施用β7整联蛋白拮抗剂的方法与流程
施用
β
7整联蛋白拮抗剂的方法
1.本技术是申请日为2012年3月30日、中国申请号为201811532372.3、发明名称为“施用β7整联蛋白拮抗剂的方法”的发明分案申请的再分案申请(原申请号为201280026685.6)。
2.相关申请的交叉参考
3.本技术要求2011年3月31日提交的临时美国申请61/470,360号和2011年10月21日提交的临时美国申请61/550,216号的优先权,二者在此以其整体引入作为参考。
4.序列表
5.本技术包含已通过efs-web以ascii格式提交的序列表,在此以其整体引入作为参考。2012年3月15日创建的该ascii拷贝命名为p4622r1wo_sequencelisting.txt,大小为17,964字节。
技术领域
6.提供治疗诸如包括溃疡性结肠炎和克隆病的炎性肠病的胃肠炎性障碍的方法。还提供施用诸如抗-β7抗体的整联蛋白β7拮抗剂的方法。此外,提供具体的给药方案,包括含有皮下施用和使用自注射装置的施用的给药方案。
背景技术:
7.炎性肠病(ibd)是胃肠(gi)道的慢性炎性自身免疫病症,其在临床上表现为溃疡性结肠炎(uc)或克隆病(cd)。cd是具有影响整个胃肠道的任意部分的潜能的慢性透壁性炎性疾病,而uc是结肠的黏膜炎症。两种病症在临床上都表征为频繁排便、营养不良和脱水,伴随日常生活活动的破坏。cd常并发吸收不良、狭窄和瘘的发展,可以需要反复手术。较少见的uc可以并发严重的血性腹泻和中毒性巨结肠,也需要手术。两种ibd病症都与提高的胃肠道恶性肿瘤风险相关。ibd的病因复杂,发病机制的许多方面仍不清楚。
8.中度至严重ibd的治疗向治疗医生提出了巨大的挑战,因为使用皮质类固醇的常规疗法和免疫调节剂疗法(例如硫唑嘌呤、6巯基嘌呤和氨甲喋呤)与副作用和不耐受相关,且未在维持疗法(类固醇)中显示经证明的益处。靶向肿瘤坏死因子α(tnf-α)的单克隆抗体(如英利昔单抗(infliximab)(嵌合抗体)和阿达木单抗(adalimumab)(全人抗体))目前用于cd的治疗。英利昔单抗还已显示有效性,并被批准用于uc。但是,约10%-20%的cd患者对抗tnf疗法是初级无响应者,另外~20%-30%的cd患者随时间推移而失去响应(schnitzler等,gut58:492-500(2009))。与抗tnf相关的其他不良事件(ae)包括细菌感染(包括结核病)及更罕见的淋巴瘤和脱髓鞘的比例提高(chang等,nat clin pract gastroenterol hepatology 3:220(2006);hoentjen等,world j.gastroenterol.15(17):2067(2009))。目前没有可用的疗法在超过20%-30%患有慢性疾病的ibd患者中达到持续缓解(hanauer等,lancet359:1541-49(2002);sandborn等,n engl j med 353:1912-25(2005))。此外,大多数患者未达到持续的无类固醇缓解和黏膜愈合(与真实的疾病改变相关的临床结果)。因此,存在发展针对慢性用途优化的靶向性更强的ibd疗法的需要:改善的
具有持续缓解(尤其是无类固醇缓解)的安全性谱及在更大比例的患者(包括从未响应抗tnf治疗剂或随时间推移失去响应的那些患者)中预防长期并发症。
9.整联蛋白是在包括白细胞黏附、信号发放、增殖和迁移的许多细胞过程中,以及在基因调节中发挥作用的α/β异二聚体细胞表面糖蛋白受体(hynes,r.o.,cell,1992,69:11-25;和hemler,m.e.,annu.rev.immunol.,1990,8:365-368)。它们由特异性结合内皮、上皮上的不同细胞黏附分子(cam)和胞外基质蛋白的两个异二聚体、非共价相互作用并跨膜的亚基组成。在这种方式中,整联蛋白可以作为组织特异性细胞黏附受体发挥作用,该受体辅助以高度调节的方式从血液招募白细胞进入几乎所有组织部位,在白细胞返回正常组织和炎症部位中发挥作用(von andrian等,n engl j med 343:1020
–
34(2000))。在免疫系统中,整联蛋白在炎症过程中涉及白细胞运输、黏附和浸润(nakajima,h.等,j.exp.med.,1994,179:1145-1154)。整联蛋白的差异表达调节细胞的黏附特性,不同整联蛋白涉及不同的炎症应答。(butcher,e.c.等,science,1996,272:60-66)。含有β7的整联蛋白(即α4β7和αeβ7)主要在单核细胞、淋巴细胞、嗜酸性粒细胞、嗜碱性粒细胞和巨噬细胞上表达,而不在嗜中性粒细胞上表达(elices,m.j.等,cell,1990,60:577-584)。
10.α4β7整联蛋白是在细胞向肠黏膜及相关淋巴组织(如小肠中的淋巴集结、大肠中的淋巴小结和肠系膜淋巴结)迁移中重要的白细胞归巢受体。在肠中,白细胞翻滚并紧紧黏附于黏膜内皮由来自趋化因子的信号起始,并由黏膜地址素细胞黏附分子(madcam)-1相关唾液酰lewis x介导。趋化因子信号发放诱导α4β7整联蛋白经历从低madcam-1结合亲和力至高madcam-1结合亲和力的变化。然后白细胞停滞,并开始通过血管内皮外渗至下层组织的过程。认为此外渗过程在正常免疫细胞再循环状态中及在炎性病症中都发生(von andrian等,上文)。浸润物中α4β7
+
细胞的数目及配体madcam-1的表达在诸如uc或cd患者的肠道的慢性炎症部位处更高(briskin等,am j pathol 151:97
–
110(1997);souza等,gut45:856-63(1999))。α4β7优先结合表达madcam-1和血管细胞黏附分子(vcam)-1的高内皮小静脉,以及胞外基质分子纤连蛋白片段cs-1(chan等,j biol chem 267:8366
–
70(1992);ruegg等,j cell biol17:179
–
89(1992);berlin等,cell 74:185
–
95(1993))。α4β7整联蛋白与肠黏膜血管中组成性表达的madcam-1一起在白细胞肠向性中发挥选择性作用,但似乎不促成白细胞返回外周组织或cns。相反,外周淋巴运输与α4β1与vcam-1的相互作用相关(yednock等,nature 356:63-6(1992);rice等,neurology 64:1336
–
42(2005))。
11.仅在t淋巴细胞上表达且与黏膜组织相关的β7整联蛋白家族的另一成员是αeβ7整联蛋白,或称为cd103。αeβ7整联蛋白选择性结合上皮细胞上的e钙黏着蛋白,已提出其在黏膜组织中的t细胞在上皮内淋巴细胞区室中的停留中发挥作用(cepek等,j immunol 150:3459-70(1993);karecla等eur j immunol 25:852
–
6(1995))。已报道固有层中的αeβ7
+
细胞对应激或感染的上皮细胞显示细胞毒性(hadley等,j immunol159:3748
–
56(1997);buri等,j pathol 206:178
–
85(2005))。cd中αeβ7的表达提高(elewaut等,acta gastroenterol belg 61:288
–
94(1998);oshitani等,int j mol med 12:715
–
9(2003)),已报道抗-αeβ7抗体治疗在小鼠中减弱实验性结肠炎,暗示αeβ7
+
淋巴细胞在ibd的实验模型中的作用(ludviksson等,j immunol 162:4975
–
82(1999))。
12.据报道,施用抗αeβ7的单克隆抗体在il-2-/-小鼠中预防和改善免疫诱发的结肠炎,表明炎性肠病的发病和保持依赖于表达αeβ7的固有层cd4
+
淋巴细胞的结肠定位
(ludviksson等,j immunol.1999,162(8):4975-82)。据报道,抗-α4抗体(natalizumab)在cd患者的治疗中具有有效性(sandborn等,n engl j med 2005;353:1912-25),据报道,抗-α4β7抗体(mln-02,mln0002,vedolizumab)在uc患者中有效(feagan等,n engl j med 2005;352:2499-507)。这些发现将α4β7确认为治疗靶点,并支持α4β7和madcam-1之间的相互作用介导ibd的发病机制的观点。因此,β7整联蛋白的拮抗剂具有在治疗ibd中作为治疗剂的巨大潜能。
13.之前已描述了靶向β7整联蛋白亚基的人源化单克隆抗体。见例如国际专利公开号wo2006/026759。一个这种抗体rhumabβ7(etrolizumab)衍生自大鼠抗-小鼠/人单克隆抗体fib504(andrew等1994)。将它改造为包含人igg1重链和κ1轻链的构架。国际专利公开号wo2006/026759。
14.rhumabβ7结合在肠黏膜中分别调节淋巴细胞亚群的运输和停留的α4β7(holzmann等,cell 56:37-46(1989);hu等,proc natl acad sci usa89:8254-8(1992))和αeβ7(cepek等,j immunol 150:3459-70(1993))。临床研究已证明抗α4抗体(natalizumab)对cd的治疗的有效性(sandborn等,n engl j med 353:1912-25(2005)),在uc的治疗中已针对抗α4β7抗体(ldp02/mln02/mln0002/vedolizumab)报道了振奋人心的结果(feagan等,n engl j med 352:2499-507(2005))。这些发现有助于将α4β7确认为潜在的治疗靶点,并支持α4β7和黏膜地址素细胞黏附分子1(madcam 1)之间的相互作用促成炎性肠病(ibd)的发病机制的假说。
15.与结合α4并因此结合α4β1和α4β7二者的natalizumab不同,rhumabβ7特异性结合α4β7和αeβ7的β7亚基,而不结合α4或β1整联蛋白单个亚基。这通过该抗体在高达100nm的浓度下也不能抑制α4β1+α4β7
–
ramos细胞与血管细胞黏附分子1(vcam 1)的黏附得到了证明。重要的是,rhumabβ7的此特征表明选择性:表达α4β1但不表达β7的t细胞亚群应不直接受rhumabβ7影响。
16.rhumabβ7对淋巴细胞归巢的肠特异性作用的支持来自几项体内非临床研究。在用cd45rb
高
cd4+t细胞重建的严重联合免疫缺陷(scid)小鼠(结肠炎的动物模型)中,rhumabβ7阻断放射性标记的淋巴细胞向发炎的结肠归巢,但不阻断向外周淋巴器官脾归巢。见例如国际专利公开号wo2006/026759。此外,大鼠-小鼠嵌合抗-鼠β7(抗β7,mufib504)不能在患有实验性自身免疫性脑炎(eae)的髓鞘碱性蛋白t细胞受体(mbp-tcr)转基因小鼠(多发性硬化的动物模型)中降低中枢神经系统(cns)炎症的组织学程度或改善疾病存活。id.此外,在食蟹猕猴(cynomolgus monkey)中的两项安全性研究中,rhumabβ7诱导外周血淋巴细胞数目的中度提高,其主要由cd45ra-β7
高
外周血t细胞(在表型上类似于人类中的肠归巢记忆/效应t细胞的亚群)的显著(约3至6倍)提高引起。见例如国际专利公开号wo2009/140684;stefanich等,br.j.pharmacol.,已接受的文章,doi:10.1111/j.1476-5381.2011.01205.x。相反,rhumabβ7对cd45ra+β7
