型的读段深度增加一致。(f)平衡易位,检测为影响携带sv的细胞中相同配对的基因组区域的相关模板链转变。(g)用于sv发现的bayesian框架。所描述的概率分布表示h1上的invdup(对于单倍型1(h1),片段在两条链上可见,而对于h2仅显示w链上);(h)单体型识别sv分类的bayesian图形模型。所示模型用于单细胞中的单倍型识别sv发现。该图形模型采用常用的图版表示法:圆圈表示随机变量,正方形表示模型参数,灰色(白色)对象表示观察的(潜在的)变量,箭头表示相关性,大矩形表示围住的变量存在多次。该模型描述了j个单细胞、k个片段和h=2单倍型。随机变量:片段长度l、基态t、单倍型sv状态v(待推断)、w/c读段的拷贝数n
w/c
、w/c方向的读段计数x
w/c
以及单倍型标记的w/c方向的读段计数x
w/ctag
。请注意,读段计数不是通过它们的单倍型(h框内的白色圆圈)观察到的,而是在没有单倍型信息(h框外的灰色圆圈)的情况下观察到的。通过单倍型观察与杂合snp重叠的读段的分数(h框内标记的灰色读段计数变量)。模型参数:背景读段的分数α,负二项式参数ρ和r,以及杂合比例h。
[0242]
图2显示出sctrip显示上皮细胞中的缺失、重复、倒位和染色体非整倍体。(a)通过dna链和单倍型分离的分箱的读段计数显示单细胞中sv的存在(w,watson链(橙色);c,crick(绿色))。左图:3p上的单倍型解析重复(dup),存在于rpe-1中但在c7中缺失。右图:3q上的单倍型解析缺失(del),存在于c7中但在rpe-1中缺失。“深度”框描述读段计数;“链”描述w:c分数;“定相”显示单倍型定相snp的位置,棒糖方向反映了包含snp的读段的链状态(核型模式图左侧是w,右侧是c)。(b)染色体17p单倍型解析倒位(inv)在c7和rpe-1中共享。(c)单倍体染色体的诊断足迹。描述的模板链状态模式来自c7,其具有核型定义
30
的单倍体13。左图显示了来自两个单细胞的染色体13链模式,具有可见的1:0模式特征的单倍体(1n)。右图总结了154个测序细胞中观察的w和c读段的分数。(d)三倍体区域的诊断足迹。所描述的模板链状态模式来自于rpe-1细胞,表现出核型定义的10q三体区域
27
。左图显示了来自四个单细胞的染色体10链模式。右图总结了80个测序细胞中观察的三倍体(3n)10q区域的w和c读段的分数,显示了三倍体的2:1和3:0链比例特征(表2)。
[0243]
图3显示了单细胞中的易位发现。(a)在bm510中,来自染色体10、13、15、17和22的片段未能与它们起源的相应染色体共分离,表明可能参与易位(使用“tr”,如“h2-tr”或“chr10tr”,表示这些片段的候选易位状态)。(b)中心金字塔:bm510易位的无偏分析。成对热图描绘了每个单倍型的片段模板链相关值,突出了易位的片段共分离诊断足迹(相关值在此表示为benjamini-hochberg调整的p值)(图1f)。带有黑色轮廓的橙色框描述了四种情况下的显著相关性(p《0.01;fisher精确检验)——对应于我们在bm510中发现的四条衍生染色体。左侧和右侧的图:彩色框举例说明了非相互易位der(x)t(x;10)和t(15;17)相互易位的片段的单倍型解析模板链状态。(在每种情况下,为可视化目的仅描绘了几个细胞。)框颜色:w(橙色);c(绿色)。灰色箭头突出显示片段之间的成对相关性,其中成对片段始终表现出相同的链状态(例如chrx和chr10tr),或始终表现出反向的链状态(例如chr15tr和chr17;反映这些易位伙伴(partner)的反向方向)。17p的易位部分内的倒位用圆形箭头表
示。(c)中心:四条推断的衍生染色体的卡通表示。虚线对应于近端着丝染色体13和15上的未装配区域。(d)circos图描绘了基因组窗口
77
中的易位和平均基因表达值,其由bm510(此处表示为“b”)、rpe-1(“r”)和c7(“c”)生成的rna-seq数据计算。图s11通过单倍型解析表达。(e)bm510中基因融合的验证。c7、rpe-1和bm510描绘了ntrk3(绿色)、ntrk3-as1(黄色)和tp53(蓝色)基于rna-seq的读段深度。紫色虚线:检测的融合连接。左下角:推断的融合转录本。紫色框显示起始密码子位置。右下角:bm510中ntrk3调节异常。r1-3,rpe-1的rna-seq重复。例如,外显子。
[0244]
图4显示了复杂重排过程的单细胞特征。(a)c7细胞的链特异性读段深度,其10p上有invdup介导的扩增的区域,其具有相同单倍型的相邻末端缺失(delter),其由bfb循环产生。(b)从154个c7细胞聚合读段数据。颜色表示扩增子内识别的六个拷贝数片段(红色、蓝色、绿色、紫色、橙色和黄色)。灰色:扩增子两侧的区域。(c)描绘了三个c7细胞,红色表示的10p扩增子区域的估计最大拷贝数(cn)为1(上图)、cn为~110(中图)和cn为~440(下图)。在15q上获得的片段,sctrip推断该片段与扩增子区发生了不平衡易位,如下所示(缺少扩增子的细胞中缺乏该sv;上图)。w(绿色)和c(橙色)的读段计数上限为50(*,饱和读段计数)。tr,易位。(d)10p的遗传多样性。cn(x轴)在154个已测序的c7细胞(y轴)显示,提供(b)中每个片段的逐个细胞的cn估计值。至少有3个不同的组是容易辨别的:高cn、中等cn和10p区域缺失(与图(c)相比)。误差线反映95%的置信区间。箭头表示在10p扩增子处cn=1且cn为~440的细胞。(e)ssv模型导致观察的“主要克隆”结构。通过bfb循环的扩增通常以2n个拷贝数步骤进行,表明发生了~7个连续的bfb循环。根据我们的模型,15q末端序列的易位稳定在10p。dbs,双链断裂。(f)bfb损伤,对应于在同一单倍型上两侧为delter的invdup,在单个bm510细胞中鉴定(g)单个bm510细胞中涉及del和inv的聚集重排。所示为分为三个典型的sctrip数据通道的分箱的读段数据(左)。所有聚集的sv影响单个单倍型(h1,红色)。
[0245]
图5显示了基于单细胞测序的pdx衍生t-all复发的核型。(a)利用sctrip产生的基于单细胞测序的sv调用,由41个测序细胞构建的单倍型解析共有p33核型。杂合子sv仅在其已映射到的单倍型上描述。纯合子sv(根据定义)出现在两种单倍型上。cnn-loh,拷贝中性的杂合性缺失(显示在两种单倍型上)
78
。粉红色的染色体反映了重复的同系物。该t-all患者携带两个x染色体单倍型(见图s16)和一个y染色体,表明x和y染色体从父系传递,而母亲将其x染色体贡献给该核型(klinefelter或xxy综合征)。受影响的白血病相关基因以红色突出显示。“bcl11b-enh”表示先前在bcl11b基因的3
′
中描述的增强子区域。(b)使用ward方法对p33中sv基因型可能性进行分层聚类的sv排列的“热图”,显示在t-all复发中单个显性克隆的存在,以及导致核型多样性的另外的体细胞dna改变很少的证据。(c)另外的t-tall样本p1中称为sv事件的“热图”。红色虚线框描绘了样本中清晰的亚克隆群体,由25个细胞表示。
[0246]
图6显示了pdx衍生的t-all复发p1的单细胞测序揭示了以前未识别的sv。(a)使用sctrip在p1中推断的单倍型解析平衡14q32 inv。最左边的断点(浅蓝色粗线)位于tcl1a附近,而最右边的断点(浅蓝色细线)位于bcl11b的3
′
处。(b)最右边的inv断点位于3
′
bcl11b中的一个“基因沙漠”区域,包含几个增强子。黑色箭头显示了最近一项研究
45
中导致t-all癌基因调节异常的易位断点。彩色箭头:t-all供体p1和p33中的sv断点。(c)tcl1a与14q32 inv的调节异常。较大的条形图显示与五个任意选择的t-all相比p1中的tcl1a调节异常。插
入的条形图显示等位基因特异性rna-seq分析,表明tcl1a调节异常仅发生在倒位(h2)单倍型上。(d)通过sctrip重建6q处的亚克隆聚集dna重排。(e)对聚集在6q处的sv进行单倍型解析分析,所有这些都属于单倍型h2。(f)结合聚集sv检测杂合缺失和loh保留,表明dna重排突变
41
。(loh,如方法中所述,以大量红点表示。参考杂合子snp密度正常(红色)但另外检测的杂合子snp密度降低(黑色)的区域,表示loh。)(g)通过批量长插入尺寸配对末端测序
75
至165x物理覆盖度验证6q处的亚克隆聚集重排爆发。sctrip推断的断点以虚线示出,sctrip推断的片段用字母a到l表示。彩色断点连接线描绘了基于配对末端映射的重排图(即缺失型、串联重复型和倒位型配对末端)。使用批量全外显子组和配对测序,这些断点的读段深度偏移很微小,因此,这种亚克隆复杂重排避开了先前在批量测序数据中进行的从头sv检测。
实施例
[0247]
本发明的某些方面和实施方案现在将通过实施例和参考本文阐述的描述、图和表的方式来说明。本发明的方法、应用和其他方面的此类实施例仅是代表性的,并且不应将本发明的范围限制为仅此类代表性实施例。
