一种基于低深度测序检测SNP标记位点的方法及装置
本发明涉及遗传学领域,尤其涉及一种基于低深度测序检测snp标记位点的方法及装置。
背景技术:
单核苷酸多态性(singlenucleotidepolymorphisms,snp)是目前最主流的遗传标记,在基因组中数量众多,分布广泛,遗传稳定性好。snp在人类和动植物研究中被广泛用于各类性状遗传机制的解析、选择进化研究和基因组预测等研究方向。
不同的研究内容对遗传标记的数量需求有所不同,其中需要使用全基因组高密度标记进行研究的内容主要包括全基因组关联分析和动植物基因组选择分析。在全基因组关联分析中,使用更高密度的全基因组遗传标记可以更准确地鉴定到目标表型真正的致因突变;而近年来动植物遗传育种中新兴的基因组选择(genomicselection,gs)技术,其利用的也是覆盖整个基因组的高密度snp,以此构建亲缘关系系数矩阵来计算个体的基因组估计育种值并选种。值得一提的是,基因组选择属于应用研究,利用基因组选择进行选种育种的样本规模正逐年大幅度递增,实际生产中其对标记的准确性、时效性和价格三个因素非常敏感,简言之,基因组选择技术对如何经济高效地获取全基因组snp分型数据提出了更高的要求。
目前的全基因组snp分型方法主要可分为商业化snp芯片和基因组测序两大类。商业化snp芯片因其标准化程度高,准确性好,操作简便而成为早期全基因组分型的主流方法。但随着研究的扩展,其不足也逐渐显现。例如,芯片所含snp标记数目多为几万至几十万,难以满足所有类型的研究需求;一种snp芯片也只能检测特定的突变位点,拓展性较差;商业化芯片在位点设计时使用特定的部分主流品种,这会造成部分标记位点在特定群体中失效;此外,随着测序技术的不断发展,芯片分型的成本优势也已逐渐消失。另一方面,尽管全基因组测序成本在不断下降,但距离大规模群体育种应用仍有不小的距离,这衍生出来众多靶向测序的替代方法,以简化基因组测序为例,这类方法通过富集并测序基因组中很小比例的片段达到降低成本的目的。这类方法相比芯片技术在标记密度和成本优化上已有很大的进步,但靶向测序并未真正实现覆盖全基因组,且分析流程需要较高的生物信息基础,因此在育种实践的全基因组分型技术中并未实现质的突破。
为了实现更高密度的基因分型,现阶段采取的主要策略是基因型填充,例如利用高密度芯片填充低密度芯片,利用高深度测序数据填充芯片数据等。但是这些方法极度依赖高质量的参考单倍型数据集(referencepanel),高质量不仅意味着该数据集群体规模大、自身分型结果具有高可信度,还要求该数据集与要填充的群体具有较近的遗传关系。目前畜禽物种中,大部分研究都是依靠小样本高深度的测序数据来构建参考单倍型数据库,已有大量的研究报道,这种panel的质量并不能保证高准确度的填充,这意味着标记数量在百万级别时,也将存在数万甚至数十万个错误的分型结果,并且该策略的计算复杂度较高,时效性较差,这依然不利于育种实践。
技术实现要素:
为了解决现有技术存在的问题,本发明提供一种基于低深度测序检测snp标记位点的方法及装置。本发明利用待测样本的低深度测序数据,基于参考单倍型数据库进行基因分型不仅可以在较大程度上缩短snp位点分型的时间,并且具有极高的准确率。
第一方面,本发明提供一种基于低深度测序检测snp标记位点的方法,包括:
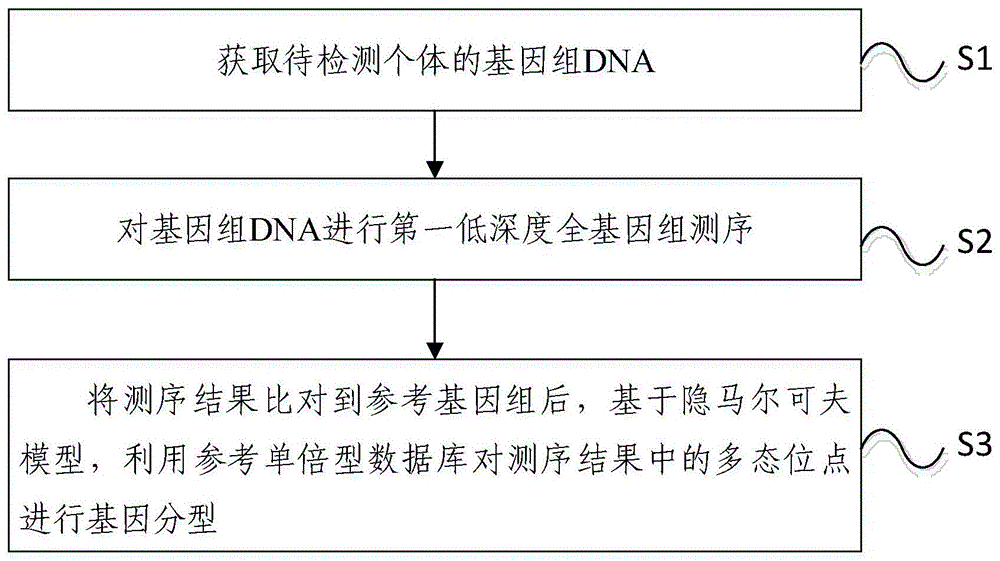
获取待检测个体的基因组dna;
对所述基因组dna进行第一低深度全基因组测序,将测序结果比对到参考基因组后进行基因分型;
