基于Python语言的基因变异检测方法及系统
本发明属于基因检测技术领域,具体涉及一种基于python语言的基因变异检测方法及系统。
背景技术:
基因组序列包含数亿计碱基对,生物多样性不仅反映在表型上的多样,更包含了碱基对排列顺序的不同。物种表型的变化与分子序列的特征具有密切关系。随着测序技术的进步,在数小时内可测定一个物种的基因组序列,但分析不同物种基因组序列的变异特征成为揭示物种表型与遗传关系的重要内容,目前已有多种序列比对方法,例如blast,exonerate,genewise,blat,fasta等,不同方法其侧重点各不相同。
基于blast的序列比对方法能够快速有效的分析query序列和target序列相似的部分,根据打分矩阵自动筛选出得分大于阈值的区段,但是blast序列比对的结果包含了所有比对信息,这部分结果中存在着较大的冗余,需要花费较长的时间确定和筛选有效的信息。
基于exonerate的序列比对能够较为准确的得到query序列与target序列相似的部分,并且能够预测出较为准确的基因结构特征,但是exonerate序列比对在target序列较长时,序列比对速度呈指数倍下降,直接利用全基因组序列进行比对,时间周期太长,没有现实意义。
技术实现要素:
本发明的目的之一在于提供一种基于python语言的基因变异检测方法,该方法能高效准确的检测基因变异。
为实现上述目的,本发明的技术方案为:一种基于python语言的基因变异检测方法,包括以下步骤:
s1:确定比对所需的query蛋白序列以及所要比对的多个物种的基因组序列信息,并根据所述基因组序列信息构建序列比对库;
s2:blast比对分析query蛋白序列与一个物种的序列比对库,根据比对分析结果将每个query蛋白序列分别比对到序列比对库中的对应目标基因组相邻区域进行整合,得到每个query蛋白序列在序列比对库中的基因组中的比对区域信息;
s3:创建并初始化列表与集合,依次比对query蛋白序列中的每个query蛋白与集合,当集合中不存在当前query蛋白时,将当前query蛋白写入集合中,并将当前query蛋白对应比对得分记为第一比对得分与比对区域信息存入列表中,如果集合中存在当前query蛋白,则计算正在比对的当前query蛋白的第二比对得分,在当前query蛋白已存入列表中的第一比对得分小于第二比对得分时,去除列表中已有的当前query蛋白的所有信息,将正在比对的当前query蛋白的第二比对得分与比对区域信息写入列表,反之,则不操作,直到列表写入每个query蛋白的最佳比对结果信息;
s4:比对经步骤s3后得到的列表与query蛋白序列,去除query蛋白序列中没有比对到的query蛋白,得到更新query蛋白序列;
s5:将更新的query蛋白序列与下一个物种的序列比对库比对,并循环步骤s2-s4,直到获得query蛋白序列的每个物种的最佳比对结果信息;
s6:设置抽取长度基数,将所述每个物种的最佳比对结果信息及其对应query蛋白序列分别通过exonerate,根据exonerate运算结果抽取query蛋白序列在不同物种的每个外显子的数目信息和变异情况并记录。
进一步地,所述比对区域信息包括比对区域的起始位置、结束位置,比对率和比对区域中的相似度信息。
进一步地,所述第一比对得分为已存入集合的当前query蛋白的比对率与相似度的乘积,所述第二比对得分正在比对的当前query蛋白的比对率与相似度的乘积。
进一步地,所述步骤s2中的所述blast比对分析query蛋白序列与一个物种的序列比对库的步骤具体包括:
使用parallel计算是否支持多线程,如果支持,则分割分析query蛋白序列与一个物种的序列比对库的数据,进行多线程blast比对分析;
否则,单线程blast比对分析query蛋白序列与一个物种的序列比对库。
进一步地,所述步骤s6中设置抽取长度基数为50000bp。
本发明的目的之二在于提供一种基于python语言的基因变异检测系统,该系统用于检测基因比对中的变异信息。
为实现上述目的,本发明的技术方案为:一种基于python语言的基因变异检测系统,包括:
数据存储模块,用于存储比对所需的query蛋白序列以及所要比对的多个物种的基因组序列信息构建成的序列比对库;
blast比对模块,与所述数据存储模块相连,用于通过blast分别比对分析query蛋白序列与每个物种的序列比对库,根据比对分析结果将每个query蛋白序列分别比对到序列比对库中的对应目标基因组相邻区域进行整合,得到每个query蛋白序列在各物种序列比对库中的基因组中的比对区域信息;
优化模块,与所述blast比对模块相连,设置有初始化的列表与集合,用于依次比对query蛋白序列中的每个query蛋白与集合,当集合中不存在当前query蛋白时,将当前query蛋白写入集合中,并将当前query蛋白对应比对得分记为第一比对得分与比对区域信息存入列表中,如果集合中存在当前query蛋白,则计算正在比对的当前query蛋白的第二比对得分,在当前query蛋白已存入列表中的第一比对得分小于第二比对得分时,去除列表中已有的当前query蛋白的所有信息,将正在比对的当前query蛋白的第二比对得分与比对区域信息写入列表,反之,则不操作,直到列表写入每个query蛋白的最佳比对结果信息,并用于去除query蛋白序列中与最终获得的列表没有比对上的序列,获得更新query蛋白序列;
exonerate比对模块,与所述优化模块相连,并与所述数据存储模块相连,用于设置抽取长度基数,将最佳比对结果信息及其对应query蛋白序列分别通过exonerate,根据exonerate运算结果抽取query蛋白序列在不同物种的每个外显子的数目信息和变异情况并记录。
进一步地,blast比对模块获得的所述比对区域信息包括比对区域的起始位置、结束位置,比对率和比对区域中的相似度信息。
进一步地,所述优化模块的所述第一比对得分为已存入集合的当前query蛋白的比对率与相似度的乘积,所述第二比对得分正在比对的当前query蛋白的比对率与相似度的乘积。
进一步地,所述blast比对模块还设置有线程单元,用于计算所述blast比对模块是否支持多线程,如果支持,则分割分析query蛋白序列与一个物种的序列比对库的数据,进行多线程blast比对分析
进一步地,所述exonerate比对模块中设置抽取长度基数为50000bp。
与现有技术相比,本发明具有如下优点:
本发明提供一种基于python语言的基因变异检测方法及系统,本方法针对目前基因比对基因组过程中,其比对信息冗余大,比对速率慢的缺陷的问题,提出一种基因变异的检测方法,能缩短比对时间,提高有效比对速率,同时能够较为准确的整合比对结果,将基因比对和变异的信息整合到结果中,以表的形式真实可靠的呈现,从而能轻松挑选在某些物种中特异性变异的基因,为了解基因功能和后续的功能验证带来极大的便利。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作一简单地介绍。显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。