[0248]
方法和材料
[0249]
细胞系和培养。从atcc(crl-4000)购买htert rpe-1细胞,并检查支原体污染。bm510细胞是使用cast方案产生的,来源于rpe-1亲本系(如mardin et al.2015先前所述)。c7细胞从riches et al 2001获得。细胞系保存在补充有10%胎牛血清和抗生素(life technologies)的dmem-f12培养基中。道德声明。本研究中使用的方案得到了相关机构审查委员会和道德委员会的批准。t-all患者样本均获基尔大学伦理委员会批准,并从临床试验all-bfm 2000(p33;年龄:诊断14年)或aieop-bfm all 2009(p1;年龄:诊断12年)中获得。从这些患者获得书面知情同意书,实验符合赫尔辛基的wma宣言和卫生与公共服务部贝尔蒙特报告中规定的原则。体内动物实验由苏黎世州兽医局批准,符合动物研究的道德规范。
[0250]
rpe和t-all细胞的单细胞dna测序。rpe细胞和pdx衍生的t-all细胞使用先前建立的方案
28,66
培养。发明人将brdu(40μm;sigma,b5002)并入生长细胞中18-48小时,然后使用bd facsmelody细胞分类器将单个细胞核分类到96孔板中,并使用前面描述的strand-seq方案
21,67
生成链特异性dna测序文库。最近发现,所使用的brdu浓度对姐妹染色单体交换
24
没有可测量的影响,是衡量dna完整性和基因组不稳定性
24
的敏感指标。为了大规模生成文库,在biomek fx
p
液体处理机器人系统上实施strand-seq方案,该系统需要两天才能生成96个有条形码的单细胞库。在nextseq5000(mid模式,75bp配对末端方案)上对文库进行测序,去重(demultiplexed)并与grch38参考组件(bwa0.7.15)比对。如
21,67
所述,选择高质量文库(从经历完整一轮dna复制并掺入brdu的细胞中获得)。简而言之,在分析之前对显示非常低、不均匀覆盖度或“背景读段”过量从而产生嘈杂的单细胞数据的文库进行过滤。在一个典型的实验中,~80%的细胞产生高质量的库,反映了在恰好单个细胞周期中brdu并入。brdu掺入不完全的细胞或在brdu暴露下经历一个以上dna合成阶段的细胞在细胞分选过程中被识别,因此在链序列实验
21,67
中很少被测序,通常占测序细胞的不到10%。因此,这种“无法使用的文库”不会明显增加实验成本。
[0251]
杂合snp的染色体长度单倍型定相。本文所述的发明人的sv发现框架使用
strandphaser
22
对模板链进行定相。基本原理是,对于“wc染色体”(其中一个亲本同源物作为w模板链遗传,另一个同源物作为c模板链遗传的染色体),杂合snp可以立即被定向为染色体长度的单倍型(链特异性dna测序特有的特征)。为了最大限度地增加完整单倍型构建的信息性snp数量,发明人汇总了所有单细胞测序文库和内部100个细胞对照的读段,并通过使用freebayes
69
对1000基因组项目(1000gp)snp位点
68
进行重新基因分型来进行snp发现。qual》=10的所有杂合snp用于单倍型重构和单细胞单倍型标记(如下所述)。
[0252]
在单细胞中发现缺失、重复、倒位和反向重复。发明人开发了发明人方法的核心工作流程,以实现dup、del、inv和invdup sv的单细胞发现。工作流程的输入数据是来自供体样本的一组单细胞bam文件,与参考基因组比对。核心工作流程执行分箱的读段计数、覆盖度标准化、分段、链状态和姐妹染色单体交换(sce)检测,以及单倍型识别sv分类。下面提供了每个步骤的简要说明,有关更多详细信息,请参阅补充信息。
[0253]
分箱的读段计数。将每个细胞、染色体和链的读段分箱到100kb的窗口中。去除pcr重复、不正确配对和低映射质量(《10)的读段,以仅计数独特的高质量片段。
[0254]
覆盖度的标准化。执行标准化以调整系统读段深度波动。为了得出合适的比例因子,发明人对通过hgsvc项目获得的9个1000gp淋巴母细胞系产生的1,058个单细胞的strand-seq数据进行了分析(http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/hgsv_sv_discovery/worki ng/20151203_strand_seq/),并采用线性模型进行标准化,用于推断每个基因组箱的比例因子。
[0255]
群体中单细胞的联合片段化。片段化是通过联合处理跨样本的所有单细胞的链解析分箱的读段深度数据来执行的,用作具有平方误差假设
70
的多元输入信号。在给定多个允许的变化点k的情况下,采用动态规划算法确定具有最小的误差平方和的k变化点的离散位置。以这种方式联合分析所有细胞,即使是相对较小的sv(~200kb),只要在单细胞数据集中有足够的证据(例如在足够多的细胞中看到),也能够检测。为每个染色体分别选择断点的数量作为最小k,这样使用k+1断点只会产生边际改善,因为平方误差项的差异低于预先选择的阈值。
[0256]
单细胞中的链状态和sce检测。链特异性分箱的读段计数的解释依赖于对给定染色体(ww、cc或wc)模板链的潜在状态的了解。这些“基态”在每个单细胞的每个染色体的长度上保持不变,除非它们通过sces
21,71
改变。为了检测sce,发明人分别在每个细胞中执行了上述相同的片段化过程(对于分割而言,与跨所有细胞联合进行相反)。然后,发明人通过识别单细胞中的链状态的变化来推断假定的sce,这些变化与联合片段化所发现的断点不兼容(“补充信息”)。利用这些假定的sce,发明人随后将基态分配至每个片段(“补充信息”)。为了促进单倍型解析sv调用,发明人使用strandphaser
72
从基态cw区分具有基态wc的片段,其中单倍型1由watson(w)读段表示,单倍型2由crick(c)读段表示,反之亦然。
[0257]
单倍型识别sv分类。发明人开发了一种bayesian框架来计算每个sv诊断足迹的后验概率,并获得单倍型解析的sv基因型可能性。为此,发明人使用负二项式(nb)分布对链特异性读段计数进行建模,该分布捕获了大规模并行测序数据
54
的典型过度分散。nb分布有两个参数,p和r;参数p控制平均值和方差的关系,并在所有细胞中联合估计,而r与平均值成正比,因此因细胞而异,以反映每个单细胞文库的不同总读段计数。在估算p和r后,发明人计算了每个单细胞中每个片段的单倍型识别sv基因型可能性:对于给定的基态(参见上
文),每个sv诊断足迹转化为有助于基因组片段的w和c方向测序的预期拷贝数(表s1),这就产生了nb模型的可能性。发明人的模型将wc与cw基态区分开来(参见上文的strand-state和sce检测),这一事实使得发明人的模型隐含了全染色体单倍型识别——对单细胞中体细胞变异调用的任何先前方法都无法达到的关键特征。除此之外,发明人还将能通过重叠snp分配至单个单倍型的w或c读段的计数并入可能性计算中,并将此过程称为“单倍型标记”(因为它涉及由特异性单倍型“标记”的读段)。发明人使用多项式分布对标记的读段的各个计数进行建模。输出是对每个单细胞的有概率分数的预测的sv的矩阵。
[0258]
细胞群体中sv调用。发明人的工作流程估计每个sv的vaf水平,并使用它们定义每个sv的先验概率(经验bayes)。通过这种方式,该框架受益于在多于一个细胞中观察sv,其导致先验增加,从而使sv发现更可信。发明人的框架调整了细胞中一致可见的灵敏地调用亚克隆sv和准确识别sv之间的权衡。发明人将这种权衡参数化为“严格”和“宽松”sv调用方法(caller),其中“严格”调用方法优化了vaf≥5%的sv的精度,并且“宽松”调用方法靶向所有sv,包括仅存在于单细胞中的sv。除非另有说明,本研究中提出的sv调用是使用“严格”参数化生成的,以实现将假阳性sv最小化的调用集。发明人使用模拟,通过将del、dup和inv随机植入计算机模拟的单个细胞中,探索这些参数化的局限性。应用strand-seq
21
的典型覆盖度水平(每个细胞400,000个读段片段),发明人在每个模拟中分析200个单细胞。当vaf≥40%时,发明人观察到尺寸≥1mb的sv具有极好的再调用和精确度(图s5)。当发明人发现较低vaf的事件的再调用和准确率下降时,发明人能够将较小的sv和具有较低的vaf的sv恢复至单个细胞。
[0259]
易位的单细胞剖析。通过搜索表现出与这些片段起源的染色体不一致的链状态,同时与基因组的另一个片段(即其易位伙伴)一致(相关或反相关)的链状态的片段,发明人发现了单细胞中的易位(“补充信息”)。为了推断易位,发明人以同源解析的方式确定了每条染色体的链状态。如果链状态在单倍型中出现变化(因为该单倍型表现sv或sce),发明人使用多数链状态(即“基态”,参见上文)来进行易位推断。发明人通过生成统计具有等效链状态和不具有等效链状态的细胞的数量的列联表来检查模板链共分离(参见图3b)。发明人采用fisher精确检验来推断列联表中计数分布的概率,然后进行p值调整