所述基因分型为基于隐马尔可夫模型,利用参考单倍型数据库对测序结果中的多态位点进行基因分型;
所述参考单倍型数据库包括由第二低深度全基因组测序得到的,所述待检测个体归属的育种群体的单倍型信息。
进一步地,所述第一低深度全基因组测序的测序深度在0.1×~1×之间。
进一步地,所述基因分型为:
针对所述测序结果中的每一个突变位点,通过隐马尔可夫模型预测该突变位点属于所述参考单倍型数据库中每种单倍型来源的概率,依据概率最大的单倍型的信息输出该突变位点的基因分型结果。
进一步地,所述参考单倍型数据库的构建方法包括如下步骤:
获取所述育种群体的多个个体的的基因组dna,进行所述第二低深度测序后得到测序数据;
将所述测序数据比对到参考基因组并进行群体多态位点的判定和筛选,得到所述育种群体中各多态位点的位置信息;
通过em迭代算法处理所述育种群体的突变位点信息构建参考单倍型数据库。
进一步地,所述第二低深度全基因组测序的群体测序深度在300×~600×之间。
进一步地,所述多个个体为1500个以上的个体。
进一步地,所述参考单倍型数据库的构建方法还包括:
在完成snp标记位点的检测之后,将检测结果得到的单倍型数据并入所述参考单倍型数据库。
本发明提供的基于低深度测序检测snp标记位点的方法与育种选育的实际过程相匹配:选育的前提需要有大规模样本的参考群体,这与构建参考单倍型数据库的流程相匹配;需要选育测定的个体数据是少量、多次逐渐累积的,这与检测snp标记位点的流程中以单样本为单位进行分析相匹配。
第二方面,本发明提供一种电子设备,包括:
至少一个处理器;以及
与所述处理器通信连接的至少一个存储器,其中:
所述存储器存储有可被所述处理器执行的程序指令,所述处理器调用所述程序指令能够执行如第一方面所提供的的方法的步骤。
第三方面,本发明提供一种非暂态计算机可读存储介质,所述非暂态计算机可读存储介质存储计算机指令,所述计算机指令使所述计算机执行如第一方面所提供的的方法的步骤。
本发明具备如下有益效果:
本发明通过利用低深度测序数据,低成本建立适用于目标群体大规模样本来源的参考单倍型数据库,将数据库构建环节和检测环节独立运行,实现单一低深度样本快速、经济、精准、覆盖全基因组的高密度snp基因分型。
此外,本发明提供的参考单倍型数据库亦存在更新迭代,即在检测获取的样本达到一定数量后,一次性将新样本的信息更新进入参考单倍型数据库,保证后续生产样本分型的高准确性。
附图说明
图1为本发明提供的基于低深度测序检测snp标记位点的方法的流程图。
图2为本发明提供的电子设备的实体结构示意图。
图3为本发明实施例3提供的不同参考样本量和测序深度与基因分型准确性的关系结果图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明中的附图,对本发明中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
图1为本发明提供的基于低深度测序检测snp标记位点的方法的流程示意图,如图1所示,本发明提供一种基于低深度测序检测snp标记位点的方法,包括:
s1,获取待检测个体的基因组dna;
具体地,在实际应用中,可以通过本领域常见的方式获取待检测个体的基因组dna,例如通过酶切的方式将全基因组随机打断,或者通过超声的方式将全基因组随机打断,此类可以实现全基因组随机打断的任意方法得到的基因组dna片段均可适用于后续的测序等流程。
s2,对基因组dna进行第一低深度全基因组测序;
具体地,在上述方案的基础上,可以通过本领域常规的方式在二代测序平台进行低深度全基因组测序,测序深度优选在0.1×~1×之间。