中间
外周血t细胞(在表型上类似于人类中的幼稚t细胞的亚群)的数目具有最小的影响或无影响,且对cd45ra-β7
低
外周血t细胞(在表型上类似于人类中的外周归巢记忆/效应t细胞的亚群)的数目无影响,确认了rhumabβ7对肠归巢淋巴细胞亚群的特异性。国际专利公开号wo2009/140684;stefanich等,br.j.pharmacol.,已接受的文章,doi:10.1111/j.1476-5381.2011.01205.x.。
17.虽然临床研究已证明抗-α4抗体(natalizumab)对cd的治疗的有效性(sandborn
no:5);或seq id no:5的变体(seq id no:29),其中氨基酸e2选自y、f、v和d,和/或氨基酸e6选自s和g,和/或氨基酸e10选自s和y,和/或氨基酸e12选自n、t、a和d,和/或氨基酸13选自p、h、d和a,和/或氨基酸e15选自l和v,和/或氨基酸e17选自s和g;和
28.(vi)hvr-h3包含氨基酸序列f2-f11,其中f2-f11是mtgssgyfdf(seq id no:6)或rtgssgyfdf(seq id no:19);或包含氨基酸序列f1-f11,其中f1-f11是amtgssgyfdf(seq id no:16);artgssgyfdf(seq id no:17);或aqtgssgyfdf(seq id no:18);或seq id no:6、16、17、18或19的变体(seq id no:30),其中氨基酸f2是r、m、a、e、g、q、s,和/或氨基酸f11选自f和y。在某些这类实施方案中,该抗-β7抗体包含三个重链可变区(hvr-h1-h3)序列和三个轻链可变区(hvr-l1-l3)序列,其中:
29.(i)hvr-l1包含seq id no:7、seq id no:8或seq id no:9;
30.(ii)hvr-l2包含seq id no:2;
31.(iii)hvr-l3包含seq id no:3;
32.(iv)hvr-h1包含seq id no:4;
33.(v)hvr-h2包含seq id no:5;和
34.(vi)hvr-h3包含seq id no:6、seq id no:16、seq id no:17或seq id no:19。
35.在另一方面,提供按基于体重的剂量皮下施用治疗有效量的抗-β7抗体的方法。在某些实施方案中,按0.5mg/kg的剂量施用该抗-β7抗体。在某些实施方案中,按1.5mg/kg的剂量施用该抗-β7抗体。在某些实施方案中,按3.0mg/kg的剂量施用该抗-β7抗体。在某些实施方案中,按4.0mg/kg的剂量施用该抗-β7抗体。在一方面,随时间按规律间隔施用该抗-β7抗体。在某些实施方案中,每周施用该抗-β7抗体一次。在某些实施方案中,每2周施用该抗-β7抗体一次。在某些实施方案中,每4周施用该抗-β7抗体一次。在某些实施方案中,每6周施用该抗-β7抗体一次。在某些实施方案中,每8周施用该抗-β7抗体一次。在某些实施方案中,施用该抗-β7抗体2个月的时间。在某些实施方案中,施用该抗-β7抗体3个月的时间。在某些实施方案中,施用该抗-β7抗体6个月的时间。在某些实施方案中,施用该抗-β7抗体12个月的时间。在某些实施方案中,施用该抗-β7抗体18个月的时间。在某些实施方案中,施用该抗-β7抗体24个月的时间。在某些实施方案中,该个体终生施用该抗-β7抗体。在某些实施方案中,该抗-β7抗体是rhumabβ7(etrolizumab)。
36.还在另一方面,提供按固定(flat)剂量皮下施用治疗有效量的抗-β7抗体的方法。在某些实施方案中,按50mg和450mg之间的固定剂量施用该抗-β7抗体。在某些实施方案中,该固定剂量是50mg。在某些这类实施方案中,该固定剂量是100mg。在某些这类实施方案中,该固定剂量是150mg。在某些这类实施方案中,该固定剂量是200mg。在某些这类实施方案中,该固定剂量是300mg。在某些实施方案中,该固定剂量是350mg。在某些这类实施方案中,该固定剂量是400mg。在某些这类实施方案中,该固定剂量是420mg。在某些这类实施方案中,该固定剂量是450mg。在一方面,随时间按规律间隔施用该抗-β7抗体。在某些这类实施方案中,每周施用该抗-β7抗体一次。在某些实施方案中,每2周施用该抗-β7抗体一次。在某些实施方案中,每4周施用该抗-β7抗体一次。在某些实施方案中,每6周施用该抗-β7抗体一次。在某些实施方案中,每8周施用该抗-β7抗体一次。在某些实施方案中,施用该抗-β7抗体2个月的时间。在某些实施方案中,施用该抗-β7抗体3个月的时间。在某些实施方案中,施用该抗-β7抗体6个月的时间。在某些实施方案中,施用该抗-β7抗体12个月的时间。在某些实
施方案中,施用该抗-β7抗体18个月的时间。在某些实施方案中,施用该抗-β7抗体24个月的时间。在某些实施方案中,该个体终生施用该抗-β7抗体。在某些实施方案中,该抗-β7抗体是rhumabβ7(etrolizumab)。
37.还在另一方面,提供按固定剂量施用治疗有效量的抗-β7抗体的方法,其中该施用包括施用第一负荷剂量和施用至少一个后续维持剂量,其中按固定剂量施用该负荷剂量和该至少一个维持剂量中的每一个。在某些实施方案中,静脉内施用负荷剂量的该抗-β7抗体。在某些实施方案中,皮下施用负荷剂量的该抗-β7抗体。在某些实施方案中,静脉内施用该抗-β7抗体的至少一个后续维持剂量。在某些实施方案中,皮下施用该抗-β7抗体的至少一个后续维持剂量。在某些实施方案中,静脉内施用该负荷剂量和至少一个后续维持剂量中的每一个。在某些实施方案中,皮下施用该负荷剂量和至少一个后续维持剂量中的每一个。在某些实施方案中,静脉内施用该负荷剂量,皮下施用该至少一个后续维持剂量中的每一个。在某些实施方案中,该负荷剂量在400mg和450mg之间,该维持剂量在50mg和350mg之间。在某些实施方案中,该负荷剂量是400mg。在某些实施方案中,该负荷剂量是420mg。在某些实施方案中,该负荷剂量是430mg。在某些实施方案中,该负荷剂量是450mg。在某些实施方案中,该维持剂量是50mg。在某些实施方案中,该维持剂量是100mg。在某些实施方案中,该维持剂量是150mg。在某些实施方案中,该维持剂量是200mg。
38.在某些实施方案中,该维持剂量是300mg。在某些实施方案中,该维持剂量是350mg。在某些实施方案中,该负荷剂量是400mg,该维持剂量是50mg。在某些实施方案中,该负荷剂量是400mg,该维持剂量是100mg。在某些实施方案中,该负荷剂量是400mg,该维持剂量是150mg。在某些实施方案中,该负荷剂量是400mg,该维持剂量是200mg。在某些实施方案中,该负荷剂量是400mg,该维持剂量是300mg。在某些实施方案中,该负荷剂量是400mg,该维持剂量是350mg。在某些实施方案中,该负荷剂量是420mg,该维持剂量是50mg。在某些实施方案中,该负荷剂量是420mg,该维持剂量是100mg。在某些实施方案中,该负荷剂量是420mg,该维持剂量是150mg。在某些实施方案中,该负荷剂量是420mg,该维持剂量是200mg。在某些实施方案中,该负荷剂量是420mg,该维持剂量是300mg。在某些实施方案中,该负荷剂量是420mg,该维持剂量是350mg。在某些实施方案中,该负荷剂量是430mg,该维持剂量是50mg。在某些实施方案中,该负荷剂量是430mg,该维持剂量是100mg。在某些实施方案中,该负荷剂量是430mg,该维持剂量是150mg。在某些实施方案中,该负荷剂量是430mg,该维持剂量是200mg。在某些实施方案中,该负荷剂量是430mg,该维持剂量是300mg。在某些实施方案中,该负荷剂量是430mg,该维持剂量是350mg。在某些实施方案中,该负荷剂量是450mg,该维持剂量是50mg。在某些实施方案中,该负荷剂量是450mg,该维持剂量是100mg。在某些实施方案中,该负荷剂量是450mg,该维持剂量是150mg。在某些实施方案中,该负荷剂量是450mg,该维持剂量是200mg。在某些实施方案中,该负荷剂量是450mg,该维持剂量是300mg。在某些实施方案中,该负荷剂量是450mg,该维持剂量是350mg。在某些实施方案中,该抗-β7抗体是rhumabβ7(etrolizumab)。
39.在另一方面,提供按负荷剂量后一个或多个维持剂量皮下施用治疗有效量的抗-β7抗体的方法。在某些实施方案中,与该负荷剂量同一天施用第一维持剂量。在某些实施方案中,在该负荷剂量后一天施用第一维持剂量。在某些实施方案中,在该负荷剂量后一周施用第一维持剂量。在某些实施方案中,在该负荷剂量后两周施用第一维持剂量。在某些实施
方案中,在该负荷剂量后四周施用第一维持剂量。在某些实施方案中,每两周施用第二及每个后续维持剂量。在某些实施方案中,每四周施用第二及每个后续维持剂量。在某些实施方案中,每八周施用第二剂及每个后续维持剂量。在某些实施方案中,在该负荷剂量后两周施用第一维持剂量,在该第一维持剂量后两周施用第二维持剂量,每四周施用每个后续维持剂量。在某些实施方案中,施用该抗-β7抗体2个月的时间。在某些实施方案中,施用该抗-β7抗体3个月的时间。在某些实施方案中,施用该抗-β7抗体6个月的时间。在某些实施方案中,施用该抗-β7抗体12个月的时间。在某些实施方案中,施用该抗-β7抗体18个月的时间。在某些实施方案中,施用该抗-β7抗体24个月的时间。在某些实施方案中,该个体终生施用该抗-β7抗体。在某些实施方案中,该抗-β7抗体是rhumabβ7(etrolizumab)。
40.还在另一方面,提供包含整联蛋白β7拮抗剂的制品。在某些实施方案中,该制品包含含有2ml(150mg)整联蛋白β7拮抗剂的预装注射器或自动注射装置。在某些实施方案中,该制品包含含有1ml(180mg)整联蛋白β7拮抗剂的预装注射器或自动注射装置。在某些实施方案中,该整联蛋白β7拮抗剂是抗-β7抗体或rhumabβ7(etrolizumab)。
41.还在另一方面,提供与至少一种其他化合物一起皮下施用治疗有效量的整联蛋白β7拮抗剂的方法。在某些实施方案中,该至少一种其他化合物选自5-氨基水杨酸(5-asa)、硫唑嘌呤(azathioprine,aza)、6-巯基嘌呤(6-mp)和氨甲喋呤。
42.在另一方面,提供皮下施用治疗有效量的整联蛋白β7拮抗剂的方法,其中该个体或该患者之前至少一种生物活性剂(治疗)失败。在某些实施方案中,该生物活性剂选自阿达木单抗、依那西普(etanercept)、英利昔单抗、戈利木单抗(golimumab)、赛妥珠单抗(certolizumab pegol)、natalizumab和vedolizumab。在某些实施方案中,该整联蛋白β7拮抗剂是抗-β7抗体或rhumabβ7(etrolizumab)。
43.还在另一方面,提供用自注射装置皮下施用治疗有效量的整联蛋白β7拮抗剂的方法。在某些实施方案中,该自注射装置选自预装注射器、微针装置、自动注射装置和无针注射装置。在某些实施方案中,该整联蛋白β7拮抗剂是抗-β7抗体或rhumabβ7(etrolizumab)。