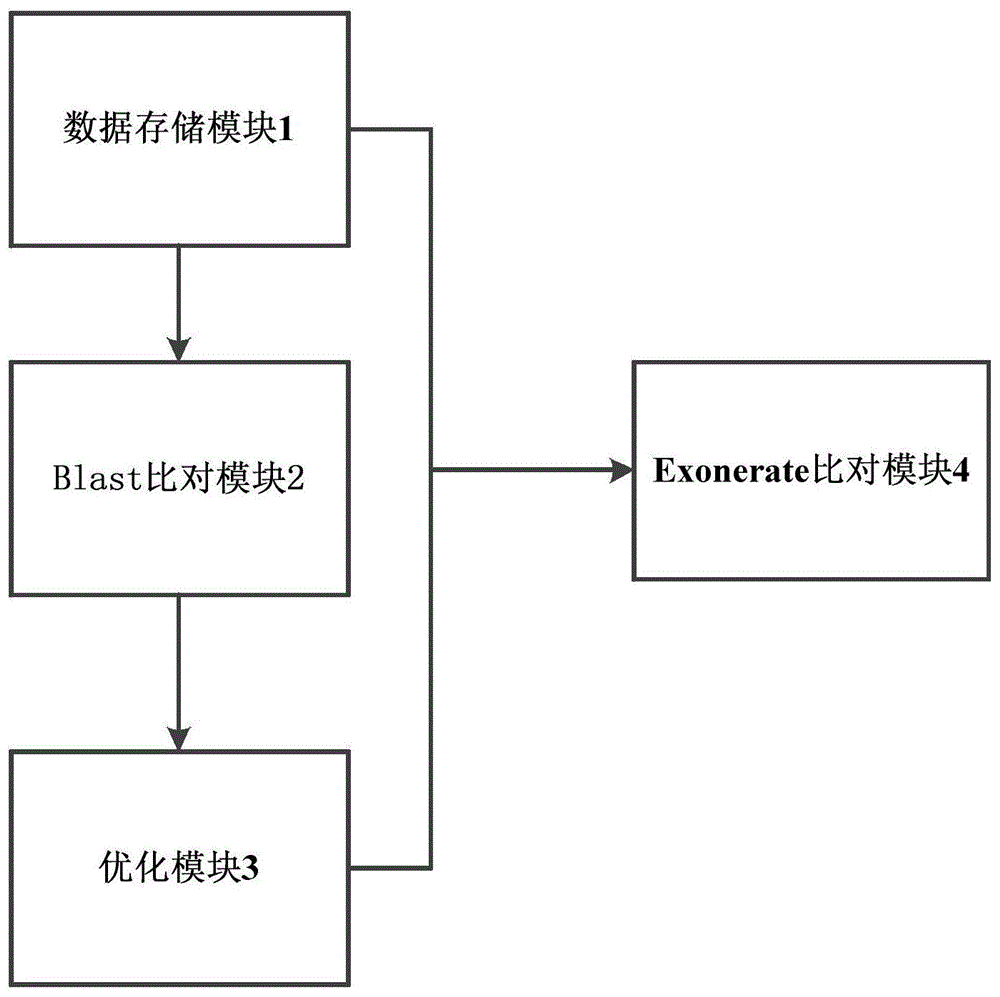
图1为本发明一种基于python语言的基因变异检测系统的一实施例结构示意图;
图2为本发明一种基于python语言的基因变异检测方法的一实施例流程图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
所举实施例是为了更好地对本发明进行说明,但并不是本发明的内容仅局限于所举实施例。所以熟悉本领域的技术人员根据上述发明内容对实施方案进行非本质的改进和调整,仍属于本发明的保护范围。
实施例1
参考图1,为本发明一种基于python语言的基因变异检测系统的结构示意图,具体地,该系统包括:
数据存储模块1,用于存储比对所需的query蛋白序列以及所要比对的多个物种的基因组序列信息构建成的序列比对库;
blast比对模块2,与数据存储模块1相连,用于通过blast分别比对分析query蛋白序列与每个物种的序列比对库,根据比对分析结果将每个query蛋白序列分别比对到序列比对库中的对应目标基因组相邻区域进行整合,得到每个query蛋白序列在各物种序列比对库中的基因组中的比对区域信息;
进一步地,blast比对模块2还设置有第一线程单元,用于计算blast比对模块是否支持多线程,如果支持,则分割分析query蛋白序列与一个物种的序列比对库的数据,进行多线程blast比对分析;
本实施例中的blast比对模块2获得的比对区域信息包括比对区域的起始位置、结束位置,比对率和比对区域中的相似度信息。
优化模块3,与blast比对模块2相连,设置有初始化的列表与集合,用于依次比对query蛋白序列中的每个query蛋白与集合,当集合中不存在当前query蛋白时,将当前query蛋白写入集合中,并将当前query蛋白对应比对得分记为第一比对得分与比对区域信息存入列表中,如果集合中存在当前query蛋白,则计算正在比对的当前query蛋白的第二比对得分,在当前query蛋白已存入列表中的第一比对得分小于第二比对得分时,去除列表中已有的当前query蛋白的所有信息,将正在比对的当前query蛋白的第二比对得分与比对区域信息写入列表,反之,则不操作,直到列表写入每个query蛋白的最佳比对结果信息;
具体地,优化模块3的第一比对得分、第二比对得分为各自的query蛋白的比对率与相似度的乘积,如第一比对得分为已存入集合的当前query蛋白的比对率与相似度的乘积,第二比对得分为当前query蛋白的比对率与相似度的乘积。
exonerate比对模块4,与优化模块3相连,并与数据存储模块1相连,用于设置抽取长度基数,将最佳比对结果信息及其对应query蛋白序列分别通过exonerate,根据exonerate运算结果抽取query蛋白序列在不同物种的每个外显子的数目信息和变异情况并记录。
优选地,exonerate比对模块中设置抽取长度基数为50000bp。
实施例2
基于实施例1的系统,本实施例中提供一种基于python语言的基因变异检测方法,参考图2,方法包括以下步骤:
s1:确定比对所需的query蛋白序列以及所要比对的多个物种的基因组序列信息,并根据基因组序列信息构建序列比对库;
本实施例中最好选用完整且错误较少的高质量query蛋白序列;
s2:blast比对分析query蛋白序列与一个物种的序列比对库,根据比对分析结果将每个query蛋白序列分别比对到序列比对库中的对应目标基因组相邻区域进行整合,得到每个query蛋白序列在序列比对库中的基因组中的比对区域信息;