s3,将测序结果比对到参考基因组后进行基因分型;
基因分型具体为,基于隐马尔可夫模型,利用参考单倍型数据库对测序结果中的多态位点进行基因分型;
进一步地,参考基因组选择与待检测个体同源的参考基因组即可,例如针对猪的基因组dna进行基因分型,参照猪的参考基因组,针对鸡的基因组dna进行基因分型,参照鸡的参考基因组。
进一步地,将测序数据比对到参考基因组可以得到每个个体的比对结果(bam文件)。
进一步地,所述基因分型为:
针对所述测序结果中的每一个突变位点,通过隐马尔可夫模型预测该突变位点属于所述参考单倍型数据库中每种单倍型来源的概率,依据概率最大的单倍型的信息输出该突变位点的基因分型结果。
本发明提供的参考单倍型数据库由如下方法构建得到:获取所述育种群体的多个个体的的基因组dna,进行所述第二低深度测序后得到测序数据;将所述测序数据比对到参考基因组并进行群体多态位点的判定和筛选,得到所述育种群体中各多态位点的位置信息;通过em迭代算法处理所述育种群体的突变位点信息构建参考单倍型数据库。
在这一步骤中,总体上,参考单倍型数据库构建环节的样本量应保证1500个以上,对一个多态位点的群体测序深度(用于构建数据库的样本量×每个个体的测序深度)应保证在300×以上,可以保证检测的准确性。在实际应用时,可根据样本数量调整测序深度,例如样本量1500个时,应保证平均测序深度达到0.2×以上,样本量3000个时,应保证平均测序深度达到0.1×以上。
进一步地,此步骤中可采取现有技术常规的软件,例如采用basevar软件进行群体多态位点的判定和筛选得到相应的多态位点信息,并可设置一定的筛选标准,例如eaf≥0.01。
需要说明的是,在此步骤中涉及的em迭代算法可采用现有技术已有的软件实现em迭代,例如stitch软件或fastphase等。
进一步地,在完成snp标记位点的检测之后,还可以将检测结果得到的单倍型数据并入参考单倍型数据库。例如在实际应用中,因为构建参考单倍型数据库是限速步骤,所以优选在每次检测累积到一定数量的样本后将检测的单倍型数据并入参考单倍型数据库,比如累积1500个样本后一次性并入,这可以保证检测流程的快速化。
图2为本发明提供的电子设备的实体结构示意图,参照图2,所述电子设备包括:处理器(processor)31、存储器(memory)32和总线33;其中,所述处理器31和存储器32通过所述总线33完成相互间的通信;所述处理器31用于调用所述存储器32中的程序指令,以执行上述各方法实施例所提供的方法,例如包括:获取待检测个体的基因组dna;将所述基因组dna进行个体低深度全基因组测序,并将测序结果比对到参考基因组得到多态位点信息;基于神经网络模型,利用参考单倍型数据库对所述多态位点信息进行基因分型。
此外,上述的存储器32中的逻辑指令可以通过软件功能单元的形式实现并作为独立的产品销售或使用时,可以存储在一个计算机可读取存储介质中。基于这样的理解,本发明的技术方案本质上或者说对现有技术做出贡献的部分或者该技术方案的部分可以以软件产品的形式体现出来,该计算机软件产品存储在一个存储介质中,包括若干指令用以使得一台计算机设备(可以是个人计算机,服务器,或者网络设备等)执行本发明各个实施例所述方法的全部或部分步骤。而前述的存储介质包括:u盘、移动硬盘、只读存储器(rom,read-onlymemory)、随机存取存储器(ram,randomaccessmemory)、磁碟或者光盘等各种可以存储程序代码的介质。