73
。
[0260]
单细胞中断裂融合桥(bfb)循环的特征。为了推断和表征单细胞中的bfb循环,发明人首先采用了发明人的框架和宽松的参数化以推断在相同同源物/单倍型上两侧有delter事件的invdup。通过搜索一个单倍型上的invdup的两侧是另一个单倍型上的delter的结构(例如,invdup(h1)-delter(h2)事件,其中h1和h2表示不同的单倍型),发明人测试了由bfb循环产生的invdup-delter足迹是否可能偶然出现在单细胞中。没有检测到这样的结构并且invdup-delter足迹总是出现在相同单倍型上,与bfb循环形成一致。为了确保图s14中所示的发明人的基于单细胞的定量的高灵敏度,发明人对单细胞数据进行另外的人工检查以获得以下重排类别中的至少一种的证据:(i)invdup,(ii)导致在其他二倍体染色体上的拷贝数=1的delter。根据图1中定义的诊断足迹,检查这些细胞是否存在指示bfb的invdup-delter模式。
[0261]
基于单细胞的cnn-loh发现。对于cnn-loh检测,发明人的框架首先通过使用strandphaser
22
分析可用于样本的所有单细胞strand-seq文库,为每个样本组装共有单倍型。然后将每个单细胞在二倍体环境中与这些共有单倍型进行比较,以确定与cnn-loh足迹
相匹配的差异。为了检测克隆出现的cnn-loh事件,发明人使用1000gp
68
参考snp面板对每个样本中聚合的单细胞文库进行重新基因分型。然后将这些重新基因分型的(观察到的)snp与1000gp参考集进行比较,以识别显示cnn-loh的杂合snp显示中显著缺失的基因组区域。为此,发明人将1000gp参考变体的样本减少到单细胞数据中观察的snp数量,然后合并两个数据集(观察的和参考变体),按基因组位置对所有snp进行排序。发明人通过这些排序的snp执行滑动窗口搜索,一次移动一个snp,并通过计算比例r=观察的snp/参考snp来比较每个窗口中观察的snp和参考snp的数量。在杂合子二倍体区域中,预计r值为~1,而偏差表明cnn-loh。窗口尺寸(由窗口中snp的数量决定)定义为每500kb窗口的中值snp计数。发明人采用循环二进制分段(cbs)
74
来检测r中的变化,并基于r的平均值为每个片段分配状态。尺寸≥2mb且表现平均值r≤0.15的片段被报告为cnn-loh。
[0262]
批量基因组dna测序。使用dna血液微型试剂盒(qiagen,hilden,germany)提取基因组dna。使用covaris s2仪器(lgc genomics)将300ng高分子量基因组dna片段化至100
–
700bp(平均大小为300bp),并使用agencourt ampre xp(beckman coulter,brea,usa)进行纯化。使用nebnext ultra ii dna文库制备试剂盒(new england biolabs,ipswich,usa)进行dna文库制备。发明人使用15ng衔接子连接的dna,并用10个pcr循环进行扩增。在0.75%琼脂糖凝胶上选择dna大小,长度范围在400-500bp之间。使用quabit 2.0荧光计(thermo fisher scientific,waltham,usa)和2100生物分析仪平台(agilent technologies,santa clara,usa)进行文库定量和质量控制。wgs采用illumina hiseq4000(illumina,san diego,usa)平台,使用150bp配对末端读段。如前所述
75
,采用大插入尺寸(~5kb)的配对测序。使用delly2
31
在批量dna序列数据中进行sv检测。rpe-1wgs数据按32
×
覆盖度测序。
[0263]
批量rna-seq。使用rneasy minelute cleanup试剂盒(qiagen,hilden,germany)从rpe细胞中提取总rna。使用2100生物分析仪平台(agilent technologies,santa clara,usa)进行rna质量控制。利用beckman biomek fx自动液体处理系统(beckman coulter,brea,usa),使用truseq-ht-chemistry(illumina,san diego,usa)以200ng的起始材料进行文库制备。使用定制的6个碱基对条形码制备样本,以实现合并(pooling)。使用片段分析仪(advanced analytics technologies,ames,usa)进行文库定量和质量控制。rna-seq在illumina hiseq 2500平台(illumina,san diego,usa)上进行,使用50个碱基对的单个读段。对于t-all中的rna测序,使用trizol(invitrogen life technologies)提取总rna。然后用turbo dnase(thermo fisher scientific,darmstadt,germany)处理rna,并使用rna clean&concentrator-5(zymo research,freiburg,germany)纯化rna。发明人要求使用具有agilent rna6000纳米试剂盒的生物分析仪(agilent,santa clara,ca)测量的最小rin(rna完整性数)为7。使用ribo-zero rrnaremoval试剂盒(illumina,san diego,ca)去除细胞质核糖体rna,并使用truseq rna library prep(illumina,san diego,ca)从1μg rna制备文库。这些样本在illumina hiseq 2000泳道上以75bp单末端测序。使用star aligner
76
检测融合连接。
[0264]
定量实时pcr(qpcr)。根据制造商的说明(cat 74106,qiagen,hombrechtikon,switzerland),使用rneasy mini kit从pdx衍生的t-all样本中提取rna,并使用high capacity cdna reverse transcription kit(applied biosystems,foster city,usa)生
genomic.fna)一致。为了以后实现单倍型定相和单倍型解析sv分配,该框架对1000基因组项目(1000gp;阶段3)提供的snp进行重新基因分型以从单细胞输入数据中检测杂合位点。当使用该框架时,将提供带有这些1000gp snp位点的vcf文件作为输入。替代地,sctrip通道能够直接从单细胞数据调用snp,或者对给定样本使用外部生成的snp调用,例如基于批量wgs。此外,使用制表符分隔文件作为框架的输入,该文件在整个基因组的每个箱有标准化因子(见下文)。
[0273]
单细胞中的分箱的读段计数。首先,针对每一条链,将所有单细胞中的读段进行分箱。箱有固定的宽度(默认值:100kb),从位置0开始一直到染色体末端。根据映射的读段的起始位置将其分配给箱,并根据以下标准进行过滤:排除非主要和辅助比对;排除与qc故障标志比对;排除pcr重复;排除映射质量《=10的读段。在配对末端数据的情况下,仅使用每对的第一读段(基于bam标志0x40)来避免重复计数。默认情况下,删除覆盖度太小(每个箱的中位数为3或更少)的细胞。nb分布的参数p和r的确定方式与sv分类相同(参见下文相应部分)。在参数估计过程中,如果箱对所有细胞的平均覆盖度非常低(《0.1,其中覆盖度先前标准化为1),或者箱显示出细胞间的wc/(wc+cc+ww)分数(wc
frac
)高度异常,则箱从参数估计过程中排除。如果箱显示wc
frac
《0.05或wc
frac
》0.95,则认为箱是异常的,表明从未显示wc状态或始终显示wc状态(例如在着丝粒内或附近区域常见的情况)的箱。
[0274]
单细胞中的覆盖度标准化。该框架在sv调用之前追求读段覆盖度的标准化。为了估计合适的标准化参数,对人类基因组结构变异联合会(hgsvc)最近生成的strand-seq数据进行了分析,包括来自1000基因组项目(1000gp)的9个淋巴母细胞系(即样本na19238、na19239、na19240、hg00731、hg00732、hg00733、hg00512、hg00513和hg00514)。发明人使用从ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/hgsv_sv_discovery/working/20151203_strand_seq/获得的通过strand-seq测序的这些hgsvc样本中的1058个细胞,并将这些细胞置于上述相同的分箱方案中。对这个1000gp样本中的几个的分析显示,这些样本不携带≥200kb的任何生殖系拷贝数变异(cnv)。为了确定标准化的比例因子,对这些hgsvc strand-seq序列数据进行了聚合,并首次使用以下“排除标准”中的任何一个掩蔽(mask)区域:观察的平均覆盖度《50%,观察到的平均覆盖度》200%,或观察的标准偏差大于平均覆盖度。然后,使用剩余的箱,假设与平均hgsvc箱覆盖度呈线性关系,对测试样本中观察的平均分箱覆盖度进行建模,这解释了66%的方差,斜率为~0.6。该线性关系被用于推导每个箱的比例因子,随后应被用于本研究的所有细胞。