本实施例中,首先分析运行环境,确定是否能够进行多线程运行程序,具体地,使用parallel进行并行计算,或者能够调用linux服务器的计算节点进行多线程计算,检查是否支持多线程,如果支持,则分割分析query蛋白序列与一个物种的序列比对库的数据,进行多线程blast比对分析;否则,单线程blast比对分析query蛋白序列与一个物种的序列比对库;
然后使用solar对于一个基因比对到目标基因组相邻区域进行整合,整合过程中目标基因组第一比对区域相间隔不超过10000bp,最终得到该蛋白在基因组中的比对区域,比对区域信息包括比对区域的起始位置、结束位置,比对率和比对区域中的相似度信息;其中,比对率和相似度来源于solar整合的结果,比对率定义为蛋白的比对区域与自身长度的比值,如果比对区域覆盖了全长比对率即为1;相似度定义为比对区域中所有一致性位点占比对区域总位点的比例,将每个基因在目标基因组的比对区域进行保存,用于后续抽取该区域的核苷酸序列信息;
s3:创建并初始化列表与集合,依次比对query蛋白序列中的每个query蛋白与集合,当集合中不存在当前query蛋白时,将当前query蛋白写入集合中,并将当前query蛋白对应比对得分记为第一比对得分与比对区域信息存入列表中,且将存入列表中的当前query蛋白的比对得分设为第一比对得分,如果集合中存在当前query蛋白,则计算正在比对的当前query蛋白的第二比对得分,在当前query蛋白已存入列表中的第一比对得分小于第二比对得分时,去除列表中已有的当前query蛋白的所有信息,将正在比对的当前query蛋白的第二比对得分与比对区域信息写入列表,反之,则不操作,直到列表写入每个query蛋白的最佳比对结果信息;
在经过步骤s2中的整合后,仍然存在较多的blast比对冗余信息,本步骤中用于挑选和每条最佳蛋白比对得分最高的信息,对冗余信息进行进一步去除,本步骤中具体操作为:具体地,在python环境中创建一个列表和集合,先初始化该列表与集合,列表与集合存在n行,并定义比对得分为比对率与相似度的积,按照步骤s2中的blast比对结果的顺序,从第1到第n行运用以下设计的算法依次将query蛋白序列中的每个query蛋白与集合进行比对,如果不存在当前query蛋白,则将该query蛋白写入集合,并将当前query蛋白对应比对得分记为第一比对得分与当前query蛋白比对信息(即比对区域信息,solar程序整合后的该基因在基因组中的比对结果)一同存入列表中,(且将存入列表中的当前query蛋白的比对得分设为第一比对得分,);如果在集合第n行与query蛋白序列进行比对时,集合中存在比对的当前query蛋白,则证明列表中已写入了当前query蛋白对应的比对区域,也即是存在一个query蛋白拥有两个比对信息,这两个比对信息为存在的blast比对冗余信息,此时,计算正在比对的当前query蛋白(集合中第n行的query蛋白)的第二比对得分,判断当前query蛋白已存入列表中的第一比对得分是否小于第二比对得分时,如果是,去除列表中已有的当前query蛋白的所有信息,将正在比对的当前query蛋白的第二比对得分与比对区域信息写入列表,同时更新第一比对得分为第二比对得分,如果不是,则跳过该循环,直到列表写入每个query蛋白的最佳比对结果信息,通过该算法可以在线性时间内得到每个基因的最佳比对结果信息;
进一步地,第一比对得分、第二比对得分为各自的query蛋白的比对率与相似度的乘积,如第一比对得分为已存入集合的当前query蛋白的比对率与相似度的乘积,第二比对得分为当前query蛋白的比对率与相似度的乘积。
