从离体血清中分析HBVDNA整合事件的系统及方法
从离体血清中分析hbv dna整合事件的系统及方法
技术领域
1.本发明涉及生物技术领域,具体涉及一种从离体血清中分析hbv dna整合事件的系统及方法。
背景技术:
2.病毒性肝炎一直是困扰全球的重要公共卫生问题。世界卫生组织2016年提出全球病毒性肝炎战略:发病率减少死亡率降低65%,这一战略对中国意义尤其重大。我国是乙肝大国,据统计,2016年我国hbv感染人数8600万,需要治疗人数3200万另外,病毒性肝炎还可以导致肝硬化、肝衰竭和肝癌等终末期肝病的发生,因此国内的乙肝防控形势仍然十分严峻。
3.对于乙型肝炎的治疗,目前有核苷酸类似物和干扰素两大类药物,而核苷酸类似物治疗为目前乙肝一线治疗的主要手段。核苷酸类似物治疗通常需要长期服药,达到一定标准后才能够停止用药,2017年easl乙肝防治指南中,关于核苷类似物治疗的停药标准,总体上基于两个方面,第一是慢乙肝病人外周血乙肝表面抗原hbsag清除,第二是获得持续性治疗应答。以外周血表面抗原清除为基础停药标准可能存在不足之处慢乙肝病人血清中hbsag存在双重来源,一是来源于hbv cccdna转录产生的s mrna,二是来源于整合至宿主基因组hbv dna转录产生s mrna。对外周血或者肝组织进行hbv dna整合检测,进而分辨外周血表面抗原来源对于优化慢乙肝治疗的停药标准是有非常重要的临床意义的。目前对hbv dna进行检测可以通过全基因组测序以及hbv dna捕获测序的方式来完成,前者所需费用较高,不太适合临床大规模应用,而hbv dna捕获测序能有效获取整合位点信息,能较高效的回答标本中hbv dna整合情况,具有更好的实用性。如何对hbv dna捕获测序结果进行有效的生物信息学处理还缺乏系统方法,我们建立了一种能准确灵敏的处理hbv dna捕获测序数据的生物信息学方法。
技术实现要素:
4.有鉴于此,本发明目的之一是基于上述问题,建立一种从血清hbv dna整合事件分析鉴定系统,采用该分析鉴定系统分析血清cfdna中hbv的整合事件,以区分血清cfdna中的hbsag是否来源于整合事件。
5.为了解决上述技术问题,本发明采用以下技术方案:
6.从离体的血清样本中分析hbv dna整合事件的系统,所述系统包括数据获取模块和数据分析模块;所述数据获取模块基于捕获测序技术获得血清样本高通量测序序列数据;所述数据分析模块包括数据过滤模块、高质量序列拼接模块、序列比对模块、整合事件鉴定模块和整合事件注释分析模块;
7.所述数据过滤模块用于过滤高通量测序得到的序列数据中的低质量序列;采用trimmomatic软件,切除序列头尾碱基质量值小于15的碱基,丢弃平均碱基质量值小于13的序列,丢弃序列长度小于30bp的序列;
8.所述高质量序列拼接模块用于将所述数据过滤模块过滤后的高质量序列进行去冗余,合并完全相同的序列,将去冗余后的序列根据序列之间的overlap(共有重叠序列区段)拼接成一致性序列片段;
9.所述序列比对模块用于利用bwa mem将所述高质量序列拼接模块拼接成的一致性序列与人和hbv基因组序列进行比对,比对至人基因组序列和hbv基因组序列,得到初步比对结果;
10.所述整合事件鉴定模块用于分析样本中包含的hbv dna整合到人基因组dna上的整合事件;提取比对结果中,包含断点clip(即同一条序列比对时分成了两段,这两段分别比对上不同的参考序列)的比对结果,并且将clip的两段其中一段比对到人基因组,另一段比对上hbv基因组;
11.所述整合事件注释分析模块用于解析整合事件发生在人类基因组的位置、整合事件发生频率、整合事件与人基因的关系;利用基因组注释文件,对整合事件位点进行定位,根据支持整合事件的序列数,计算整合事件发生频率,根据已报道热点整合事件,对整合事件进行功能注释。
12.具体的,所述人基因组序列为decoy序列hs37d5版本。
13.具体的,所述hbv基因组序列为hbv a/b/c/d/e/f/g/h亚型的参考基因组序列的合并序列。
14.进一步,所述所述整合事件鉴定模块处理程序包括如下内容:
15.1)比对中clip(即同一条序列比对时分成了两段,这两段分别比对上不同的参考序列)的处理;
16.2)比对中discordant(即一对pair end测序序列的两条序列分别比对上不同的参考序列)的处理;
17.3)clip位点两侧含有poly(即连续相同碱基,基因组该位置为低复杂度区段)的处理;
18.4)邻近breakpoint(即序列比对断裂点)位点的处理;
19.5)重复区段的处理。
20.进一步,所述步骤5)包括:
21.a.选取hotspot list中存在的已报道的时间;
22.b.选取基因上的时间;
23.c.选取在染色体上基因间区比对reads(测序序列)数最多的事件;
24.d.其他序列上比对reads数最多的事件。
25.进一步,所述数据过滤模块处理步骤如下:
26.1)切除序列头尾碱基质量值小于15的碱基;
27.2)从序列5’至3’方向,以5bp为窗口滑动,当窗口平均质量值小于15时,切除该窗口至序列3’末位的序列;
28.3)去掉序列凭借质量值低于15的序列;
29.4)去掉trim之后序列长度小于30bp的序列。
30.进一步,所述高质量序列拼接模块用于将所述数据过滤模块过滤后的高质量(pair end)双端测序序列拼接成一致性序列片段(即consensus序列);
31.具体的,所述高质量序列拼接模块处理步骤如下:
32.1)去冗余,将相同的序列根据数据比对进行合并,得到特异唯一的序列(即unique序列);
33.2)根据参考基因组对所述unique序列进行聚类并拼接得到consensus序列。
34.进一步,所述序列比对模块用于将拼接后的所述consensus序列与人类基因组合乙肝病毒基因组进行比对,得到初步比对结果,进行过滤筛选;
35.进一步,所述整合事件鉴定模块用于根据所述初步比对结果,进行hbv dna整合事件的鉴定;
36.具体的,所述整合事件鉴定模块处理程序包括如下内容:
37.1)比对中clip的处理;
38.如果一条序列是来源于两个片段整合,在比对结果中将出现clip比对结果中的clip分为hard clip(比对不上并且不会存在于比对结果中的序列)和soft clip(指虽然比对不到基因组,但是还是存在于比对结果中的序列)两大类,这两大类的共同点是,都是由于一条序列在比对的时候,序列断开了两个片段,分别比对到基因组上的不同位置,区别是hard clip在比对结果中,不展示clip的碱基,而softclip在比对结果中保留了clip的碱基。因此在我们的整合事件中,这两大类都应该保留。
39.对clip进行处理时,首先对于一条read的5’和3’均发生clip,保留clip序列较长的一端的clip。当clip的两端,一条read的clip一段比对上人的基因组序列,clip的另一段比对上hbv基因组,为可能包含整合位点信息的比对,clip的两段均比对上hbv或者hg19则不包含整合位点信息(见附图1)。
40.2)比对中discordant的处理;
41.在比对中,discordant比对是指:pair
‑
end reads一对双端测序序列中,其中一条read双端序列比对到hg19,相对应的pair read另一条双端序列比对到hbv基因组,表明在对应比对的方向不远处可能存在clip位点,但本对pe
‑
reads双端序列未覆盖该clip位点,这样的比对支持发生整合事件。
42.3)clip位点两侧含有poly的处理;
43.由于人基因组上包含很多的短串联重复区段,如polyt(即该位置为连续相同的碱基t),当检测的clip位于此类序列附近时,因这样的区段在人基因组上的其他位置也存在,因此这条read的比对可能不太可信,应舍弃。
44.4)邻近breakpoint位点的处理;
45.由于hbv整合过程中可能存在宿主的dna修复,以及比对过程中clip对断点的判断误差,同一个整合事件在clip上体现的具体breakpoint位点可能存在偏移,因此对检测到邻近的breakpoint进行整合,如两个breakpoint在基因组上距离在200bp以内,则认为是同一整合事件。