本发明还提供一种非暂态计算机可读存储介质,其上存储有计算机程序,该计算机程序被处理器执行时实现以执行上述各实施例提供的检测方法,例如包括:获取待检测个体的基因组dna;将所述基因组dna进行个体低深度全基因组测序,并将测序结果比对到参考基因组得到多态位点信息;基于神经网络模型,利用参考单倍型数据库对所述多态位点信息进行基因分型。
以上所描述的装置实施例仅仅是示意性的,其中所述作为分离部件说明的单元可以是或者也可以不是物理上分开的,作为单元显示的部件可以是或者也可以不是物理单元,即可以位于一个地方,或者也可以分布到多个网络单元上。可以根据实际的需要选择其中的部分或者全部模块来实现本实施例方案的目的。本领域普通技术人员在不付出创造性的劳动的情况下,即可以理解并实施。
通过以上的实施方式的描述,本领域的技术人员可以清楚地了解到各实施方式可借助软件加必需的通用硬件平台的方式来实现,当然也可以通过硬件。基于这样的理解,上述技术方案本质上或者说对现有技术做出贡献的部分可以以软件产品的形式体现出来,该计算机软件产品可以存储在计算机可读存储介质中,如rom/ram、磁碟、光盘等,包括若干指令用以使得一台计算机设备(可以是个人计算机,服务器,或者网络设备等)执行各个实施例或者实施例的某些部分所述的方法。
以下基于更具体的实施例来进一步说明本发明。
实施例1
1、实验材料
使用杜洛克核心种猪群3000个个体耳组织样本,提取基因组,并稀释至40ng/μl。
2、实验方法
2.1低深度dna文库构建及测序
本实施例以tn5酶切进行dna文库构建说明,具体为:
(1)将tn5原酶与特定的tn5me-a/tn5merev以及tn5me-b/tn5merev接头72℃包埋2h,获得具有剪切-粘贴活性的tn5工作酶,将工作酶稀释至16.5ng/μl,在4μl5×taps-mgcl2,2μldimethylformamide(dmf)和nuclease-freewater的反应体系下酶切50ng基因组,条件为55℃酶切10min。
(2)在每个反应中加入3.5μl0.2%sds,再次在55℃条件下孵育10min。随后进行pcr反应,引物中包括96种不同的index来区分个体。
pcr程序为:1×(72℃,9min);1×(98℃,30sec);9×(98℃,30sec;63℃,30sec;72℃,3min)。
(3)每个体的pcr产物经qubitfluorometricquantitation(invitrogen)定量后,96个体各取等量混池,用ampurexpbeads(beckmann)在0.55×留上清,0.1×留磁珠的条件下进行纯化,纯化产物进行浓度检测后,用agilentbioanalyzer2100检测文库片段大小,确保文库质量合格。
对所有样本在mgiseq2000平台进行双端2×100bp全基因组重测序,每个样本平均测序深度为0.7×。
2.2多态位点鉴定筛选
经过过滤的原始测序数据使用基于fpga加速的服务器进行基因组比对,参考基因组使用猪sscrofa11.1(ftp://ftp.ensembl.org/pub/release-99/fasta/sus_scrofa/dna/)版本,比对软件使用bwa。每个样本的比对时间约为2-3min。本实施例中采用basevar软件进行多态位点鉴定,筛选位点的标准为eaf≥0.01,采用箱线图评估每个位点群体测序深度,保留测序深度≥1.5iqr的位点作为本群体突变位点集合。本实施例共获得猪全基因组11.6m的候选多态位点。
2.3参考单倍型数据库构建