44.还在另一方面,提供在溃疡性结肠炎患者中诱导缓解的方法。这类方法包括按上文所示剂量或给药方案中的任一种施用抗-β7抗体。在某些实施方案中,该抗-β7抗体是rhumabβ7(etrolizumab)。在某些实施方案中,在绝对mayo临床评分≤2且没有单个小分》1时,确定在该患者中诱导缓解(也称为临床缓解)。在某些实施方案中,在起始抗-β7抗体治疗后至少第10周在患者中诱导缓解。在某些实施方案中,按上文所述施用抗-β7抗体的患者从诱导开始保持临床缓解,并继续缓解某段时间,也称为持续缓解。在某些这类实施方案中,持续缓解在诱导缓解后维持8周的时间,或诱导缓解后维持30周的时间,或诱导缓解后维持50周的时间,或诱导缓解后维持54周或更长的时间。在某些实施方案中,按上文所示剂量或给药方案中的任一种施用抗-β7抗体的患者经历持续的无类固醇缓解。在某些实施方案中,经历持续的无类固醇缓解的患者在诱导缓解后第30周停用皮质类固醇,并保持缓解至诱导缓解后第50周或第54周或更长。在某些实施方案中,按上文所述施用抗-β7抗体的患者在给定的时期内经历减少的发病时间或降低的发病比例。在某些实施方案中,按上文所述施用抗-β7抗体的患者经历黏膜愈合。在某些实施方案中,将经历黏膜愈合的患者确定为具有通过可屈性乙状结肠镜检查评估的0或1的内窥镜检查小分。在某些实施方案中,该抗-β7抗体是rhumabβ7(etrolizumab)。
45.提供下述实施方案。
46.1.在患者中治疗胃肠炎性障碍的方法,所述方法包括对所述患者施用治疗有效量的整联蛋白β7拮抗剂,其中所述整联蛋白β7拮抗剂皮下施用。
47.2.实施方案1的方法,其中所述整联蛋白β7拮抗剂是单克隆抗-β7抗体。
48.3.实施方案2的方法,其中所述抗-β7抗体按0.5mg/kg、1.5mg/kg、
49.3.0mg/kg或4.0mg/kg的剂量施用。
50.4.实施方案3的方法,其中所述抗-β7抗体每四周施用一次。
51.5.实施方案4的方法,其中所述抗-β7抗体施用两个月、三个月、六个月、12个月、18个月或24个月的时期,或施用患者的终生。
52.6.实施方案1的方法,其中所述患者是人。
53.7.实施方案1的方法,其中所述胃肠炎性障碍是炎性肠病。
54.8.实施方案7的方法,其中所述炎性肠病是溃疡性结肠炎或克隆病。
55.9.实施方案2的方法,其中所述抗-β7抗体选自嵌合抗体、人抗体和人源化抗体。
56.10.实施方案9的方法,其中所述抗-β7抗体是抗体片段。
57.11.实施方案9的方法,其中所述抗-β7抗体包含六个高变区(hvr),其中:
58.(i)hvr-l1包含氨基酸序列a1-a11,其中a1-a11是rasesvdtylh(seq id no:1);rasesvdsllh(seq id no:7);rasesvdtllh(seq id no:8);或rasesvddllh(seq id no:9);或seq id no:1、7、8或9的变体(seq id no:26),其中氨基酸a2选自a、g、s、t和v,和/或氨基酸a3选自s、g、i、k、n、p、q、r和t,和/或a4选自e、v、q、a、d、g、h、i、k、l、n和r,和/或氨基酸a5选自s、y、a、d、g、h、i、k、n、p、r、t和v,和/或氨基酸a6选自v、r、i、a、g、k、l、m和q,和/或氨基酸a7选自d、v、s、a、e、g、h、i、k、l、n、p、s和t,和/或氨基酸a8选自d、g、n、e、t、p和s,和/或氨基酸a9选自l、y、i和m,和/或氨基酸a10选自l、a、i、m和v,和/或氨基酸a11选自h、y、f和s;
59.(ii)hvr-l2包含氨基酸序列b1-b8,其中b1-b8是kyasqsis(seq id no:2);ryasqsis(seq id no:20);或xaayasqsis(seq id no:21,其中xaa代表任意氨基酸);或seq id no:2、20或21的变体(seq id no:27),其中氨基酸b1选自k、r、n、v、a、f、q、h、p、i、l、y和xaa(其中xaa代表任意氨基酸),和/或氨基酸b4选自s和d,和/或氨基酸b5选自q和s,和/或氨基酸b6选自s、d、l和r,和/或氨基酸b7选自i、v、e和k;
60.(iii)hvr-l3包含氨基酸序列c1-c9,其中c1-c9是qqgnslpnt(seq id no:3);或seq id no:3的变体(seq id no:28),其中氨基酸c8选自n、v、w、y、r、s、t、a、f、h、i l和m;
61.(iv)hvr-h1包含氨基酸序列d1-d10,其中d1-d10是gffitnnywg(seq id no:4);
62.(v)hvr-h2包含氨基酸序列e1-e17,其中e1-e17是gyisysgstsynpslks(seq id no:5);或seq id no:5的变体(seq id no:29),其中氨基酸e2选自y、f、v和d,和/或氨基酸e6选自s和g,和/或氨基酸e10选自s和y,和/或氨基酸e12选自n、t、a和d,和/或氨基酸13选自p、h、d和a,和/或氨基酸e15选自l和v,和/或氨基酸e17选自s和g;和
63.(vi)hvr-h3包含氨基酸序列f2-f11,其中f2-f11是mtgssgyfdf(seq id no:6)或rtgssgyfdf(seq id no:19);或包含氨基酸序列f1-f11,其中f1-f11是amtgssgyfdf(seq id no:16);artgssgyfdf(seq id no:17);或aqtgssgyfdf(seq id no:18);或seq id no:6、16、17、18或19的变体(seq id no:30),其中氨基酸f2是r、m、a、e、g、q、s,和/或氨基酸f11选自f和y。
64.12.实施方案11的方法,其中所述抗-β7抗体包含三个重链高变区(hvr-h1-h3)序列和三个轻链高变区(hvr-l1-l3)序列,其中:
65.(i)hvr-l1包含seq id no:7、seq id no:8或seq id no:9;
66.(ii)hvr-l2包含seq id no:2;
67.(iii)hvr-l3包含seq id no:3;
68.(iv)hvr-h1包含seq id no:4;
69.(v)hvr-h2包含seq id no:5;和
70.(vi)hvr-h3包含seq id no:6、seq id no:16、seq id no:17或seq id no:19。
71.13.在患者中治疗胃肠炎性障碍的方法,所述方法包括对所述患者施用治疗有效量的整联蛋白β7拮抗剂,其中所述整联蛋白β7拮抗剂按固定剂量皮下施用。
72.14.实施方案13的方法,其中所述整联蛋白β7拮抗剂是单克隆抗-β7抗体。
73.15.实施方案14的方法,其中所述抗-β7抗体按50mg和450mg之间的固定剂量施用。
74.16.实施方案15的方法,其中所述固定剂量是50mg。
75.17.实施方案15的方法,其中所述固定剂量是100mg。
76.18.实施方案15的方法,其中所述固定剂量是150mg。
77.19.实施方案15的方法,其中所述固定剂量是200mg。
78.20.实施方案15的方法,其中所述固定剂量是300mg。
79.21.实施方案15的方法,其中所述固定剂量是350mg。
80.22.实施方案15的方法,其中所述固定剂量是400mg。
81.23.实施方案15的方法,其中所述固定剂量是420mg。
82.24.实施方案15的方法,其中所述固定剂量是450mg。
83.25.实施方案15的方法,其中每周一次、每两周一次、每四周一次、每六周一次或每八周一次施用所述抗-β7抗体。
84.26.实施方案25的方法,其中所述抗-β7抗体施用两个月、三个月、六个月、12个月、18个月或24个月的时期,或施用患者的终生。
85.27.实施方案13的方法,其中所述患者是人。
86.28.实施方案13的方法,其中所述胃肠炎性障碍是炎性肠病。
87.29.实施方案28的方法,其中所述炎性肠病是溃疡性结肠炎或克隆病。
88.30.实施方案14的方法,其中所述抗-β7抗体选自嵌合抗体、人抗体和人源化抗体。
89.31.实施方案30的方法,其中所述抗-β7抗体是抗体片段。
90.32.实施方案30的方法,其中所述抗-β7抗体包含六个高变区(hvr),其中:
91.(i)hvr-l1包含氨基酸序列a1-a11,其中a1-a11是rasesvdtylh(seq id no:1);rasesvdsllh(seq id no:7);rasesvdtllh(seq id no:8);或rasesvddllh(seq id no:9);或seq id no:1、7、8或9的变体(seq id no:26),其中氨基酸a2选自a、g、s、t和v,和/或氨基酸a3选自s、g、i、k、n、p、q、r和t,和/或a4选自e、v、q、a、d、g、h、i、k、l、n和r,和/或氨基酸a5选自s、y、a、d、g、h、i、k、n、p、r、t和v,和/或氨基酸a6选自v、r、i、a、g、k、l、m和q,和/或氨基酸a7选自d、v、s、a、e、g、h、i、k、l、n、p、s和t,和/或氨基酸a8选自d、g、n、e、t、p和s,和/或氨基酸a9选自l、y、i和m,和/或氨基酸a10选自l、a、i、m和v,和/或氨基酸a11选自h、y、f和s;
92.(ii)hvr-l2包含氨基酸序列b1-b8,其中b1-b8是kyasqsis(seq id no:2);
ryasqsis(seq id no:20);或xaayasqsis(seq id no:21,其中xaa代表任意氨基酸);或seq id no:2、20或21的变体(seq id no:27),其中氨基酸b1选自k、r、n、v、a、f、q、h、p、i、l、y和xaa(其中xaa代表任意氨基酸),和/或氨基酸b4选自s和d,和/或氨基酸b5选自q和s,和/或氨基酸b6选自s、d、l和r,和/或氨基酸b7选自i、v、e和k;
93.(iii)hvr-l3包含氨基酸序列c1-c9,其中c1-c9是qqgnslpnt(seq id no:3);或seq id no:3的变体(seq id no:28),其中氨基酸c8选自n、v、w、y、r、s、t、a、f、h、i l和m;
94.(iv)hvr-h1包含氨基酸序列d1-d10,其中d1-d10是gffitnnywg(seq id no:4);
95.(v)hvr-h2包含氨基酸序列e1-e17,其中e1-e17是gyisysgstsynpslks(seq id no:5);或seq id no:5的变体(seq id no:29),其中氨基酸e2选自y、f、v和d,和/或氨基酸e6选自s和g,和/或氨基酸e10选自s和y,和/或氨基酸e12选自n、t、a和d,和/或氨基酸13选自p、h、d和a,和/或氨基酸e15选自l和v,和/或氨基酸e17选自s和g;和
96.(vi)hvr-h3包含氨基酸序列f2-f11,其中f2-f11是mtgssgyfdf(seq id no:6)或rtgssgyfdf(seq id no:19);或包含氨基酸序列f1-f11,其中f1-f11是amtgssgyfdf(seq id no:16);artgssgyfdf(seq id no:17);或aqtgssgyfdf(seq id no:18);或seq id no:6、16、17、18或19的变体(seq id no:30),其中氨基酸f2是r、m、a、e、g、q、s,和/或氨基酸f11选自f和y。