46.对clip检出的原始的breakpoint位点,支持reads数小于3的点,认为是极低频的检出点,为噪音的可能性较大,先进行过滤。
47.5)重复区段的处理;
48.从人基因组的clip位点上下游分别提取500bp碱基,作为flank序列,用blat软件对所有的clip上下游flank序列进行比对,比对可信度identity>=85%,且比对块长度大
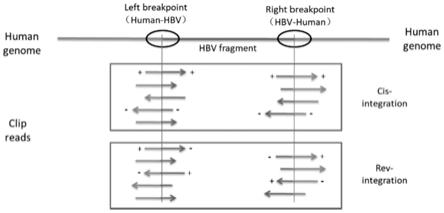
小/flank片段大小>=80%&match block/min(query length,subject length)的两个片段认为是重复片段,这两个clip可能来源于同一个整合事件,只是由于在比对的时候reads无法唯一的比对到基因组上某个位置而产生的。
49.具体的,所述步骤5)对所述两个clip可能来源于同一个整合事件进行整合,按照以下规则顺序选取其包括:
50.a.选取hotspot list中存在的已报道的事件;
51.b.选取基因上的事件;
52.c.选取在染色体上基因间区比对reads数最多的事件;
53.d.其他序列上比对reads数最多的事件。
54.进一步,所述整合事件注释分析模块用于对生和事件进行注释,分析整合事件与关键基因,整合类型、支撑整合事件reads数的标准化。
55.具体的,所述整合事件注释分析模块处理步骤如下:
56.1)使用自编代码对整个事件进行注释,根据hg19基因组基因注释gtf信息,对整合事件进行注释;
57.2)根据序列比对的方向(fr/rf),以及clip两端序列的5
’‑3’
顺序,区分整合类型为human
‑
hbv或hbv
‑
human;根据比对方向,确认是顺式整合(cis
‑
intergration)还是反式整合(rev
‑
intergration);),即hbv是正向插入还是反向插入到人基因组上(示意图见附图2);
58.3)使用高质量碱基数量对支持整合事件的reads数进行标准化,应用公式如下:
[0059][0060]
其中,nsur为number of support unique reads,即支持该整合事件的unique reads唯一片段数量(均包含clip信息),efr为去除低质量后用于与数据库比对监测clip的高质量pair
‑
end reads数。
[0061]
本发明目的之二是提供一种利用目的一所述的基于捕获测序技术分析鉴定hbv dna整合事件的系统对所述hbv dna整合事件进行分析鉴定的方法。
[0062]
为了解决上述技术问题,本发明采用以下技术方案:
[0063]
基于捕获测序技术分析鉴定hbv dna整合事件的方法,利用目的一所述的系统分析鉴定hbv dna整合事件。
[0064]
本发明的有益效果在于:本发明提供的基于捕获测序技术分析鉴定hbv dna整合事件的非诊断目的的系统和方法有效获取整合位点信息,能较高效的回答标本中hbv dna整合情况,具有更好的实用性,更利于实际应用。
附图说明
[0065]
所举实施例是为了更好地对本发明进行说明,但并不是本发明的内容仅局限于所举实施例。所以熟悉本领域的技术人员根据上述发明内容对实施方案进行非本质的改进和调整,仍属于本发明的保护范围。
[0066]
图1:对clip处理示意图;
[0067]
图2:hbv正向和反向插入示意图。
具体实施方式
[0068]
所举实施例是为了更好地对本发明进行说明,但并不是本发明的内容仅局限于所举实施例。所以熟悉本领域的技术人员根据上述发明内容对实施方案进行非本质的改进和调整,仍属于本发明的保护范围。
[0069]
实施例1
[0070]
利用模拟数据检测hbv dna整合事件。
[0071]
一、样品准备及数据获取
[0072]
人类参考基因组及hbv参考基因组数据库的建立
[0073]
人参考基因组有多个版本,我们认为decoy序列有助于提升比对的准确率,因此我们选择hs37d5版本作为我们的人基因组参考序列(decoy sequence诱饵序列,来自huref、bac或者质粒克隆和na12878)。并下载对应版本的注释文件。
[0074]