[0275]
还创建了表现出强烈测序/映射异常的区域的“黑名单”,以避免假阳性的体细胞变异调用。为了构建所述黑名单,从独立hgsvc样本中具有异常覆盖度的“掩蔽区域”开始(参见上一段)。然后,如果间隔显示出500kb或更小的距离,则逐步合并这些间隔(这避免了高度碎片化的黑名单的生成)。最后,通过从我们的黑名单中移除所有与hgsvc报告的尺寸大于100kb的生殖系倒位重叠的间隔,确保没有已知的多态倒位被意外掩蔽。产生的黑名单被用于以下所有分析,这些分析考虑了单细胞sv调用黑名单间隔之外的区域。
[0276]
单细胞的联合片段化。关于片段化,huber et al.提出的策略被应用于使用平方误差假设
35
对多变量输入执行片段化。因此,样本中所有单细胞的分箱的读段计数数据被同时用作输入,其基本原理是在多个细胞中重复出现的sv可以相互增强。给定多个允许的变化点k,动态规划算法以最小平方误差和(sse)找到k变化点的离散位置。通过动态规划,利
用关于一组k-1最佳变化点的知识计算k级的变化点。该算法使用成本矩阵来确定每个可能连续片段的成本(平方误差总和)。虽然在huber et al.最初的实施中,假设所有样本的变化方向相同,但我们调整了算法以分别计算每个细胞和链的成本矩阵。发明人另外修改了成本矩阵以惩罚尺寸低于200kb的片段,作为避免过度片段化的一种手段。片段化程序(马赛克片段)对每个染色体分别执行片段化,并输出产生的变化点,最多允许变化点的最大数量。通过评估与实际计数数据相比,根据分段常数函数的平方和误差(sse)增加变化点数量(k)的益处,选择合适的片段化参数。令ssek为将染色体分成k个片段的残差。然后选择最小的数字k,使sse
k-sse
k+1
低于用户设置的参数(本研究中使用的默认值:0.1)以调整染色体的变化点k的数量。
[0277]
单细胞中的链状态和sce检测。sv诊断特征的检测取决于单细胞中的相应片段是否遵循有丝分裂分离的ww、cc、wc或cw模式(表1)。在本发明的上下文中,将沿着染色体的w和c读段的基本基线分布称为“基态”(参见方法)。虽然基态通常在染色体长度上保持不变,但它可以通过姐妹染色单体交换(sce)来改变,这是与结构变异无关的重组的有丝分裂模式的基础。源于有丝分裂重组事件/sce的strand-seq数据中的变化点表示本发明方法能够校正的“噪声”源。幸运的是,sce在每个单细胞2中独立发生,并且与sv不同,sce不会克隆传递给子细胞(即,仅在发生sce的细胞中可检测到2)。因此,sce引起的变化点不太可能在样本
1,2
的》1个细胞中的相同位置再出现。本发明使用转换点重复作为区分sce和sv的关键标准。为了识别sce,如上所述,采用了相同的片段化策略,但对每个单细胞单独地而不是联合地进行片段化。为此,选择断点数k(见上文)的阈值被设置为0.5。发明人通过计算分数f
wc
=w/(w+c)将观察的状态分配至每个所得片段,以及如果f
wc
>0.8分配状态ww,如果f
wc
<0.2分配状态cc以及其他情况分配状态wc/cw。相邻片段的状态相互比较,如果状态不变,则中间的变化点被丢弃,而剩余的变化点随后被进一步视为推定的sce。注意,此处“wc/cw”被用于表示在该步骤中这两种状态之间没有区别,在随后的链定相步骤中区分这两种状态。
[0278]
一个重要的考虑因素是,在某些情况下,以这种方式检测的变化点可能对应于sv,而不是sce。采用以下策略来选择sce的高置信度列表:首先选择距离联合片段化期间识别的任何断点较远(》500kb)的变化点(参见上一段);这些变化点可能代表真正的sce。有了这组临时的候选sce,三个基态ww、cc、wc/cw中的每一个被认为是确定一个合理的“基态”。采用的假设是染色体起始处的给定状态和一组sce位置(其改变状态)唯一地确定染色体上每个片段的状态。为了评估染色体开始拾取的三个基态(ww、cc或wc/cw)中的哪一个,计算了不一致的长度,定义为观察的状态与预测的基态不同的基因组间隔的总长度。虽然极不可能,但在极少数情况下,sce变化点可能与sv断点重合。为了使本发明的方法能够恢复这种少见的sce,分析了联合片段化中至断点小于500kb的所有推定的sce。如果添加这些推定的sce中的一个减少不一致的长度20mb或更多,则本发明的方法分配这些sce状态。如此,本发明的方法能够避免错过的sce引起沿着染色体的较大部分错误地分配基态。请注意,添加最多一个这种另外的sce阻止掩蔽大多数真正的sv,sv有两个断点,而sce通常仅导致沿着染色体的w和c状态中的单个“改变”(转换点)。此外,应注意的是,由于sce从不与拷贝数改变相关联,因此对于许多sv种类(即del、dup、invdup和复杂重排),sce与sv混淆的可能性接近“零”,即使这些sv仅存在于单个细胞中。因此,在现实中,sce很少被错误地分配sv状态(我们的实验验证数据也证明了这一点)。
[0279]
使用单细胞测序数据进行染色体长度单倍型定相。为了促进单倍型识别sv调用,发明人使用strandphaser对所有可用染色体进行定相。在为样本构建全染色体单倍型时,区域被分配为w和c链如每个细胞为wc或cw。也就是说,读段被用于重叠杂合snp以确定单倍型h1是否由w读段表示,以及h2是否由c读段表示(这种情况我们称为wc),或者反之亦然(称为cw)(参见方法)。除了基态的精细表征外,strandphaser还将染色体水平单倍型输出为vcf文件,发明人随后在“单倍型标记”步骤中使用了该文件。这个框架的定相步骤要求每个染色体至少有几十个snp。为了确保足够的snp的可用性,发明人使用具有选项
“‑
@《1000gp-snps.vcf》
‑‑
only-use-input-alleles《input.bam》
‑‑
genotype-qualities”的freebayes,对之前在1000gp中识别的生殖系变体进行了重新基因分型。所有杂合snp保留qual》=10。替代地,本框架可以使用外部提供的snp。为了提高snp调用的可用覆盖度,我们进行了细胞分选实验,对每个样本中的100个细胞(称为“100细胞对照”)进行独立地分选,然后进行短读段全基因组测序,平均覆盖度达到1.9x。
[0280]
先前显示的映射至基因组窗口(或箱)的高通量测序读段的数量与负二项式(nb)分布
37
一致,其可以解释过度分散。发明人采用nb分布作为bayesian框架的基础。nb分布有两个参数:p和r,其根据观察的读段计数进行估计,如下所示。n值表示为样本中分析的单细胞的数量。假设从每个单细胞以固定的箱尺寸采样的读段的数量是nb随机变量。事实上,单细胞的覆盖度是不同的,从而导致每个细胞的nb参数不同。参数估计的关键不仅是单个单细胞的覆盖度,而且所有单细胞的总覆盖度都来自nb分布。这意味着所有单细胞应该具有相同的p,因此有n+1个自由参数需要估计(一个p参数和n个分散参数)。
[0281]
在nb分布中,平均值与方差的比值等于1-p。在所有单细胞上具有相同的p参数意味着所有单细胞的平均值与方差的比值是恒定的。因此,单细胞之间的分箱的读段计数的平均值和方差共享线性关系,其中连接单细胞的这些平均值-方差的点的直线通过原点坐标,斜率决定p参数。这种关系允许估计共享的p参数:对于每个单细胞,发明人计算整个基因组中固定尺寸的箱中观察的读段计数的经验平均值和方差。如果用(m1,s
21
),(m2,s
22
),...,和(mn,s
2n
)表示经验平均值-方差对的集合,则p参数估计如下:
[0282][0283]
在获得p后,通过将分布平均值设置为单细胞的每个箱的平均读段计数来估计每个单细胞j的分散参数rj。发明人采用修整平均值来估计发散参数(修整参数设置为0.05),以消除异常高或零读段计数的影响(例如,在低可映射性区域中看到的)。
[0284]
sv诊断足迹。每个sv诊断足迹(图1)可以转化为在w和c方向测序的预期拷贝数,这有助于所考虑的基因组片段。表1显示了每个sv类的这种关系,既适用于两个单倍型均由不同模板链表示的染色体(此处称为“wc/cw染色体”),也适用于两个单倍型均由相同模板链表示的染色体(“ww染色体”和“cc染色体”)。每个单倍型解析sv分别暗示在wc、cw、ww和cc染色体中一个特定的片段链模式。例如,如果染色体区域中单细胞的基态为ww,而该区域某个片段中的sv状态为“w链上所代表的父系单倍型的反向重复”,则观察到的片段链模式将是该给定单细胞中的wwc。相比之下,如果基态为wc(h1单倍型为w),sv状态为h1单倍型的缺失,则观察到的片段链模式为c(见表1)。这些期望在我们的bayesian模型中被形式化,我们
将在下面描述该模型。
[0285]