97.33.实施方案32的方法,其中所述抗-β7抗体包含三个重链高变区(hvr-h1-h3)序列和三个轻链高变区(hvr-l1-l3)序列,其中:
98.(i)hvr-l1包含seq id no:7、seq id no:8或seq id no:9;
99.(ii)hvr-l2包含seq id no:2;
100.(iii)hvr-l3包含seq id no:3;
101.(iv)hvr-h1包含seq id no:4;
102.(v)hvr-h2包含seq id no:5;和
103.(vi)hvr-h3包含seq id no:6、seq id no:16、seq id no:17或seq id no:19。
104.34.实施方案33的方法,其中所述抗-β7抗体按每四周100mg的固定剂量施用,其中所述患者患有溃疡性结肠炎,且其中所述患者随时间推移经历黏膜愈合或降低的发作率。
105.35.实施方案33的方法,其中所述抗-β7抗体按每四周300mg的固定剂量施用,其中所述患者患有溃疡性结肠炎,且其中所述患者随时间推移经历黏膜愈合或降低的发作率。
106.36.在患者中治疗胃肠炎性障碍的方法,所述方法包括对所述患者施用治疗有效量的整联蛋白β7拮抗剂,其中所述施用包括施用第一负荷剂量和施用至少一个后续维持剂量,其中所述负荷剂量和所述至少一个维持剂量中的每一个按固定剂量皮下施用。
107.37.实施方案36的方法,其中所述整联蛋白β7拮抗剂是单克隆抗-β7抗体。
108.38.实施方案37的方法,其中所述负荷剂量在400mg和450mg之间,且所述维持剂量在50mg和350mg之间。
109.39.实施方案38的方法,其中所述负荷剂量是400mg、420mg、430mg或450mg。
110.40.实施方案38的方法,其中所述维持剂量是50mg、100mg、150mg、200mg、300mg或350mg。
111.41.实施方案38的方法,其中所述负荷剂量是400mg,且所述维持剂量是50mg、100mg、150mg、200mg、300mg或350mg。
112.42.实施方案38的方法,其中所述负荷剂量是420mg,且所述维持剂量是50mg、100mg、150mg、200mg、300mg或350mg。
113.43.实施方案38的方法,其中所述负荷剂量是430mg,且所述维持剂量是50mg、100mg、150mg、200mg、300mg或350mg。
114.44.实施方案38的方法,其中所述负荷剂量是450mg,且所述维持剂量是50mg、100mg、150mg、200mg、300mg或350mg。
115.45.实施方案38的方法,其中所述负荷剂量是400mg、420mg、430mg或450mg,且所述维持剂量是50mg。
116.46.实施方案38的方法,其中所述负荷剂量是400mg、420mg、430mg或450mg,且所述维持剂量是100mg。
117.47.实施方案38的方法,其中所述负荷剂量是400mg、420mg、430mg或450mg,且所述维持剂量是150mg。
118.48.实施方案38的方法,其中所述负荷剂量是400mg、420mg、430mg或450mg,且所述维持剂量是200mg。
119.49.实施方案38的方法,其中所述负荷剂量是400mg、420mg、430mg或450mg,且所述维持剂量是300mg。
120.50.实施方案38的方法,其中所述负荷剂量是400mg、420mg、430mg或450mg,且所述维持剂量是350mg。
121.51.实施方案49的方法,其中所述负荷剂量是420mg。
122.52.实施方案49的方法,其中所述负荷剂量是450mg。
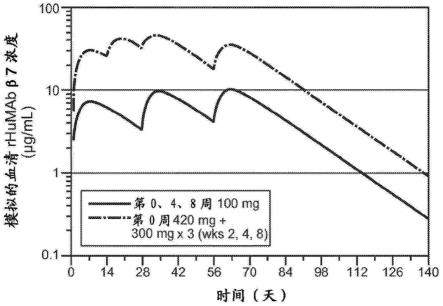
123.53.实施方案38的方法,其中所述第一维持剂量在所述负荷剂量后一天、一周、两周或四周后施用。
124.54.实施方案38的方法,其中所述维持剂量与所述负荷剂量在同一天施用。
125.55.实施方案53或实施方案54的方法,其中所述第二及每个后续维持剂量每两周、每四周或每八周施用。
126.56.实施方案38的方法,其中所述第一维持剂量在所述负荷剂量后两周施用,所述第二维持剂量在所述第一维持剂量后两周施用,且每个后续维持剂量每四周施用。
127.57.实施方案38的方法,其中所述抗-β7抗体施用两个月、三个月、六个月、12个月、18个月或24个月的时期,或施用患者的终生。
128.58.实施方案36的方法,其中所述患者是人。
129.59.实施方案36的方法,其中所述胃肠炎性障碍是炎性肠病。
130.60.实施方案59的方法,其中所述炎性肠病是溃疡性结肠炎或克隆病。
131.61.实施方案37的方法,其中所述抗-β7抗体选自嵌合抗体、人抗体和人源化抗体。
132.62.实施方案61的方法,其中所述抗-β7抗体是抗体片段。
133.63.实施方案61的方法,其中所述抗-β7抗体包含六个高变区(hvr),其中:
134.(i)hvr-l1包含氨基酸序列a1-a11,其中a1-a11是rasesvdtylh(seq id no:1);rasesvdsllh(seq id no:7);rasesvdtllh(seq id no:8);或rasesvddllh(seq id no:9);或seq id no:1、7、8或9的变体(seq id no:26),其中氨基酸a2选自a、g、s、t和v,和/或氨基酸a3选自s、g、i、k、n、p、q、r和t,和/或a4选自e、v、q、a、d、g、h、i、k、l、n和r,和/或氨基酸a5
选自s、y、a、d、g、h、i、k、n、p、r、t和v,和/或氨基酸a6选自v、r、i、a、g、k、l、m和q,和/或氨基酸a7选自d、v、s、a、e、g、h、i、k、l、n、p、s和t,和/或氨基酸a8选自d、g、n、e、t、p和s,和/或氨基酸a9选自l、y、i和m,和/或氨基酸a10选自l、a、i、m和v,和/或氨基酸a11选自h、y、f和s;
135.(ii)hvr-l2包含氨基酸序列b1-b8,其中b1-b8是kyasqsis(seq id no:2);ryasqsis(seq id no:20);或xaayasqsis(seq id no:21,其中xaa代表任意氨基酸);或seq id no:2、20或21的变体(seq id no:27),其中氨基酸b1选自k、r、n、v、a、f、q、h、p、i、l、y和xaa(其中xaa代表任意氨基酸),和/或氨基酸b4选自s和d,和/或氨基酸b5选自q和s,和/或氨基酸b6选自s、d、l和r,和/或氨基酸b7选自i、v、e和k;
136.(iii)hvr-l3包含氨基酸序列c1-c9,其中c1-c9是qqgnslpnt(seq id no:3);或seq id no:3的变体(seq id no:28),其中氨基酸c8选自n、v、w、y、r、s、t、a、f、h、i l和m;
137.(iv)hvr-h1包含氨基酸序列d1-d10,其中d1-d10是gffitnnywg(seq id no:4);
138.(v)hvr-h2包含氨基酸序列e1-e17,其中e1-e17是gyisysgstsynpslks(seq id no:5);或seq id no:5的变体(seq id no:29),其中氨基酸e2选自y、f、v和d,和/或氨基酸e6选自s和g,和/或氨基酸e10选自s和y,和/或氨基酸e12选自n、t、a和d,和/或氨基酸13选自p、h、d和a,和/或氨基酸e15选自l和v,和/或氨基酸e17选自s和g;和
139.(vi)hvr-h3包含氨基酸序列f2-f11,其中f2-f11是mtgssgyfdf(seq id no:6)或rtgssgyfdf(seq id no:19);或包含氨基酸序列f1-f11,其中f1-f11是amtgssgyfdf(seq id no:16);artgssgyfdf(seq id no:17);或aqtgssgyfdf(seq id no:18);或seq id no:6、16、17、18或19的变体(seq id no:30),其中氨基酸f2是r、m、a、e、g、q、s,和/或氨基酸f11选自f和y。
140.64.实施方案63的方法,其中所述抗-β7抗体包含三个重链高变区(hvr-h1-h3)序列和三个轻链高变区(hvr-l1-l3)序列,其中:
141.(i)hvr-l1包含seq id no:7、seq id no:8或seq id no:9;
142.(ii)hvr-l2包含seq id no:2;
143.(iii)hvr-l3包含seq id no:3;
144.(iv)hvr-h1包含seq id no:4;
145.(v)hvr-h2包含seq id no:5;和
146.(vi)hvr-h3包含seq id no:6、seq id no:16、seq id no:17或seq id no:19。
147.65.实施方案1-54或56-64中任一项的方法,其中所述整联蛋白β7拮抗剂与至少一种选自5-氨基水杨酸(5-asa)、硫唑嘌呤(aza)、6-巯基嘌呤(6-mp)和氨甲喋呤的其他化合物一起施用。
148.66.实施方案1-54或56-64中任一项的方法,其中所述患者之前的至少一种生物活性剂治疗失败。
149.67.实施方案66的方法,其中所述生物活性剂选自阿达木单抗、依那西普、英利昔单抗、戈利木单抗、赛妥珠单抗、natalizumab和vedolizumab。
150.68.实施方案1-54或56-64中任一项的方法,其中所述整联蛋白β7拮抗剂用自注射装置施用。
151.69.实施方案68的方法,其中所述自注射装置选自预装注射器、微针装置、自动注射装置和无针注射装置。
152.70.制品,其包含含有整联蛋白β7拮抗剂的液体制剂的预装注射器,其中所述制剂的体积是2ml,整联蛋白β7拮抗剂的量是150mg。
153.71.实施方案70的制品,其中所述整联蛋白β7拮抗剂是etrolizumab。
154.72.制品,其包含含有整联蛋白β7拮抗剂的液体制剂的预装注射器,其中所述制剂的体积是1ml,整联蛋白β7拮抗剂的量是180mg。
155.73.实施方案72的制品,其中所述整联蛋白β7拮抗剂是etrolizumab。
156.74.在患有溃疡性结肠炎的患者中诱导缓解的方法,其包括对所述患者施用治疗有效量的etrolizumab,其中按每四周100mg或300mg的固定剂量皮下施用etrolizumab。
157.75.实施方案74的方法,其进一步包括测定mayo临床评分和mayo临床小分,其中所述患者测定为具有《2的mayo临床评分,且没有单个小分》1。
158.76.实施方案75的方法,其中在第一次施用etrolizumab后至少第10周诱导缓解。
159.77.在患有溃疡性结肠炎的患者中诱导持续缓解的方法,其包括对所述患者施用治疗有效量的etrolizumab,其中按每四周100mg或300mg的固定剂量皮下施用etrolizumab,其中缓解保持诱导缓解后8周的时间、诱导缓解后30周的时间、诱导缓解后50周的时间,或诱导缓解后54周或更长的时间。
160.78.实施方案77的方法,其中所述缓解是无类固醇缓解。