本实施示例选取stitch软件进行em算法迭代计算,奠基者单倍型数目预设为10,采用预分型结果作为数据库单倍型过滤标准,具体参数为imputationinfoscore>0.4,hardyweinbergequilibrium(hwe)p-value>1e-6。
2.4候选样本突变分型及准确性评估
待分型的样本采用上述相同的dna建库、测序、比对方法。使用构建好的参考单倍型数据库,读取该分型样本原始测序数据,采用hmm隐马尔科夫模型进行所有候选多态位点的基因型鉴定分型。最终获得该个体全基因组11.6m的snp分型结果。随后采用geneseekgenomicprofilerporcine80ksnparray芯片对分型的结果进行准确性判定,共采集42个样本的分型结果进行评估,挑选13号染色体为例,结果显示两种方法重合位点的基因分型的一致性达到99.67%,证明该方法具有极高的准确性。
实施例2
本实施例用于说明本发明所提供的检测snp标记位点的方法的准确性和时效性。
1、实验材料
使用惠阳胡须鸡和岭南黄鸡远源深度杂交家系中3000个个体的血液样本提取基因组并稀释至40ng/μl。
2、实验方法
低深度dna文库构建及测序步骤、多态位点鉴定筛选、参考单倍型数据库构建、候选样本突变分型及准确性评估基本方法同实施例1。不同点包括:每个个体的平均测序深度约为0.8×;参考基因组使用鸡grcg6a(insdcassemblygca_000002315.5,mar2018)版本;由于杂交群体的基因组杂合度和复杂度远高于纯系群体,因此本例奠基者单倍型数目预设为24;参考单倍型数据库中共获得鸡常染色体上7.9m个候选多态位点(snp间距约为平均96bp/snp,基因组分布均与);随后以鸡chr11的结果为例进行准确性的评估,本实例共分析了28个个体,所有个体均成功获得所有chr11上288895个snp位点的分型结果;将该28个个体额外进行了超高深度的全基因组测序(平均每个样本的测序深度为80×)并使用gatk4.1标准化snp鉴定流程进行基因分型。
本实施例中构建参考单倍型数据库所使用计算资源为40个核心,每个样本全基因组测序数据比对基因组的时间约为1-2min,3000个样本用于构建数据库共计耗时4h。在检测流程中,每100个样本从原始测序数据到产出一条染色体数十万级别的snp基因分型结果仅需要8-10min,而产出全基因组所有snp(千万数量级)可通过不同染色体并行计算完成。28个用于评估准确性的个体的分型结果显示,其高深度数据结果与本专利方法基因分型的一致性超过99.71%,证明该方法在杂交群体中依然具有极高的准确性。
综上,本发明方法实现了利用单一样本的低深度测序数据,在极短的时间内进行全基因组千万数量级的snp位点的高准确度的,标准化的基因分型。
实施例3
本实施例用于说明参考单倍型数据库构建环节,每个样本测序深度和样本量对基因分型准确性的影响。
本实施例所用实验材料和实验方法同实施例2。在参考单倍型数据库构建环节,抽取不同参考样本量(200,500,1000,1500,2000,3000,4000)以及每个样本的测序深度(0.05×,0.1×,0.2×,0.3×,0.5×),利用最终获得的基因分型结果与高深度数据进行比较来评估准确性。
结果如图3所示。图中可知,每个样本平均测序深度达到0.2×以上,样本量超过1500时,基因分型的准确性基本达到稳定(保持在98.78%以上),不再随着测序深度和样本数目的增加而发生明显变化;在0.2×测序条件时,样本量超过2000,准确性即超过99%,达到99.13%。
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!