发明人利用bayesian模型(图1h)计算每个单细胞中每个片段的单倍型解析sv基因型可能性。发明人将v(将要推断的sv类型)建模为一对其中给出了正向上该段的拷贝数,并给出了反向上该段的拷贝数(即,当存在倒位时)。也就是说,该对(1,0)编码了单倍型的参考状态(一个正向拷贝和零个反向拷贝)。如图1h所示,单细胞j∈j中的每个片段k∈k和单倍型h∈h={h1,h2}都具有对于该sv状态的变量v,我们称之为v
j,k,h
。与基态t一起,每个sv状态v确定地导致在crick方向nc和watson方向nw观察到的相应“拷贝数”,如上一节sv诊断特征所述(另见表1)。以两种单倍型的crick和watson拷贝数之和为条件,假设相应的覆盖率xc和xw遵循负二项式分布
[0286][0287][0288]
对于每个单细胞j和片段k。这里,p是nb分布的估计公共p参数(见上文“估计负二项式参数”),并且和与估计参数rj(也见上文)、分段大小lk以及watson和crick段拷贝数(和)成比例,因此计算如下(对于d∈{w,c}):
[0289][0290]
在这个公式中,α是我们模型中的一个参数,表示“背景读段”的比例,它表示strand-seq数据中的噪声(例如,由于brdu未完全合并或移除的区域)
1,2
)。通过假设α=0.1将这些背景读段考虑在内,这反映了实践中观察到的大量此类背景读段的上限。注意,上述公式中的系数用于将分散参数缩放到拷贝数1(rj如上估计来反映拷贝数2的二倍体状态)。总之,片段中每个单倍型解析sv种类(v)和基态(t),定义了用于计算观察到的读段计数的nb可能性的watson和crick拷贝数(n)。通过该机制,获得了表1中所有诊断特征的可能性。
[0291]
结合单倍型特异性测序读段(“单倍型标记”)。sctrip的关键优势之一是能够利用通过链特异性测序获得的单倍型信息。在上一段描述的基础模型中,这种单倍型识别是通过区分wc和cw基态来实现的(也可参见“使用单细胞测序数据的染色体长度单倍型定相”)。本框架还能够利用由于与单倍型定相的snp重叠而未直接分配给单倍型的读段(即ww和cc区域中的那些)。这一特性可以进一步促进仅在少数甚至单细胞中看到的假定sv的验证和伪造。发明人利用使用strandphaser
36
生成的全染色体单倍型,以使用whatshap
38,39
的“单倍型标记”命令,通过单倍型标记读段,从而在每个单细胞库中产生一个“单倍型标记”的bam文件。然后,这些bam文件用于计算每个片段和每个单个细胞的watson/crick读段数量,这些读段数量可分别由单倍型h1/h2标记。产生的单倍型标记发读段计数作为随机变量和合并到bayesian模型中(见图1h)。发明人采用多项式分布来模拟给定(单倍型和链特异性)拷贝数nc和nw的这些标记读段计数的条件分布。更准确地说,我们为每个片段k和单个细胞j定义了多项式分布的参数和使得它们与相应的拷贝
数成比例:
[0292][0293]
其中d∈{w,c}和先前一样。这里,α同样是背景读段的比率(设置为α=0.1),并且被标准化为总和为1。给定读段的总数和(单倍型和链特异性)拷贝数nc和nw,标记的读段是多项式分布的:
[0294][0295]
采用bayesian模型进行sv调用。为了利用我们的bayesian模型进行sv调用,发明人定义了先验概率,并将其与每个单细胞和分段的基于模型的可能性相结合。发明人首先对原始概率进行正则化,在所有概率中添加小的常数(设置为10-6
),然后再进行正则化。这确保避免了非常小的值(或零),并与错误假设(即每个sv基因型在给定的小概率下都是可能的,无论数据表明什么)相对应。然后,使用了两种形式的先验值。首先,生物学知识是关于观察某些事件类型的合理性获得的。为此,将先验值定义为与每种sv类型的预先指定常数成比例,并按如下方式选择这些常数:ref=200,del/inv/dup=100,invdup=90,其他/复杂=1。虽然这种选择有些随意,但它鼓励sv调用过程来选择参考状态(ref)而不是规范sv(del/inv/dup/invdup)或不是更奇特的sv类(例如涉及一个单倍型的倒位和另一个单倍型的缺失(其他/复杂))——除非模型观察到足够的证据压倒这些先验值。因此,要求调用者收集更多被认为不可信的sv类型的证据。应用的第二类先验值分别作用于每个片段,并使用模型计算的所有细胞的原始可能性来计算所有sv类型的概率分布。也就是说,对于每个片段,将所有细胞中每种sv类型的可能性相加,并标准化为1,这对应于估计该片段的每种sv基因型的频率。这一过程背后的直觉是,我们需要鼓励sv调用者选择存在于许多细胞中的sv类型,而不是仅存在于少数细胞中的sv类型——除非基因型可能性的固有证据足以压倒这些先验值。在应用这些先验值之前,如果每个sv基因型的估计频率低于阈值,则该基因型的先验值被设置为零,该阈值被称为gtcutoff(严格调用集设置为0.05,宽松调用集设置为0)。实际上,这意味着如果所有细胞的可能性表明它以至少5%的预期频率存在于细胞群体中,则严格的参数化只考虑sv基因型。相比之下,宽松的调用集通过将其设置为零来禁用此临界值,因此很容易允许仅单细胞中存在sv基因型。最后,发明人使用产生的后验概率来计算对数优势比(sv基因型相比于参考状态的),并且如果对数优势比至少为4,则接受sv调用。丢弃具有》20%黑名单箱的片段中的sv调用。
[0296]
调用集后处理:过滤:开发了过滤程序,以仅与严格的参数化结合使用,其主要目标是为vaf大于5%的所有sv获得高置信度的sv调用集。该过滤程序去除仅1或2个细胞中出现的罕见倒位,因为罕见倒位可能偶尔对应于sce。此程序进一步删除显示特定偏差的sv调用,最重要的是,被偏差来主要发生在特定基态的背景下的sv调用。特别是,sv可以在所有四种基态(ww、cc、wc和cw;见表1)的情况下检测到。调用ww或cc染色体上的缺失或重复确实在概念上与先前开发的拷贝数分析方法有关;即,在ww或cc染色体上调用的sv将不会受益于sctrip基于链特异性读段深度增益或损耗调用这些sv的能力(图1,表1)。
[0297]
实现了以下硬过滤器,用于严格的参数化:
[0298]
(i)去除在少于3个细胞中看到的倒位。
[0299]
(ii)去除多个细胞中看到的缺失,如果这些缺失显示主要发生在ww和cc染色体
上,在wc或cw区域出现的少于三分之一(对数优势比≥50的缺失将不会被此硬过滤器去除)。如上所述,我们实施了该过滤器,因为在ww或cc基态中重复出现但在wc基态中不出现或很少出现的缺失的可信度较低(根据我们的经验)。
[0300]
(iii)去除多个细胞中出现的重复,如果这些重复显示主要发生在ww和cc染色体上,在wc或cw染色体中出现的少于三分之一(对数优势比≥50的重复将不会被此硬过滤器去除)。如上所述,我们实施了该过滤器,因为根据我们的经验,出现在ww或cc基态中但在wc基态中不出现或很少出现的重复的可信度较低。
[0301]
(iv)将基因组(文件:segdups_hg38_ucsctrack.bed.gz)中与ucsc注释的分片段的重复重叠的sv去除50%以上(我们发现此类sv调用的可信度较低)。
[0302]
合并:开发了合并程序,与严格的参数化结合使用,将相邻sv与类似的vaf(其中vaf≥0.1)转换为单个sv调用,以避免过度分割,并生成最终的高置信度sv位点列表。为此,发明人认为如果vaf
sv1
/vaf
sv2
≥0.75(对于vaf
sv2
》vaf
sv1
的情况)或vaf
sv2
/vaf
sv1
≥0.75(对于vaf
sv1
》vaf
sv2
的情况),则将相邻sv的vaf视为相似的,并将通过该相似性标准选择的所有紧邻sv分组。该程序合并的sv几乎总是与验证实验中的单个结构变异事件相对应。
[0303]
我们的单细胞sv发现框架的严格和宽松的参数化。如上所述,我们的框架能够在敏感地调用低vaf下出现的sv和准确识别细胞间一致出现的sv之间进行折衷。发明人将这种折衷参数化为“严格”和“宽松”的sv调用者,从而“严格”调用者优化了vaf≥5%的sv的精度,而“宽松”调用者的目标是所有sv,包括仅在单细胞中存在的sv。这些参数化在三个设置中有所不同:gtcutoff(请参阅“采用bayesian模型进行sv调用”),是否合并单倍型标记的读段计数(请参阅“结合单倍型特异性测序读段”),以及是否启用过滤(请参阅“调用集后处理”)。严格调用者使用gtcutoff=0.05,而宽松调用者使用gtcutoff=0。对于严格调用者,我们禁用了单倍体标记功能,而对宽松调用者启用了单倍体标记——理由是单倍体标记对于解决具有低vaf的假定sv最有价值。最后,我们对严格调用者使用了上一段中描述的过滤,而发明人对宽松调用者使用了未过滤的集。建议使用严格的调用者来可靠检测低至5%vaf的亚克隆sv。宽松的调用者应用于分析整个vaf谱直至单个细胞的sv。
[0304]
实施例2:sctrip未发现的rpe细胞的sv图谱
[0305]
为了使用sctrip研究单细胞sv图谱,发明人接下来从端粒酶永生化视网膜色素上皮(rpe)细胞生成了链特异性dna测序库。htert rpe细胞(rpe-1)通常用于研究基因组不稳定性的模式