161.79.实施方案78的方法,其中所述无类固醇缓解在诱导缓解后维持20周,或在诱导缓解后维持24周或更长。
162.附图简述
163.图1a-1c显示实施例1中所述的i期研究中rhumabβ7的组平均血清浓度-时间谱。(图1a)在该研究的单递增剂量阶段接受研究药物iv的患者中的血清rhumabβ7;(图1b)在该研究的单递增剂量阶段按3.0mg/kg iv接受研究药物的患者中的血清rhumabβ7,与接受相同剂量的研究药物sc的患者相比较;(图1c)该研究的多递增剂量阶段的患者中的血清rhumabβ7。各图在垂直轴上显示血清rhumabβ7浓度(μg/ml),在水平轴上显示时间(天)。
164.图2a-2d显示实施例1中所述的整联蛋白β7占据(occupancy)。(图2a)来自群组g中的各患者的样品中的占据;四名患者(v、w、x、y)接受0.5mg/kg rhumabβ7sc,而一名患者接受安慰剂(p6);(图2b)来自群组h中的各患者的样品中的占据;三名患者(aa、ba、da)接受1.5mg/kg rhumabβ7sc,而一名患者接受安慰剂(p8);(图2c)来自群组i中的各患者的样品中的占据;四名患者(ea、fa、ga、ha)接受3.0mg/kg rhumabβ7sc,而一名患者接受安慰剂(p9);(图2d)来自群组j中的各患者的样品中的占据;五名患者(ia、ja、ka、la、ma)接受4.0mg/kg rhumabβ7iv,而一名患者接受安慰剂(p10)。各图在垂直轴上显示占据,在水平轴上显示研究日。某些患者在研究期间具有发作,这按各患者,在括号中指出发作日。
165.图3a-3e显示实施例1中所述的mayo临床评分的个体变化。(图3a)群组g(0.5mg/kg sc);(图3b)群组h(1.5mg/kg sc);(图3c)群组i(3mg/kg sc);(图3d)群组j(4mg/kg iv);(图3e)安慰剂。对于各图,mayo临床评分显示在垂直轴上,时间(天)显示在水平轴上。箭头指示施用研究药物(图3a-3d)或安慰剂(图3e)的天。
166.图4显示用衍生自实施例2中所述的初步的i期药物动力学数据的药物动力学模型模拟的血清rhumabβ7浓度-时间谱(中间水平)。模拟的血清rhumabβ7浓度(μg/ml)在垂直轴上,时间(天)在水平轴上。
167.图5针对实施例2中所述的基于体重的剂量对固定剂量显示群体模拟预测的稳态暴露(c
max
和auc
0-84
)和变异性的比较。
168.图6a和6b针对以下显示可变轻链和重链的序列的比对:轻链人亚型κi共有序列(图6a,seq id no:12);重链人亚型iii共有序列(图6b,seq id no:13);大鼠抗-小鼠β7抗体(fib504)可变轻链(图6a,seq id no:10);大鼠抗-小鼠β7抗体(fib504)可变重链(图6b,seq id no:11);及人源化抗体变体:人源化hu504k移植可变轻链(图6a,seq id no:14),人源化hu504k移植可变重链(图6b,seq id no:15),变体hu504-5、hu504-16和hu504-32(对于变体hu504-5、hu504-16和hu504-32,来自人源化hu504k移植的氨基酸变异显示在图6a(轻链)(按出现的顺序分别为seq id no:22-24)和图6b(重链)(seq id no:25)中)。
169.图7显示实施例2中所述的ii期临床研究的研究方案。
170.图8显示已进入实施例2中所述的开放标签延长研究的患有中度至严重溃疡性结肠炎的患者的部分mayo临床评分(pmcs)。
171.发明详述
172.除非另有说明,本文所用的技术和科学术语具有与本发明所属领域的普通技术人员的通常理解相同的含义。singleton等,dictionary of microbiology and molecular biology第2版,j.wiley&sons(new york,n.y.1994)和march,advanced organic chemistry reactions,mechanisms and structure第4版,john wiley&sons(new york,n.y.1992)为本领域技术人员提供了本技术中所用的许多术语的一般指导。
173.定义
174.为了解释本说明书的目的,将适用以下定义,无论何时适当时,以单数形式使用的术语还将包括复数形式,反之亦然。在下文所示的任意定义与本文引入作为参考的任意文件冲突时,将以下文所示的定义为准。
175.除非文中清楚地另有说明,本说明书和所附权利要求中所用的单数形式“一”、“一个”和“该”包括复数指代物。因此,例如提到“一个蛋白质”包括多个蛋白质;提到“一个细胞”包括细胞的混合物等。
176.本说明书和所附权利要求中提供的范围包括两个重点及终点之间的所有点。因此,例如,2.0至3.0的范围包括2.0、3.0及2.0和3.0之间的所有点。
[0177]“治疗”及其语法变化形式指改变所治疗的个体或细胞的天然过程的尝试中的临床干预,且可以为了预防而进行或在临床病理的过程中进行。希望得到的疗效包括预防疾病的发生或复发、减轻症状、减少疾病的任意直接或间接的病理结果、降低疾病进展的速率、改善或缓和疾病状态及缓解或改善预后。
[0178]“治疗方案”指剂量、施用的频率、或治疗的持续时间、加入或不加入第二药物的组合。
[0179]“有效治疗方案”指将向接受该治疗的患者提供有益反应的治疗方案。
[0180]“改变治疗”指改变治疗方案,包括改变剂量、施用的频率、或治疗的持续时间、和/或加入第二药物。
[0181]
可以用显示对患者的益处的任意终点来评估“患者反应”或“患者反应性”,其非限制性地包括:(1)以某种程度抑制疾病进展,包括减慢和完全阻断;(2)疾病发作和/或症状的数目的减少;(3)损伤大小的减小;(4)抑制(即减少、减慢或完全阻止)疾病细胞浸润入邻
近的外周器官和/或组织;(5)抑制(即减少、减慢或完全阻止)疾病传播;(6)自身免疫反应的减少,其可以但并非必须导致疾病损伤的消退或消融;(7)以某种程度减轻与该障碍相关的一种或多种症状;(8)治疗后无疾病呈现的长度增加;和/或(9)治疗后给点时间点的死亡率降低。术语“反应性”指可测量的反应,包括完全反应(cr)和部分反应(pr)。
[0182]
本文所用的“完全反应”或“cr”意指炎症的所有病征响应治疗而消失或缓解。这并非必然意指该疾病已治愈。
[0183]“部分反应”或“pr”指炎症的严重度响应治疗而降低至少50%。
[0184]
患者对整联蛋白β7拮抗剂治疗的“有益反应”和类似的用词指来自诸如抗-整联蛋白β7抗体的拮抗剂治疗或由于诸如抗-整联蛋白β7抗体的拮抗剂治疗而赋予处于胃肠炎性障碍或患有胃肠炎性障碍的患者的临床或治疗益处。这种益处包括来自或由于拮抗剂治疗的患者的细胞或生物学反应、完全反应、部分反应、稳定疾病(无进展或复发)或具有较晚的复发的反应。
[0185]
治疗过程中患者的反应性不随时间降低时,“患者保持对治疗的反应性”。
[0186]
本文所用的术语“样品”指获自或衍生自目的个体的组合物,该目的个体包含有待例如基于物理、生物化学、化学和/或生理学特征表征和/或鉴定的细胞实体和/或其他分子实体。例如,短语“疾病样品”及其变化形式指获自预期或已知包含待表征的细胞和/或分子实体的目的个体的任意样品。样品可以获自目的个体的组织或获自该个体的外周血。
[0187]“β7整联蛋白拮抗剂”或“β7拮抗剂”指抑制β7整联蛋白的一种或多种生物学活性或阻断β7整联蛋白与它的一种或多种结合分子的结合的任意分子。本发明的拮抗剂可以用来调节β7结合作用的一个或多个方面,包括但不限于与α4整联蛋白亚基的结合,与αe整联蛋白亚基的结合,α4β7整联蛋白与madcam、vcam-1或纤连蛋白的结合,及αeβ7整联蛋白与e-钙黏着蛋白的结合。这些作用可以通过任意生物学上相关的机制来调节,包括破坏配体与β7亚基或与α4β7或αeβ7二聚体整联蛋白的结合,和/或通过破坏α和β整联蛋白亚基之间的结合,使得二聚体整联蛋白的形成被抑制。在本发明的一个实施方案中,该β7拮抗剂是抗-β7整联蛋白抗体(或抗-β7抗体)。在一个实施方案中,该抗-β7整联蛋白抗体是人源化抗-β7整联蛋白抗体,更具体而言,重组人源化单克隆抗-β7抗体(或rhumabβ7)。在一些实施方案中,本发明的抗-β7抗体是抗-整联蛋白β7拮抗抗体,其抑制或阻断β7亚基与α4整联蛋白亚基的结合,与αe整联蛋白亚基的结合,α4β7整联蛋白与madcam、vcam-1或纤连蛋白的结合,及αeβ7整联蛋白与e-钙黏着蛋白的结合。
[0188]“beta7亚基”或“β7亚基”意指人β7整联蛋白亚基(erle等,(1991)j.biol.chem.266:11009-11016)。β7亚基与α4整联蛋白亚基(如人α4亚基)结合(kilger和holzmann(1995)j.mol.biol.73:347-354)。据报道,α4β7整联蛋白在大多数成熟淋巴细胞以及一小群胸腺细胞、骨髓细胞和肥大细胞上表达(kilshaw和murant(1991)eur.j.immunol.21:2591-2597;gurish等,(1992)149:1964-1972;及shaw,s.k.和brenner,m.b.(1995)semin.immunol.7:335)。β7亚基还与αe亚基(如人αe整联蛋白亚基)结合(cepek,k.l等(1993)j.immunol.150:3459)。αeβ7整联蛋白在肠内上皮淋巴细胞(iiel)上表达(cepek,k.l.(1993)上文)。
[0189]“alphae亚基”或“alphae整联蛋白亚基”或“αe亚基”或“αe整联蛋白亚基”或“cd103”意指与上皮内淋巴细胞上的β7整联蛋白结合的整联蛋白亚基,该αeβ7整联蛋白介
导iel与表达e-钙黏着蛋白的肠上皮的结合(cepek,k.l.等(1993)j.immunol.150:3459;shaw,s.k.和brenner,m.b.(1995)semin.immunol.7:335)。
[0190]“madcam”或“madcam-1”在本发明的背景中可互换使用,指蛋白质黏膜地址素细胞黏附分子-1,其是包含短的胞质尾、跨膜区和由三个免疫球蛋白样结构域组成的胞外序列的单链多肽。已克隆了鼠、人和猕猴(macaque)madcam-1的cdna(briskin等,(1993)nature,363:461-464;shyjan等,(1996)j.immunol.156:2851-2857)。
[0191]“vcam-1”或“血管细胞黏附分子-1”或“cd106”指表达在活化的内皮上且在内皮-淋巴细胞相互作用(如炎症过程中淋巴细胞的结合和变移)中重要的α4β7和α4β1的配体。