hbv有多种型别,我们从ncbi数据库中下载hbv a/b/c/d/e/f/g/h型别的参考基因组合并后,构建参考序列比对索引库。
[0075]
人工合成32种hbv dna整合到人基因组上的整合序列;将其余人源细胞按一定浓度混合,得到待检样品。利用捕获测序技术,得到一份测序下机数据,测序为双端pair end测序,读长150bp。
[0076]
二、数据过滤
[0077]
原始fastq文件序列数为8,042,989条,过滤掉去掉序列平均质量值低于15的序列,切除接头及低质量碱基后长度小于30bp的序列后最后得到7,464,916条高质量序列用于后续分析。
[0078]
三、数据拼接
[0079]
先将序列进行去冗余,合并,具体如下:将所有r1和r2序列转换成fasta格式,并合并成一个文件;然后利用blast比对,将完全相同的序列进行合并,得到unique序列,其中包含3条以上原始序列的unique序列被称为high quality unique reads,用于后续分析然后根据参考基因组进行聚类并拼接,得到较长的consensus序列,以及支持consensus的unique序列,用于后续分析。
[0080]
四、数据比对
[0081]
使用bwa mem将上述consensus序列及unique序列与参考序列进行比对,得到比对结果文件,过滤掉比对打分mapping quality score<40的比对结果。
[0082]
五、整合事件鉴定
[0083]
根据过滤后的比对结果,对clip进行上述处理,得到整合事件及整合类型,及每种整合事件支持的unique reads数,计算均一化支持整合事件序列片段数(nnss);并根据其在基因组上的整合位置,结合基因组注释,对整合事件进行注释。
[0084]
六、方法性能评估
[0085]
为评估分析方法的性能,对样本进行整合事件检测,检测阳性阈值为nnss≥1,统计整体的真阳性(true positive)、假阳性(false positive)、假阴性(false negative)和真阴性(true negative)整合事件数。
[0086]
实际存在即为我们实际往样本中加入的整合dna分子种类数;
[0087]
实际不存在即在我们的设计的整合dna分子库中,但并未加入到样本中的整合dna
分子;
[0088]
预测存在即为自有分析方法检测出样本存在该整合事件;
[0089]
预测不存在即为自有分析方法检测样本不存在该整合事件;
[0090]
例如,在构建的5个样本中,我们总共加入了55种整合事件分子,则实际存在为55实际不存在为0构建的5个样本中,未加入的整合事件为100本方法检测出62种整合事件,包含所有55种加入的整合事件,即tp为55fn为0fp为7tn为93;由此构建二联表,如下:
[0091] 预测存在(p)预测不存在(n)合计实际存在(p)55055实际不存在(n)793100合计6293155
[0092]
根据公式:
[0093][0094][0095][0096]
得到方法的准确定(accuracy)为0.955,敏感性(sensitivity)为0.887,特异性(specificity)为0.930。
[0097]
上述结果表示,本发明方法能够良好的检出hbv dna整合事件。
[0098]
实施例2
[0099]
人工合成32种hbv dna整合事件,序列信息见下表1。
[0100]
表1 32种hbv dna整合事件记录表
[0101]
[0102][0103]
具体合成序列为seq id no:1
‑
seq id no:32。
[0104]
对30例样本进行illumina测序后,计算样本中上述32种整合事件的均一化支持整合事件序列片段数(normalized number of support segments,nnss),当其中任意一种整合事件的nnss≥1时,整合事件判断为阳性,即样本中发生了整合事件。详细结果见表2
[0105]
表2 30样本检测结果表
[0106]
[0107][0108]
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1