20,27
–
29
,此外还使用了c7 rpe细胞,其表明被用作细胞转化的指标的锚定非依赖性生长
30
。rpe-1和c7细胞均来自同一匿名女性捐献者。发明人分别对rpe-1和c7的80个和154个单细胞进行测序,平均深度为387,000个映射的非重复片段(“方法”部分)。这相当于每个细胞只有0.01x的基因组覆盖率。
[0306]
发明者首先搜索del、dup、inv和invdup。在读段标准化后,在rpe-1中鉴定出54个sv,在c7细胞中鉴定出53个sv。22种sv仅存在于rpe-1中,21种sv仅存在于c7中,因此可能对应于样本特异性sv(即在体细胞或培养细胞中形成的sv,而不是对应于生殖系变异;以下简称为“体细胞sv”)。图2a中显示了两个代表性sv,包括rpe-1中的1.4兆碱基(mb)体细胞dup和c7中检测到的800kb体细胞del。尽管除一个del和dup事件外,所有del和dup事件都是rpe-1和c7所独有的,但inv和invdup事件,包括图2b显示的17p上的inv,在很大程度上在两者之间共享。这些变异映射到已知反向多态性的位点
23
。发明人还鉴定了染色体臂水平的
cna,包括c7中13q的缺失和rpe-1中大10q区域的重复。13q臂显示了对单体的1:0的链比诊断(图2c),而获得的10q区域显示了对三体区域的2:1和3:0的链比诊断(图2d)。
[0307]
实施例3:剖析单细胞中复杂的癌症相关易位
[0308]
为了评估sctrip检测sv类别更广泛多样性的能力,发明人将rpe-1细胞进行cast方案
28
:发明人沉默了有丝分裂纺锤体机制,以构建可能表现出基因组不稳定性的锚定独立系(bm510)。发明人对145个单个bm510细胞进行测序,在搜索del、dup、inv和invdup事件时检测到总共67个sv。此外,一些dna片段没有与它们起源的相应染色体分离,表明染色体间sv形成(图3a)。发明人通过sctrip搜索诊断性共分离足迹进行易位检测(图3b),并在bm510中识别出四种易位(图3b,c)。发明人还对rpe-1和c7进行易位检测,分别识别出一个易位(图3d)。
[0309]
rpe-1和bm510之间共享一个易位,其涉及上述获得的10q片段,该片段与x染色体单倍型发生不平衡易位(图3b)。发明人利用姐妹染色单体交换事件
21
的足迹对片段进行定向和排序,将10q增益置于xq的端粒末端,与已公开的rpe-1光谱核型
27
一致(图3c)。在bm510中,sctrip还发现了涉及15q和17p的平衡相互易位(图3b,c)。值得注意的是,在同一17p单倍型上还检测到新的体细胞倒位,其与相互易位共用一个断点(图3c)。由于这些sv共享一个断点,因此很可能两者共同出现,可能涉及复杂的重排过程。对该基因座的分析表明,倒位包含tp
53
基因,在易位时,tp
53
的5
′
外显子与ntrk3癌基因
32
的编码区融合(图3e)。这表明sctrip可以利用单细胞序列数据揭示融合基因。
[0310]
批量全基因组测序(wgs)和rna-seq(“方法”部分)分析显示发明人的框架具有极好的准确性和特异性。发明人验证了所有易位(100%),其中4/5通过wgs概括,剩余的der(x)t(x;10)事件通过现有核型数据
27
概括。在深度序列数据中未检测到额外的易位,表明sctrip具有极好的敏感性。wgs未能验证der(x)t(x;10)不平衡易位,因为chrx断点位于高度重复的端粒dna中(导致不明确的比对,妨碍读段对分析),而sctrip使用不受重复断点影响的有丝分裂共分离模式。发明人还观察到在der(x)t(x;10)事件的背景下,重复单倍型的表达增加,证实了发明人的单倍型定位。最后,发明人验证了17p处存在复杂重排,并发现了bm
510
独有的表达的ntrk基因融合转录本(图3d,e)。因此,sctrip通过单细胞测序以高精度和灵敏度实现了单倍型解析的易位发现,其包括检测大量wgs缺失的易位。
[0311]
实施例4:复杂dna重排过程的单细胞剖析
[0312]
癌症基因组经常含有通过复杂重排产生的聚集sv,这有助于加速癌症进化
33
。导致此类sv的一个过程是断裂融合桥(bfb)循环
34
–
39
。bfb是由末端染色体片段的丢失引起的,这导致新复制的姐妹染色单体融合。由此产生的双着丝粒染色体将导致染色体桥,通过dna断裂分解可以启动新的bfb
14
。因此,bfb以反向方向连续复制dna片段(即生成invdup),通常具有相同单倍型的末端染色体片段的相邻缺失(即末端缺失,此处称为“delter”)。通过分析“折叠反转”(读段对在反转方向上彼此靠近排列)
34
,可以从批量wgs推断出导致高vaf的bfb。由于高覆盖要求,无法在单细胞中系统地跟踪折叠反转。但发明人认为sctrip可以提供直接研究单细胞中bfb形成的机会。
[0313]
为了研究bfb,发明人首先转向c7,其中先前描述了折叠反转
28
。sctrip在154个测序细胞中的152个细胞中定位了10p臂上的聚集invdup(图4)。对10p的进一步分析显示,具有“逐步”invdup事件的扩增子与同一单倍型上相邻的delter,与bfb一致(图4a-c和图
s12)。剩下的两个缺乏invdup的细胞尤其显示出较大的delter,影响相同的10p片段(图4c)。通过聚合细胞间的序列读段,发明人确定了沿染色体10的8个可识别片段,其包括10p扩增子(包含6个拷贝数片段)及其相邻区域(10p末端区域和着丝粒近端区域)(图4b)。为了进一步表征10p处的遗传异质性,发明人推断了所有8个片段的细胞特异性拷贝数(图4d)。这表明至少有三组不同的细胞与10p拷贝数有关:(i)一大组呈现“中间”拷贝数,最高拷贝数片段(称为“主要克隆”)检测到100-130个拷贝。(ii)通过delter失去相应10p区的两个细胞,(iii)表现出极高的拷贝数(~440个拷贝)的单细胞,可能经历了额外的bfb循环(图4c)。
[0314]
c7中鉴定的其他sv提供了主要克隆中发生的重排的进一步了解:即,发明人检测到将重复的15q片段拼接到10p扩增子上的不平衡易位(图4c)。重复片段包围了15q端粒(图4c),这可能稳定了扩增子以终止bfb过程。进一步支持含有至少三组相对于10p结构的细胞的c7的是,两个含有延伸delter的细胞中没有不平衡易位,而易位区域在10p拷贝数过多的细胞中进一步扩增(图4c)。图4e显示了导致主要克隆的重排的时间序列的模型。这些数据强调了sctrip表征bfb循环的能力,对此先前通过单细胞测序直接测量是不可能的。
[0315]
实施例5:锚定非依赖性rpe细胞中丰富的bfb形成
[0316]
体细胞中bfb介导的sv形成的频率是未知的。由于sctrip可以系统地检测invdup和delter足迹,发明人搜索了所有已测序的rpe细胞(总共379个)(“方法”部分),并鉴定了另外15个显示bfb形成特征的细胞。其中,11个显示了“经典”bfb足迹——同一同源物上的invdup两侧有delter,同源物上没有其他sv(图4f)。其余四个实例在与bfb相关的sv相同的同系物上显示出额外的重排。发明人通过搜索其中一个单倍型上的invdup两侧为另一个单倍型上的delter的结构,来测试invdup-delter足迹是否碰巧重合。在379个细胞中,invdup-delter足迹总是出现在同一个单倍型上,这与众所周知的bfb模型
38
一致。15个invdup-delter事件中有11个发生在bm510中,影响8%(11/145)的测序细胞,4个发生在c7中,影响3%(4/154)的细胞。rpe-1细胞(0%;0/80)中未出现invdup-delter足迹,因此bfb仅出现在转化的、锚定独立生长的细胞中。invdup区域的拷贝数估计值范围为3到9,表明这些细胞中发生了多达三个bfb循环(图4f)。
[0317]
有趣的是,所有这15个invdup-delter足迹都是在分离细胞中检测到的单例事件(即,没有一个在多个细胞中共享),因此可能代表具有偶发形成和潜在持续的bfb循环的染色体。发明人推断,在单细胞中识别的sv可以作为当前活跃的突变过程的代理。利用sctrip,发明人系统地在rpe细胞系中寻找其他丰富的sv突变模式,其中发明人诱导了基因组不稳定性(bm510)。发明人定位了60条染色体,有证据表明有丝分裂错误导致大量(兆碱基级)缺失或重复。其中35/60(58%)影响整个同源臂,17/60(28%)涉及同源臂末端(末端缺失或增加),但不影响整个臂,7/60(12%)对应于整个同源非整倍体(单倍体或三倍体)。这些丰富sv类别的统一特征是,它们都可能由有丝分裂分离错误引起,并反映持续的染色体不稳定性
40
。
[0318]
进一步强调这一点的是,九个细胞显示出影响同一单倍型的多个聚集sv。这包括显示invdup-delter足迹和至少一个额外的sv的四个细胞。通过采用无限位点假设
37