[0192]“cd45”指蛋白质酪氨酸磷酸酶(ptp)家族的蛋白质。已知ptp是调节包括细胞生长、分化、有丝分裂周期和致癌性转化的多种细胞过程的信号发放分子。此ptp包含胞外结构域、单跨膜区段和两个串联的胞质内催化结构域,因此隶属于受体型ptp。此基因在造血细胞中专一性表达。已显示此ptp是t细胞和b细胞抗原受体信号发放的必需调节物。它通过与抗原受体复合物的成分的直接相互作用或通过活化抗原受体信号发放所需的多种src家族激酶来发挥作用。此ptp还抑制jak激酶,因此作为细胞因子受体信号发放的调节物发挥作用。已报道了此基因的编码不同同种型的四种选择性剪接转录物变体(tchilian ez,beverley pc(2002)."cd45 in memory and disease."arch.immunol.ther.exp.(warsz.)50(2):85-93;ishikawa h,tsuyama n,abroun s等(2004)."interleukin-6,cd45 and the src-kinases in myeloma cell proliferation."leuk.lymphoma 44(9):1477-81)。
[0193]
存在多种同种型的cd45:cd45ra、cd45rb、cd45rc、cd45rab、cd45rac、cd45rbc、cd45ro、cd45r(abc)。cd45还高度糖基化。cd45r是最长的蛋白质,在从t细胞分离时按200kda迁移。b细胞也表达具有更重的糖基化的cd45r,使分子量达到220kda,因此称为b220;220kda的b细胞同种型。b220表达不限于b细胞,还可以表达在活化的t细胞、一个亚群的树突细胞及其他抗原呈递细胞上。stanton t,boxall s,bennett a等(2004)."cd45 variant alleles:possibly increased frequency of a novel exon 4cd45 polymorphism in hiv seropositive ugandans."immunogenetics 56(2):107-10。
[0194]“肠归巢淋巴细胞”指具有选择性地向肠淋巴结和组织归巢但不向外周淋巴结和组织归巢的特征的淋巴细胞亚群。此亚群的淋巴细胞表征为多种细胞表面分子的组合的独特表达模式,该组合包括但不限于cd4、cd45ra和β7的组合。通常,可以根据标记cd45ra和β7进一步划分至少两个亚群的外周血cd4
+
淋巴细胞,cd45ra-β7
高
和cd45ra-β7
低
cd4
+
细胞。cd45ra-β7
高
cd4
+
细胞优先向肠淋巴结和组织归巢,而cd45ra-β7
低
cd4
+
细胞优先向外周淋巴结和组织归巢(rott等1996;rott等1997;williams等1998;ros
é
等1998;williams和butcher1997;butcher等1999)。因此,肠归巢淋巴细胞是在流式细胞术测定中鉴定为cd45ra-β7
高
cd4
+
的不同亚群的淋巴细胞。鉴定此组淋巴细胞的方法为本领域已知,还在本技术的实施例部分中详细公开。
[0195]
本文关于细胞表面标记所用的符号“+”表示细胞表面标记的阳性表达。例如,cd4
+
淋巴细胞是在其细胞表面表达cd4的一组淋巴细胞。
[0196]
本文关于细胞表面标记所用的符号
“‑”
表示细胞表面标记的阴性表达。例如,cd45ra-淋巴细胞是未在其细胞表面表达cd45ra的一组淋巴细胞。
[0197]
本文关于细胞表面标记的表达所用的符号“低”表示淋巴细胞上的细胞表面标记
的相对低水平的表达,而“高”表示淋巴细胞上的细胞表面标记的相对高水平的表达。在流式细胞术中,β7
高
的强度比β7
低
的强度高至少约10或100倍。因此,如本文在示例性实施方案中所提供,cd45ra-β7
低
cd4
+
和cd45ra-β7
高
cd4
+
细胞定位在流式细胞术分析的点阵图或直方图的不同部分,其中x轴是cd45ar的表达强度,y轴是β7的表达强度。
[0198]“外周归巢淋巴细胞”指具有向外周淋巴结和组织归巢而不向肠淋巴结和组织归巢的特征的淋巴细胞亚群。在示例性实施方案中,如上文所解释,外周归巢淋巴细胞是在流式细胞术测定中鉴定为cd45ra-β7
低
cd4
+
细胞的不同组的淋巴细胞。鉴定此组淋巴细胞的方法为本领域已知。
[0199]“胃肠炎性障碍”是引起黏膜中的炎症和/或溃疡的一组慢性障碍。这些障碍包括例如炎性肠病(例如克隆病、溃疡性结肠炎、不确定性结肠炎和感染性结肠炎)、黏膜炎(例如口腔黏膜炎、胃肠黏膜炎、鼻黏膜炎和直肠炎)、坏死性小肠结肠炎和食管炎。
[0200]“炎性肠病”或“ibd”在本文中可互换使用,指引起炎症和/或溃疡的肠病,且非限制性地包括克隆病和溃疡性结肠炎。
[0201]“克隆病(cd)”和“溃疡性结肠炎(uc)”是未知病因的慢性炎性肠病。与溃疡性结肠炎不同,克隆病可以影响肠的任意部分。克隆病最突出的特征是肠壁的颗粒状、红紫色水肿增厚。随着炎症的发展,这些肉芽瘤常失去它们的限制边界,并与周围组织结为一体。腹泻和肠梗阻是主要的临床特征。与溃疡性结肠炎一样,克隆病的过程可以连续的或复发的,轻微的或严重的,但与溃疡性结肠炎不同,克隆病不可通过切除所累及的肠区段治愈。大多数克隆病患者需要在某个点手术,但随后的复发很常见,通常需要连续的医疗处理。
[0202]
虽然克隆病可以累及从口至肛门的消化道的任意部分,但它通常出现在回肠结肠、小肠或结肠-肛门直肠区域。在组织病理学上,该疾病表征为不连续的granulomatomas、隐窝脓肿、龟裂和口疮性溃疡。炎症性浸润物是混合的,由淋巴细胞(t和b细胞二者)、浆细胞、巨噬细胞和嗜中性粒细胞组成。分泌igm和igg的浆细胞、巨噬细胞和嗜中性粒细胞有不成比例的增加。
[0203]
抗炎药物柳氮磺吡啶和5-氨基水杨酸(5-asa)用于治疗轻度活性的结肠克隆病,并常在维持疾病的缓解的尝试中使用。metroidazole和环丙沙星的功效与柳氮磺吡啶相似,且尤其用于治疗肛周疾病。在更严重的病例中,用皮质类固醇可有效治疗活性恶化,且有时可以维持缓解。硫唑嘌呤和6-巯基嘌呤也已用于需要长期施用皮质类固醇的患者。已提出这些药物可以在长期预防中发挥作用。不幸的是,在一些患者中发挥作用前,存在非常长的延迟(长达6个月)。抗腹泻药物也可以在一些患者中提供症状减轻。营养疗法或要素膳食可以改善患者的营养状态,并诱导急性疾病的症状改善,但是它不诱导持续的临床缓解。抗生素用于治疗继发性小肠细菌过度生长及治疗化脓性并发症。
[0204]“溃疡性结肠炎(uc)”伤害大肠。该疾病的过程可以是持续的或复发的,轻微的或严重的。最早的损伤是炎性浸润,在肠腺基部形成脓肿。这些膨胀和破裂的隐窝的合并倾向于将覆盖的黏膜与其血液供应分开,导致溃疡形成。该疾病的症状包括腹部绞痛、下腹痛、直肠出血及频繁的稀排泄物,其主要由具有极少的粪便颗粒的血液、脓和黏液组成。急性、严重或慢性的持续性溃疡性结肠炎可以需要全结肠切除术。
[0205]
uc的临床特征高度可变,且发病可以是潜伏的或突然的,且可以包括腹泻、里急后重和复发性直肠出血。可能发生整个结肠的爆发性累及、中毒性巨结肠、危及生命的紧急状
况。肠外表现包括关节炎、脓皮病gangrenoum、葡萄膜炎和结节性红斑。
[0206]
uc的治疗包括用于轻微病例的柳氮磺吡啶和相关的含水杨酸的药物,用于严重病例的皮质类固醇药物。水杨酸类或者皮质类固醇的局部施用有时是有效的,尤其至在疾病局限于末端肠时,且与全身性使用相比与降低的副作用相关。有时指出需要支持性措施,如施用铁和抗腹泻剂。硫唑嘌呤、6-巯基嘌呤和氨甲喋呤有时也被建议用于顽固性皮质类固醇依赖性病例。
[0207]“有效剂量”指对在必要的剂量和时间内达到希望的治疗或预防结果的有效的量。
[0208]
本文所用的术语“患者”指希望得到治疗的任意单个个体。在某些实施方案中,本文中的患者是人。
[0209]
本文中的“个体”通常是人。在某些实施方案中,个体是非人哺乳动物。示例性非人哺乳动物包括实验动物、驯养动物、宠物动物、运动动物和家畜动物,例如小鼠、猫、狗、马和牛。通常,该个体符合治疗的条件,例如胃肠炎性障碍的治疗。
[0210]
本文所用的术语个体的“终生”指开始治疗后个体的生命的剩余时间。
[0211]
术语“抗体”和“免疫球蛋白”以最广泛的含义可互换地使用,且包括单克隆抗体(例如,全长或完整的单克隆抗体)、多克隆抗体、多价抗体、多特异性抗体(例如,双特异性抗体,只要它们显示希望的生物学活性),且也可以包括某些抗体片段(如本文更详细地描述)。抗体可以是人、人源化和/或亲和力成熟的抗体。
[0212]“抗体片段”仅包含完整抗体的部分,其中该部分优选保留与该部分存在于完整抗体中时通常相关的功能的至少一种,一般为多数或全部。在一个实施方案中,抗体片段包含完整抗体的抗原结合部位,从而保持结合抗原的能力。在另一实施方案中,抗体片段(例如包含fc区的抗体片段)保留通常与该fc区存在于完整抗体中时相关的生物学功能的至少一种,如fcrn结合、抗体半寿期调节、adcc功能和补体结合。在一个实施方案中,抗体片段是单价抗体,其具有基本上类似于完整抗体的体内半衰期。例如,这种抗体片段可以包含与能够赋予该片段体内稳定性的fc序列连接的抗原结合臂。
[0213]
本文所用的术语“单克隆抗体”指从基本上同质的抗体群体获得的抗体,即除了可以少量存在的可能的天然存在的突变外,包含该单种抗体的群体是相同的。单克隆抗体高度特异,针对单一抗原。此外,与通常包括针对不同决定簇(表位)的不同抗体的多克隆抗体制剂不同,每种单克隆抗体针对抗原上的单个决定簇。
[0214]
本文的单克隆抗体特别包括“嵌合”抗体以及这类抗体的片段,其中重链和/或轻链的一部分与衍生自特定物种或隶属于特定抗体种类或亚类的抗体中对应的序列相同或同源,一条或多条链的其余部分与衍生自另一物种或隶属于另一抗体种类或亚类的抗体中对应的序列相同或同源,只要它们显示希望的生物学活性(美国专利号4,816,567;及morrison等,proc.natl.acad.sci.usa 81:6851-6855(1984))。
[0215]
非人(例如鼠)抗体的“人源化”形式是含有来自非人免疫球蛋白的最小序列的嵌合抗体。通常,人源化抗体是人免疫球蛋白(受体抗体),其中用来自非人物种(如小鼠、大鼠、兔或非人灵长类)的具有希望的特异性、亲和力和能力的高变区(供体抗体)的残基取代来自受体的高变区的残基。在一些情况下,用对应的非人残基取代人免疫球蛋白的构架区(fr)残基。此外,人源化抗体可以包含不见于受体抗体或供体抗体中的残基。进行这些修饰来进一步改良抗体性能。通常,人源化抗体将包含至少一个、通常两个可变结构域的基本上
全部,其中全部或基本上全部高变环对应于非人免疫球蛋白的那些,且全部或基本上全部fr是人免疫球蛋白序列的那些。人源化抗体还将可选地包含至少部分免疫球蛋白恒定区(fc),通常是人免疫球蛋白的恒定区。进一步的细节见jones等,nature321:522-525(1986);riechmann等,nature 332:323-329(1988);及presta,curr.op.struct.biol.2:593-596(1992)。还见以下综述文章及其中引用的参考文献:vaswani和hamilton,ann.allergy,asthma&immunol.1:105-115(1998);harris,biochem.soc.transactions 23:1035-1038(1995);hurle和gross,curr.op.biotech.5:428-433(1994)。