s4:比对经步骤s3后得到的列表与query蛋白序列,去除query蛋白序列中没有比对到的序列,得到更新query蛋白序列;
本实施例中,通过步骤s3获得的列表为去除query蛋白序列因blast比对得到的冗余信息,本步骤中,将query蛋白序列中的冗余信息对应去除,具体地,根据步骤s3中的获得的当前物种的最佳比对结果信息列表(即列表),与query蛋白序列进行比对,去除query蛋白序列中与列表相比没有比对信息的序列,把剩余序列作为更新后的query蛋白序列,并把最佳比对结果文件保存在一个新的列表中。
s5:将更新的query蛋白序列与下一个物种的序列比对库比对,并循环步骤s2-s4,直到获得query蛋白序列的每个物种的最佳比对结果信息;
本实施例中在每次经过一个物种与query蛋白序列的比对后,均经过s3-s4的去除冗余,获得更新query蛋白序列,再将更新query蛋白序列与下一个物种进行比对,直到获得query蛋白序列的每个物种的最佳比对结果信息;
s6:设置抽取长度基数,将每个物种的最佳比对结果信息及其对应query蛋白序列分别通过exonerate,根据exonerate运算结果抽取query蛋白序列在不同物种的每个外显子的数目信息和变异情况并记录。
通过步骤s1-s5去冗余之后的solar整合blast比对结果信息,得到每个query蛋白序列在目标基因组的最佳比对区域信息,由于blast比对中有遗漏12bp长度的比对信息(blast算法默认设置比对区域大于12bp),从而没有被solar程序整合,本步骤中以50000bp为抽取长度基数,其中,比对区域增加长度=(1-比对率)*比对基数,此步骤能够动态科学的增加比对区域的长度,根据比对率线性增加了目标基因组的比对长度,既不太影响运算速度,又能够最大的保留有效信息;
然后将增加对于区域长度后的最佳比对区域信息输入exonerate同时也抽取相应的query蛋白序列根据本程序中的函数抽取该比对蛋白,将每个物种的最佳比对结果信息及其对应query蛋白序列写入运行中,再根据步骤s2中的方法分析当前环境是否能够并行计算,如果能,进行多线程exonerate分析,否则单线程分析;随后得到该物种的所有基因基于exonerate软件运行的预测结果,并根据exonerate结果文件格式设计相应算法抽取每个外显子的数目信息和变异情况,根据结果汇总当前结果中外显子的数目,所有外显子的序列插入变异、缺失变异和移码突变信息,并将信息写入当前物种的变异汇总文件中,然后对下一个物种进行以上循环操作,知道所有物种的exonerate分析结果得到,最后根据所有物种变异信息,汇总每个基因在所有物种中的外显子数目、插入变异数目、缺失变异数目和移码突变信息,将所有信息列入一个总表之中,完成基因变异的比较。
本方法通过调用solar程序对blast比对结果进行整合,将一个基因比对到基因组相近位置区域进行合并,然后对结果信息的冗余部分进行过滤,之后创建满足比较不同物种不同基因的变异情况的步骤,同时本方法支持多线程,在运算环境允许的情况下,支持多个线程的并行计算,大大提高了迅速按效率。
上面结合附图对本发明的实施例进行了描述,但是本发明并不局限于上述的具体实施方式,上述的具体实施方式仅仅是示意性的,而不是限制性的,本领域的普通技术人员在本发明的启示下,在不脱离本发明宗旨和权利要求所保护的范围情况下,还可做出很多形式,这些均属于本发明的保护之内。
- 还没有人留言评论。精彩留言会获得点赞!