,发明人推断了在这些情况下发生在相同单倍型上的sv的相对顺序,确定了额外sv的形成先于bfb形成的情况,以及额外sv的形成接替bfb形成的情况。该分析还揭示了单细胞表现出多
个重新定向和丢失的片段,所有这些片段都在同一个单倍型上,导致12个sv断点影响单个同源物。这种重排可能是由一次重排爆发(染色体碎裂)
41,42
引起的(图4g)。因此,sctrip能够系统地检测单细胞中从头sv的形成和sv突变过程,包括bfb和其他复杂的重排。
[0319]
实施例6:从41个单细胞构建pdx衍生t-all样本的核型
[0320]
为了评估sctrip的潜在诊断价值,发明人接下来分析了来自患者的白血病细胞。平衡sv和复杂sv在白血病中都很丰富,但在针对cna
26,41,43
的单细胞研究中大部分未被检测到。发明人表征了来自两名t细胞急性淋巴细胞白血病(t-all)患者的pdx衍生
44
的样本,以研究sctrip用于表征白血病样本的效用。发明者首先关注p33,这是一种pdx衍生的t-all复发,来自患有klinefelter综合征的青少年患者。发明人对41个单细胞进行测序,并使用这些数据以200kb的分辨率重建了主要克隆的单倍型解析核型(图5a)。虽然大多数染色体都是二倍体的,但发明人鉴定了典型的xxy核型(klinefelter综合征),并观察到了染色体7、8和9的三倍体。发明人进一步检测了cnn-loh的3个区域,其特征是在存在恒定读段深度和方向的情况下单倍型丢失。此外,发明人观察到6个焦点cna,其中5个影响先前报告为在t-all中发生遗传改变和/或“驱动”t-all
43,45
–
47
的基因,包括大小为300kb及以上的phf6、rpl2和ctcf的缺失,以及cdkn2a和cdkn2b的纯合缺失(图5a)。发明人还发现了一种t(5;14)(q35;q32)平衡易位(图5a)——一种已知以tlx3为靶点的t-all中的复发性重排,其致癌失调
48
。虽然很少有单细胞表现出核型多样性,但大多数细胞支持主要克隆的核型(图5b)。
[0321]
发明人试图通过诊断期间从原发性t-all获得的经典(细胞遗传学)核型来验证该核型——这是目前对t-all进行遗传学特征分析的临床标准。虽然这证实了染色体x、7、8和9的重复,但经典核型分析未能检测到所有的焦点cna,也未能捕获先前设计为“隐性”(即“核型分析无法检测到”)
49
的t(5;14)(q35;q32)易位。为了验证sctrip检测到的额外的sv,发明人接下来在诊断、缓解和复发
50
以及表达测量时通过批量捕获测序p33进行cna分析。这些实验证实了所有(6/6,100%)焦点cna,并证实tlx3失调支持t(5;14)(q35;q32)平衡易位的发生。通过sctrip推断的单倍型解析核型包括大小小至200kb的sv,定位了临床核型分析遗漏的“隐性”易位,并使用来自41个细胞的序列数据构建,达到只有约0.9
×
累积基因组覆盖率。
[0322]
实施例7:sctrip发现pdx衍生t-all中先前未识别的dna重排
[0323]
发明人接着转向从一名青少年女性患者(p1)获得的第二个t-all复发样本。发明人对79个p1的单细胞进行测序,发现了两个亚克隆,每个亚克隆至少有25个细胞(图5c)。发明人首先关注克隆sv,其包括14q32处新型2.6mb平衡倒位(图6a)。有趣的是,其中一个倒位断点落入受p33 t(5;14)(q35;q32)易位影响的完全相同的14q区域(图6b)。先前的研究表明,根据其精确的断点位置,t(5;14)易位可以通过将14q35处的增强子元件重新定位到这些癌基因
43,51
附近,从而靶向5q35处的tlx3和nkx2-5癌基因。
[0324]
观察到两名t-all患者均表现出影响同一区域的平衡sv,这激发了进一步的分析。这表明,发明人在p1中定位的新型14q32倒位将包含bcl11b的3
′
区域的增强子元件
48,51
并列到t细胞白血病/淋巴瘤1a(tcl1a)癌基因附近(图6a)。先前的研究报告了t细胞白血病/淋巴瘤以及t-all中不同的增强子并列重排,导致tcl1a过度表达
52,53
,因此发明人采用rna-seq来研究p1中的差异表达。这确实证实tcl1a是p1中高表达的基因(与五个任意选择的t-all相比,过度表达》160倍;p=1.8e22 wald test
54
,benjamini-hochberg校正;图6c,左
图)。发明人推断,如果tcl1a失调是由倒位引起的,那么tcl1a过度表达将仅限于重排的单倍型。利用sctrip的单倍型解析sv分配,发明人进行了等位基因特异性表达分析,这表明tcl1a过度表达确实只产生于携带倒位的单倍型(图6c,右图)。这些数据暗示了驱动癌基因表达的一种新的倒位。需要进一步的研究来评估这种倒位在其他t-all或t细胞恶性肿瘤中的复发情况,并研究涉及bcl11b增强子的癌基因失调sv的多样性。由于sctrip能够通过浅层测序进行平衡sv的可扩展发现,因此它将非常适合在更大的患者队伍中研究这些问题。
[0325]
发明人接下来分析了p1中的亚克隆sv,发现了一系列影响单个6q单倍型(vaf=0.32)的高度聚集的亚克隆重排。这些重排包括两个inv、一个invdup、一个dup和三个del,导致总共13个可检测断点,跨越近90mb的6q(图6d,e)。所有在6q处表现出sv的细胞都显示了13个完整断点的证据。此外,拷贝数分布仅在三种拷贝数状态
41
之间振荡,发明人观察到杂合性中的保留和丢失的岛状
41
(图6f)——一种重排模式,使人想起染色体碎裂
41,42
。为了证实这些数据,发明人进行了长(4.9kb)插入大小配对测序,批量到深度(165x)物理覆盖。虽然这种亚克隆复合物重排几乎看不出读段深度的改变,但深度配对测序证实了所有13个亚克隆sv断点,从而验证了亚克隆重排爆发与染色碎裂一致(图6g)。这些数据强调了sctrip揭示标准批量wgs
42
可能遗漏的亚克隆复杂sv的能力。
[0326]
讨论
[0327]
sctrip能够使用集成读段深度、链和单倍型定相的联合调用框架,在单细胞中系统地检测多种sv。它可以将亚克隆sv降至vaf《1%,并识别作用于单细胞的sv形成过程,解决sv检测方法
10,13,26,55,56
未满足的需求。以前研究不同sv类别的单细胞研究是通过在wga
10,17,57
之后仅对相对较少的选定细胞进行测序来实现的。虽然先前使用strand-seq进行sv检测的工作仅限于种系倒位
23
,但本文介绍的计算进展能够系统地发现cna、平衡和不平衡易位、倒位、反向重复和复杂sv形成过程的结果,包括bfb和染色体碎裂——全部在单细胞中。值得注意的是,sctrip能够进一步解析重复嵌入的sv(例如以端粒dna中显示断点的不平衡易位),这是一类大部分标准wgs无法访问的sv。此外,sctrip检测到的sv是单倍型解析的,这有助于减少误报,并允许整合等位基因特异性基因表达数据
57,58
。
[0328]
发明人展示了sctrip通过识别转化rpe细胞中多达8%的细胞中的bfb循环来测量sv形成过程的能力,表明通过bfb循环形成sv在这些细胞中明显丰富。尽管最初描述于~80年前
38
,但现在sctrip允许直接、无偏地测量单个体细胞中的bfb。bfb循环是在染色体臂水平和末端丢失/增加事件之后确定的最丰富的sv形成过程,所有这些都可能由染色体桥
40,59
引起。bfb循环发生在多种癌症
14
中,可导致其他突变过程,如染色体碎裂
37
,并与疾病预后相关
60
。根据基于杂交的单细胞分析
58
显示,在体细胞外,即体外受精后的卵裂期胚胎中,也有报道称存在bfb循环。据估计,在癌症基因组
25,26
中,20%的体细胞缺失和50%以上的体细胞sv都是由复杂的dna重排引起的。通过在单细胞中直接和可靠地测量这些重排过程,sctrip将有助于将来对复杂sv在克隆进化中的作用的研究。
[0329]
发明人的研究还通过调查来自患者的白血病细胞的平衡和不平衡sv、复杂sv和核型异质性,证明了疾病分类的潜在价值。发明人使用41个单细胞构建了分辨率为200kb的t-all样本的分子核型,仅相当于0.9x的基因组覆盖率。这揭示了亚显微cna和致癌dna重排,而在目前临床上使用的细胞遗传学方法中是不可见的。经典的细胞遗传学通常只适用于每个患者有限数量的中期扩散,通常无法捕获sctrip可获得的低水平核型异质性。在其中一
routes to metastases:tracerx renal.cell 173,581
–
594.e12(2018).
[0341]
8.sottoriva,a.et al.a big bang model of human colorectal tumor growth.nat.genet.47,209
–
216(2015).
[0342]
9.aparicio,s.&caldas,c.the implications of clonal genome evolution for cancer medicine.n.engl.j.med.368,842
–
851(2013).