[0216]“人抗体”是包含对应于人产生的抗体的氨基酸序列的氨基酸序列和/或用本文公开的制备人抗体的技术中的任意种制备的抗体。这类技术包括筛选人衍生的组合文库,如噬菌体展示文库(见例如marks等,j.mol.biol.,222:581-597(1991)及hoogenboom等,nucl.acids res.,19:4133-4137(1991));用人骨髓瘤和小鼠-人杂骨髓瘤细胞系来产生人单克隆抗体(见例如kozbor j.immunol.,133:3001(1984);brodeur等,monoclonal antibody production techniques and applications,55-93页(marcel dekker,inc.,new york,1987;及boerner等,j.immunol.,147:86(1991));及在能够在缺乏内源免疫球蛋白产生的情况下产生人抗体的所有组成成分的转基因动物(例如小鼠)中产生单克隆抗体(见例如jakobovits等,proc.natl.acad.sci usa,90:2551(1993);jakobovits等,nature,362:255(1993);bruggermann等,year in immunol.,7:33(1993))。人抗体的此定义特别排除包含来自非人动物的抗原结合残基的人源化抗体。
[0217]“分离的”抗体是已鉴定并从其天然环境的成分分离和/或回收的抗体。它的天然环境的污染成分是将干扰该抗体的诊断或治疗用途的物质,且可以包括酶、激素和其他蛋白质性质或非蛋白质性质的溶质。在某些实施方案中,将该抗体纯化至:(1)通过lowry法测定抗体大于95wt%,且常超过99wt%;(2)足以通过使用旋杯式测序仪获得n端或内部氨基酸序列的至少15个残基的程度;或(3)考马斯蓝染色或银染的还原或非还原条件下的sds-page显示同质。分离的抗体包括重组细胞内的原位抗体,因为将不存在该抗体的天然环境的至少一种成分。但是,通常将通过至少一个纯化步骤来制备分离的抗体。
[0218]
在本文中使用时,术语“高变区”、“hvr”或“hv”指抗体可变结构域的区域,该区域在序列上高变和/或形成结构上确定的环。通常,抗体包含六个高变区;三个在vh(h1、h2、h3)中,三个在vl(l1、l2、l3)中。许多高变区的描述在使用,并为本文所涵盖。kabat互补决定区(cdr)基于序列变异性是最常用(kaba等,sequences of proteins of immunological interest,第5版public health service,national institutes of health,bethesda,md.(1991))。而chothia涉及结构环的定位(chothia和lesk j.mol.biol.196:901-917(1987))。abm高变区代表kabat cdr和chothia结构环之间的折衷,并为oxford molecular的abm抗体建模软件所使用。“接触”高变区基于可用的复杂晶体结构的分析。来自这些hvr的每一个的残基在下文指出。
[0219][0220]
高变区可以包含如下的“延伸的高变区”:vl中的24-36或24-34(l1)、46-56或49-56或50-56或52-56(l2)和89-97(l3)及vh中的26-35(h1)、50-65或49-65(h2)和93-102、94-102或95-102(h3)。对于这些定义的每一个,按上文的kabat等编号可变结构域残基。
[0221]“构架”或“fr”残基是除本文定义的高变区残基外的那些可变结构域残基。
[0222]“人共有构架”是代表人免疫球蛋白vl或vh构架序列的选择中最常存在的氨基酸残基的构架。通常,人免疫球蛋白vl或vh序列的选择来自可变结构域序列的亚型。通常,该序列亚型是如kabat等中的亚型。在一个实施方案中,对于vl,该亚型是如kabat等中的亚型κi。在一个实施方案中,对于vh,该亚型是如kabat等中的亚型iii。
[0223]“亲和力成熟的”抗体是在其一个或多个cdr中具有一个或多个改变的抗体,与不具有那些改变的亲本抗体相比,该改变导致抗体对抗原的亲和力的改善。在某些实施方案中,亲和力成熟的抗体将对靶抗原具有纳摩尔或甚至皮摩尔的亲和力。通过本领域已知的方法产生亲和力成熟的抗体。marks等bio/technology 10:779-783(1992)描述了通过vh和vl结构域改组进行亲和力成熟。barbas等proc nat.acad.sci,usa91:3809-3813(1994);schier等gene 169:147-155(1996);yelton等j.immunol.155:1994-2004(1995);jackson等,j.immunol.154(7):3310-9(1995);和hawkins等j.mol.biol.226:889-896(1992)描述了cdr和/或构架残基的随机诱变。
[0224]
本文所用的短语“基本上相似”或“基本上相同”表示两个数值(通常一个与本发明的抗体相关,另一个与参考/比较抗体相关)之间的相似性程度足够高,使得本领域技术人员将认为,在通过该值度量的生物学特征(例如kd值)的背景中,这两个值之间的差异有很小的或者没有生物学和/或统计学显著性。作为参考/比较抗体的值的函数,该两个值之间的差异小于约50%、小于约40%、小于约30%、小于约20%、小于约10%。
[0225]“结合亲和力”一般指分子(例如抗体)的单个结合部位与其结合配偶体(例如抗原)之间非共价相互作用的总和的强度。除非另有说明,本文所用的“结合亲和力”指内在的结合亲和力,其反映结合对(例如抗体和抗原)的成员之间的1:1相互作用。分子x对其配偶体y的亲和力一般可通过解离常数(kd)来代表。亲和力可以通过本领域公知的方法来测量,包括本文中描述的那些。低亲和力抗体一般缓慢地结合抗原并倾向于容易解离,而高亲和力抗体一般更快地结合抗原并倾向于保持更长久的结合。多种测量结合亲和力的方法为本领域已知,其中的任一种都可以用于本发明的目的。
[0226]
术语“可变的”指这样的事实,在抗体之间,可变结构域的某些部分的序列差别很大,且用于每种具体抗体对其具体抗原的结合和特异性。但是,可变性并非均勻分布在抗体的整个可变结构域中。它集中在轻链和重链可变结构域中称为高变区的三个区段中。可变结构域的更高度保守的部分称为构架区(fr)。天然重链和轻链的可变结构域各包含四个fr,四个fr主要采用β-折叠构型,通过三个高变区连接,该高变区形成连接β-折叠结构的环,且在一些情况下形成β-折叠结构的部分。各条链中的高变区通过fr近距离保持在一起,并与来自其他链的高变区一起促成抗体的抗原结合部位的形成(见kabat等,sequences of proteins of immunological interest,第5版public health service,national institutes of health,bethesda,md.(1991))。恒定区不直接参与抗体与抗原的结合,但显示多种效应子功能,如抗体在抗体依赖性细胞毒作用(adcc)中的参与。
[0227]
抗体的木瓜蛋白酶消化产生两个相同的抗原结合片段,称为“fab”片段,每个片段具有单个抗原结合部位和残留的“fc”片段,其名称反映了其易于结晶的能力。胃蛋白酶处理产生f(ab')2片段,其具有两个抗原结合部位且仍然能够交联抗原。
[0228]“fv”是包含完整的抗原识别和抗原结合部位的最小抗体片段。此区域由紧密非共价结合的一个重链可变结构域和一个轻链可变结构域的二聚体组成。各可变结构域的三个高变区正是在此构型中相互作用来定义v
h-v
l
二聚体表面上的抗原结合部位。六个高变区共同赋予抗体抗原结合特异性。但是,甚至单个可变结构域(或fv的一半,其仅包含三个对抗原特异的高变区)也具有识别和结合抗原的能力,虽然亲和力低于整个结合部位。
[0229]
fab片段还包含轻链的恒定结构域和重链的第一恒定结构域(ch1)。fab’片段与fab片段的不同在于,在重链ch1结构域的羧基端加入了几个残基,其包括一个或多个来自抗体铰链区的半胱氨酸。fab
’‑
sh是本文对其中恒定结构域的一个或多个半胱氨酸残基具有至少一个自由巯基的fab’的命名。f(ab’)2抗体片段最初作为其间具有铰合部半胱氨酸的fab’片段对产生。还已知抗体片段的其他化学偶联。
[0230]
根据其恒定结构域的氨基酸序列,可以将来自任意脊椎动物物种的抗体的“轻链”分配至称为κ和λ的两个明显不同的类型之一。
[0231]
取决于其重链恒定结构域的氨基酸序列,可以将抗体(免疫球蛋白)分配至不同种类。存在五个主要种类的免疫球蛋白:iga、igd、ige、igg和igm,这些中的几个可以进一步划分为亚类(同种型),例如igg1、igg2、igg3、igg4、iga1和iga2。对应于不同种类的免疫球蛋白的重链恒定结构域分别称为α、δ、ε、γ和μ。不同种类的免疫球蛋白的亚基结构和三维构型众所周知,且通常描述于例如abbas等cellular and mol.immunology,第4版(w.b.saunders,co.,2000)中。抗体可以是通过该抗体与一种或多种其他蛋白质或多肽的共价或非共价结合形成的更大的融合分子的一部分。
[0232]
术语“全长抗体”、“完整抗体”和“全抗体”在本文中可互换使用,指处于其基本上完整的形式的抗体,不是下文定义的抗体片段。这些术语尤其指具有含fc区的重链的抗体。
[0233]
为了本文的目的,“裸抗体”是不与细胞毒性部分或放射性标记缀合的抗体。
[0234]
本文所的术语“fc区”用来定义免疫球蛋白重链的c-端区域,包括天然序列fc区和变体fc区。虽然免疫球蛋白重链的fc区的边界可变,但是人igg重链fc区通常定义为从cys226位或从pro230位的氨基酸残基延伸至其羧基端。fc区的c-端赖氨酸(eu编号系统的残基447)可以例如在抗体的产生或纯化期间去除,或者通过重组改造编码抗体的重链的核
酸来去除。因此,完整抗体的组合物可以包含去除了所有k447残基的抗体群体、没有去除k447残基的抗体群体及具有含和不含k447残基的抗体的混合物的抗体群体。
[0235]
除非另有说明,在本文中,免疫球蛋白重链中的残基的编号是kabat等,sequences of proteins of immunological interest,第5版public health service,national institutes of health,bethesda,md(1991)中的eu指数的编号,其在此明确引入作为参考。“kabat中的eu指数”指人iggl eu抗体的残基编号。
[0236]“功能性fc区”具有天然序列fc区的“效应子功能”。示例性“效应子功能”包括c1q结合、依赖补体的细胞毒性、fc受体结合、依赖抗体的细胞毒性(adcc)、吞噬作用、细胞表面受体(例如b细胞受体;bcr)的下调等。此类效应子功能一般需要fc区与结合结构域(例如,抗体可变结构域)组合,且可以例如用本文中公开的多种测定来评估。
[0237]“天然序列fc区”包含与见于自然界中的fc区的氨基酸序列相同的氨基酸序列。天然序列人fc区包括天然序列人igg1 fc区(非a和a同种异型);天然序列人igg2 fc区;天然序列人igg3 fc区;天然序列人igg4 fc区;及其天然存在的变体。
[0238]“变体fc区”包含由于至少一个氨基酸修饰而不同于天然序列fc区的氨基酸序列的氨基酸序列。在某些实施方案中,与天然序列fc区或与亲本多肽的fc区相比,变体fc区在天然序列fc区中或在亲本多肽的fc区中具有至少一个氨基酸取代,例如从约一个至约十个氨基酸取代,且在某些实施方案中从约一个至约五个氨基酸取代。在某些实施方案中,本文的变体fc区将与天然序列fc区和/或与亲本多肽的fc区具有至少约80%同源性,或与之具有至少约90%同源性,或与之具有至少约95%同源性。