[0343]
10.forsberg,l.a.,gisselsson,d.&dumanski,j.p.mosaicism in health and disease
ꢀ‑
clones picking up speed.nat.rev.genet.18,128
–
142(2017).
[0344]
11.stratton,m.r.exploring the genomes of cancer cells:progress and promise.science 331,1553
–
1558(2011).
[0345]
12.korbel,j.o.et al.paired-end mapping reveals extensive structural variation in the human genome.science 318,420
–
426(2007).
[0346]
13.layer,r.m.,chiang,c.,quinlan,a.r.&hall,i.m.lumpy:a probabilistic framework for structural variant discovery.genome biol.15,r84(2014).
[0347]
14.leibowitz,m.l.,zhang,c.-z.&pellman,d.chromothripsis:anew mechanism for rapid karyotype evolution.annu.rev.genet.49,183
–
211(2015).
[0348]
15.navin,n.e.cancer genomics:one cell at a time.genome biol.15,452(2014).
[0349]
16.zahn,h.et al.scalable whole-genome single-cell library preparation without preamplification.nat.methods 14,167
–
173(2017).
[0350]
17.gawad,c.,koh,w.&quake,s.r.single-cell genome sequencing:current state of the science.nat.rev.genet.17,175
–
188(2016).
[0351]
18.bakker,b.et al.single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies.genome biol.17,115(2016).
[0352]
19.voet,t.et al.single-cell paired-end genome sequencing reveals structural variation per cell cycle.nucleic acids res.41,6119
–
6138(2013).
[0353]
20.zhang,c.z.et al.chromothripsis from dna damage in micronuclei.nature 522,179
–
184(2015).
[0354]
21.falconer,e.et al.dna template strand sequencing of single-cells maps genomic rearrangements at high resolution.nat.methods 9,1107
–
1112(2012).
[0355]
22.porubsky,d.et al.dense and accurate whole-chromosome haplotyping of individual genomes.nat.commun.8,1293(2017).
[0356]
23.sanders,a.d.et al.characterizing polymorphic inversions in human genomes by single-cell sequencing.genome res.26,1575
–
1587(2016).
[0357]
24.van wietmarschen,n.&lansdorp,p.m.bromodeoxyuridine does not contribute to sister chromatid exchange events in normal or bloom syndrome cells.nucleic acids res.44,6787
–
6793(2016).
[0358]
25.yang,l.et al.diverse mechanisms of somatic structural variations in human cancer genomes.cell 153,919
–
929(2013).
[0359]
26.li,y.et al.patterns of structural variation in human cancer,
biorxiv.biorxiv 181339(2017).doi:10.1101/181339
[0360]
27.janssen,a.,van der burg,m.,szuhai,k.,kops,g.j.&medema,r.h.chromosome segregation errors as a cause of dna damage and structural chromosome aberrations.science 333,1895
–
1898(2011).
[0361]
28.mardin,b.r.et al.a cell-based model system links chromothripsis with hyperploidy.mol.syst.biol.11,828(2015).
[0362]
29.maciejowski,j.,li,y.,bosco,n.,campbell,p.j.&de lange,t.chromothripsis and kataegis induced by telomere crisis.cell 163,1641
–
1654(2015).
[0363]
30.riches,a.et al.neoplastic transformation and cytogenetic changes after gamma irradiation of human epithelial cells expressing telomerase.radiat.res.155,222
–
229(2001).
[0364]
31.rausch,t.et al.delly:structural variant discovery by integrated paired-end and split-read analysis.bioinformatics 28,i333
–
i339(2012).
[0365]
32.amatu,a.,sartore-bianchi,a.&siena,s.ntrk gene fusions as novel targets of cancer therapy across multiple tumour types.esmo open 1,e000023(2016).
[0366]
33.zhang,c.-z.,leibowitz,m.l.&pellman,d.chromothripsis and beyond:rapid genome evolution from complex chromosomal rearrangements.genes dev.27,2513
–
2530(2013).
[0367]
34.campbell,p.j.et al.the patterns and dynamics of genomic instability in metastatic pancreatic cancer.nature 467,1109
–
1113(2010).
[0368]
35.rode,a.,maass,k.k.,willmund,k.v.,lichter,p.&ernst,a.chromothripsis in cancer cells:an update.int.j.cancer 138,2322
–
2333(2016).
[0369]
36.selvarajah,s.et al.the breakage-fusion-bridge(bfb)cycle as a mechanism for generating genetic heterogeneity in osteosarcoma.chromosoma 115,459
–
467(2006).
[0370]
37.li,y.et al.constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia.nature 508,98
–
102(2014).
[0371]
38.mcclintock,b.the stability of broken ends of chromosomes in zea mays.genetics 26,234
–
282(1941).
[0372]
39.gisselsson,d.et al.chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity.proc.natl.acad.sci.u.s.a.97,5357
–
5362(2000).
[0373]
40.thompson,s.l.,bakhoum,s.f.&compton,d.a.mechanisms of chromosomal instability.curr.biol.20,r285
–
95(2010).
[0374]
41.stephens,p.j.et al.massive genomic rearrangement acquired in a single catastrophic event during cancer development.cell 144,27
–
40(2011).
[0375]
42.korbel,j.o.&campbell,p.j.criteria for inference of chromothripsis in cancer genomes.cell 152,1226
–
1236(2013).
[0376]
43.girardi,t.,vicente,c.,cools,j.&de keersmaecker,k.the genetics and molecular biology of t-all.blood 129,1113
–
1123(2017).
[0377]
44.richter-pecha
ń
ska,p.et al.pdx models recapitulate the genetic and epigenetic landscape of pediatric t-cell leukemia.embo mol.med.e9443(2018).
[0378]
45.liu,y.et al.the genomic landscape of pediatric and young adult t-lineage acute lymphoblastic leukemia.nat.genet.49,1211
–
1218(2017).
[0379]
46.wang,q.et al.mutations of phf6 are associated with mutations of notch1,jak1 and rearrangement of set-nup214 in t-cell acute lymphoblastic leukemia.haematologica 96,1808
–
1814(2011).
[0380]
47.rao,s.et al.inactivation of ribosomal protein l22 promotes transformation by induction of the stemness factor,lin28b.blood 120,3764
–
3773(2012).
[0381]
48.nagel,s.et al.activation of tlx3 and nkx2-5 in t(5;14)(q35;q32)t-cell acute lymphoblastic leukemia by remote 3
’‑
bcl11b enhancers and coregulation by pu.1 and hmga1.cancer res.67,1461
–
1471(2007).
[0382]
49.bernard,o.a.et al.a new recurrent and specific cryptic translocation,t(5;14)(q35;q32),is associated with expression of the hox11l2 gene in t acute lymphoblastic leukemia.leukemia 15,1495
–
1504(2001).
[0383]
50.kunz,j.b.et al.pediatric t-cell lymphoblastic leukemia evolves into relapse by clonal selection,acquisition of mutations and promoter hypomethylation.haematologica 100,1442
–
1450(2015).
[0384]
51.li,l.et al.a far downstream enhancer for murine bcl11b controls its t-cell specific expression.blood 122,902
–
911(2013).
[0385]
52.sugimoto,k.-j.et al.t-cell lymphoblastic leukemia/lymphoma with t(7;14)(p15;q32)[tcrγ-tcl1a translocation]:a case report and a review of the literature.int.j.clin.exp.pathol.7,2615
–
2623(2014).
[0386]
53.virgilio,l.et al.deregulated expression of tcl1 causes t cell leukemia in mice.proc.natl.acad.sci.u.s.a.95,3885
–
3889(1998).
[0387]
54.love,m.i.,huber,w.&anders,s.moderated estimation of fold change and dispersion for rna-seq data with deseq2.genome biol.15,550(2014).
[0388]
55.alkan,c.,coe,b.p.&eichler,e.e.genome structural variation discovery and genotyping.nat.rev.genet.12,363
–
376(2011).
[0389]
56.campbell,i.m.,shaw,c.a.,stankiewicz,p.&lupski,j.r.somatic mosaicism:implications for disease and transmission genetics.trends genet.31,382
–
392(2015).
[0390]
57.dou,y.,gold,h.d.,luquette,l.j.&park,p.j.detecting somatic mutations in normal cells.trends genet.34,545
–
557(2018).
[0391]
58.voet,t.et al.breakage-fusion-bridge cycles leading to inv dup del occur in human cleavage stage embryos.hum.mutat.32,783
–
793(2011).
discovery rate.nucleic acids res.40,e69(2012).
[0408]
75.rausch,t.et al.genome sequencing of pediatric medulloblastoma links catastrophic dna rearrangements with tp53 mutations.cell 148,59
–
71(2012).
[0409]
76.dobin,a.et al.star:ultrafast universal rna-seq aligner.bioinformatics 29,15
–
21(2013).
[0410]
77.fan,j.et al.linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell rna-seq data.genome res.28,1217
–
1227(2018).
[0411]
78.lapunzina,p.&monk,d.the consequences of uniparental disomy and copy number neutral loss-of-heterozygosity during human development and cancer.biol.cell 103,303
–
317(2011).
[0412]
79.vogelstein,b.et al.cancer genome landscapes.science 339,1546
–
1558(2013)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1