[0239]
取决于其重链恒定结构域的氨基酸序列,可以将完整抗体分配至不同“种类”。存在五个主要种类的完整抗体:iga、igd、ige、igg和igm,这些中的几个可以进一步划分为亚类(同种型),例如igg1、igg2、igg3、igg4、iga和iga2。对应于不同种类的抗体的重链恒定结构域分别称为α、δ、ε、γ和μ。不同种类的免疫球蛋白的亚基结构和三维构型众所周知。
[0240]“依赖抗体的细胞毒性”和“adcc”指细胞介导的反应,其中表达fc受体(fcr)的非特异性细胞毒性细胞(例如天然杀伤(nk)细胞、中性粒细胞和巨噬细胞)识别靶细胞上结合的抗体,随后引起靶细胞的裂解。用于介导adcc的主要细胞(nk细胞)仅表达fcγriii,而单核细胞表达fcγri、fcγrii和fcγriii。造血细胞上的fcr表达总结在ravetch和kinet,annu.rev.immunol 9:457-92(1991)的464页上的表3中。为了评估目的分子的adcc活性,可以进行体外adcc测定,如描述于美国专利号5,500,362或5,821,337中的体外adcc测定。用于这类测定的效应细胞包括外周血单核细胞(pbmc)和天然杀伤(nk)细胞。备选地,或此外,可以在体内,例如在诸如公开于clynes等pnas(usa)95:652-656(1998)中的动物模型中评估目的分子的adcc活性。
[0241]“人效应细胞”是表达一种或多种fcr并执行效应子功能的白细胞。在某些实施方案中,该细胞至少表达fcγriii,并执行adcc效应子功能。介导adcc的人白细胞的实例包括外周血单核细胞(pbmc)、天然杀伤(nk)细胞、单核细胞、细胞毒性t细胞和中性粒细胞。可以从其天然来源,例如从本文所述的血液或pbmc分离效应细胞。
[0242]
术语“fc受体”或“fcr”用来描述与抗体的fc区结合的受体。在某些实施方案中,fcr是天然序列人fcr。此外,fcr是结合igg抗体的fcr(γ受体),且包括fcγri、fcγrii和fcγriii亚类的受体,包括这些受体的等位基因变体和选择性剪接形式。fcγrii受体包括
fcγriia(“活化受体”)和fcγriib(“抑制受体”),其具有相似的氨基酸序列,主要在其胞质结构域中不同。活化受体fcγriia在其胞质结构域中包含基于免疫受体酪氨酸的活化基序(itam)。抑制受体fcγriib在其胞质结构域中包含基于免疫受体酪氨酸的抑制基序(itim)(见annu.rev.immunol.15:203-234(1997)中的综述)。fcr综述于ravetch和kinet,annu.rev.immunol 9:457-92(1991);capel等,immunomethods 4:25-34(1994);及de haas等,j.lab.clin.med.126:330-41(1995)中。本文的术语“fcr”涵盖其他fcr,包括有待将来鉴定的那些。该术语还包括负责将母体igg转移至胎儿(guyer等,j.immunol.117:587(1976)和kim等,j.immunol.24:249(1994))并调节免疫球蛋白的稳态的新生儿受体fcrn。wo00/42072(presta,l.)和us2005/0014934a1(hinton等)中描述了对新生儿fc受体(fcrn)具有改进的结合和延长的半衰期的抗体。这些抗体包含其中具有一个或多个取代的fc区,该取代改善了fc区与fcrn的结合。例如,fc区可以在位置238、250、256、265、272、286、303、305、307、311、312、314、317、340、356、360、362、376、378、380、382、413、424、428或434(残基的eu编号)的一个或多个上具有取代。在某些实施方案中,具有改善的fcrn结合的包含fc区的抗体变体在其fc区的位置307、380和434(残基的eu编号)的一个、两个或三个上包含氨基酸取代。
[0243]“单链fv”或“scfv”抗体片段包含抗体的vh和v
l
结构域,其中这些结构域存在于单条多肽链中。在某些实施方案中,fv多肽进一步在vh和v
l
结构域之间包含多肽接头,该多肽接头使得scfv能够形成希望的结构用于抗原结合。scfv的综述见pl
ü
ckthun in the pharmacology of monoclonal antibodies,113卷,rosenburg和moore编辑,springer-verlag,纽约,269-315页(1994)。her2抗体scfv片段公开于wo93/16185、美国专利号5,571,894和美国专利号5,587,458中。
[0244]
术语“双抗体”指具有两个抗原结合部位的小的抗体片段,该片段包含在同一条多肽链中与可变轻链结构域(v
l
)连接的可变重链结构域(vh)(v
h-v
l
)。通过使用太短而不允许同一条链上的两个结构域之间配对的接头,迫使结构域与另一条链上的互补结构域配对,并产生两个抗原结合部位。双抗体更充分地描述于例如ep 404,097;wo 93/11161;和hollinger等,proc.natl.acad.sci.usa,90:6444-6448(1993)中。
[0245]“亲和力成熟的”抗体是在其一个或多个高变区中具有一个或多个改变的抗体,与不具有那些改变的亲本抗体相比,该改变导致抗体对抗原的亲和力的改善。在某些实施方案中,亲和力成熟的抗体将对靶抗原具有纳摩尔或甚至皮摩尔的亲和力。通过本领域已知的方法产生亲和力成熟的抗体。marks等bio/technology 10:779-783(1992)描述了通过vh和vl结构域改组进行亲和力成熟。barbas等proc nat.acad.sci,usa91:3809-3813(1994);schier等gene 169:147-155(1995);yelton等j.immunol.155:1994-2004(1995);jackson等,j.immunol.154(7):3310-9(1995);和hawkins等,j.mol.biol.226:889-896(1992)描述了cdr和/或构架残基的随机诱变。
[0246]
本文的“氨基酸序列变体”抗体是具有不同于主要种类抗体的氨基酸序列的抗体。在某些实施方案中,氨基酸序列变体将与主要种类抗体具有至少约70%同源性,或者它们将与主要种类抗体具有至少约80%或至少约90%同源性。氨基酸序列变体在主要种类抗体的氨基酸序列内部或相邻的某些位置上具有取代、缺失和/或添加。本文的氨基酸序列变体的实例包括酸性变体(如脱酰胺化的抗体变体)、碱性变体、在其一条或两条轻链上具有氨
基端前导序列延伸(例如vhs-)的抗体、在其一条或两条重链上具有c-端赖氨酸残基的抗体等,且包括重链和/或轻链的氨基酸序列的变异的组合。本文的尤其重要的抗体变体是这样的抗体,相对于主要种类抗体,其在其一条或两条轻链上包含氨基端前导序列延伸,可选地进一步包含其他氨基酸序列和/或糖基化差异。
[0247]
本文的“糖基化变体”抗体是具有附着于该抗体的一个或多个糖类部分的抗体,该糖类部分不同于附着于主要种类抗体的一个或多个糖类部分。本文的糖基化变体的实例包括具有附着于其fc区的gl或g2寡糖结构而不是g0寡糖结构的抗体、具有附着于其一条或两条轻链的一个或两个糖类部分的抗体、没有附着于该抗体的一条或两条重链的糖类的抗体等,以及糖基化改变的组合。在抗体具有fc区时,寡糖结构可以例如在残基299(298,残基的eu编号)处附着于该抗体的一条或两条重链。
[0248]
本文所用的术语“细胞毒剂”指抑制或阻止细胞的功能和/或导致细胞的破坏的物质。该术语旨在包括放射性同位素(例如at
211
、i
131
、i
125
、y
90
、re
186
、re
188
、sm
153
、bi
212
、p
32
和lu的放射性同位素)、化疗剂和毒素,如细菌、真菌、植物或动物来源的小分子毒素或酶活性毒素,包括其片段和/或变体。
[0249]
术语“细胞因子”是由一个细胞群体释放的作为胞间介质作用于另一细胞的蛋白质的通用术语。这类细胞因子的实例是淋巴因子、单核因子和传统的多肽激素。细胞因子包括生长激素,如人生长激素、n-甲硫氨酰人生长激素和牛生长激素;甲状旁腺激素;甲状腺素;胰岛素;胰岛素原;松弛素;松弛素原;糖蛋白激素,如促卵泡激素(fsh)、促甲状腺激素(tsh)和黄体生成素(lh);肝生长因子;成纤维细胞生长因子;促乳素;胎盘促乳素;肿瘤坏死因子-α和-β;缪勒氏管抑制物质;小鼠促性腺激素相关肽;抑制素;激活蛋白;血管内皮生长因子;整联蛋白;血小板生成素(tpo);神经生长因子,如ngf-β;血小板生长因子;转化生长因子(tgf),如tgf-α和tgf-β;胰岛素样生长因子-i和-ii;促红细胞生成素(epo);骨诱导因子;干扰素,如干扰素-α、-β和-γ;集落刺激因子(csf),如巨噬细胞-csf(m-csf)、粒细胞-巨噬细胞-csf(gm-csf)和粒细胞-csf(g-csf);白细胞介素(il),如il-1、il-1α、il-2、il-3、il-4、il-5、il-6、il-7、il-8、il-9、il-10、il-11、il-12;肿瘤坏死因子,如tnf-α或tnf-β;及其他多肽因子,包括lif和kit配体(kl)。本文所用的术语细胞因子包括来自天然来源或来自重组细胞培养物的蛋白质及天然序列细胞因子的生物学活性等同物。
[0250]
本文针对辅助疗法所用的术语“免疫抑制剂”指发挥作用来抑制或掩蔽本文所治疗的受试者的免疫系统的物质。这将包括抑制细胞因子产生、下调或抑制自身抗原表达或掩蔽mhc抗原的物质。这类物质的实例包括2-氨基-6-芳基-5-取代的嘧啶(见美国专利号4,665,077);非类固醇抗炎药(nsaid);更昔洛韦;他克莫司;糖皮质激素,如皮质醇或醛固酮;抗炎剂,如环加氧酶抑制剂;5-脂加氧酶抑制剂;或白三烯受体拮抗剂;嘌呤拮抗剂,如硫唑嘌呤或麦考酚酸酯(mmf);烷化剂,如环磷酰胺;溴隐亭;达那唑;氨苯砜;戊二醛(其掩蔽mhc抗原,如美国专利号4,120,649中所述);mhc抗原和mhc片段的抗独特型抗体;环孢菌素;6-巯基嘌呤;类固醇,如皮质类固醇或糖皮质激素或糖皮质激素类似物,例如泼尼松、甲泼尼龙,包括solu-medrol.rtm、琥珀酸钠甲基强的松龙和地塞米松;二氢叶酸还原酶抑制剂,如氨甲喋呤(口服或皮下);抗疟疾剂,如氯喹和羟氯喹;柳氮磺吡啶;来氟米特;细胞因子或细胞因子受体抗体或拮抗剂,包括抗干扰素-α、-β或-γ抗体,抗肿瘤坏死因子(tnf)-α抗体(英利昔单抗(remicade.rtm.)或阿达木单抗),抗-tnf-α免疫黏附素(依那西普)、抗-tnf-β
抗体、抗白介素-2(il-2)抗体和抗-il-2受体抗体,及抗白介素-6(il-6)受体抗体和拮抗剂;抗-lfa-1抗体,包括抗-cdlla和抗-cd18抗体;抗-l3t4抗体;异源抗淋巴细胞球蛋白;泛-t抗体,抗-cd3或抗-cd4/cd4a抗体;含有lfa-3结合结构域的可溶性肽(w0 90/08187,1990年7月26日公开);链激酶;转化生长因子-β(tgf-β);链道酶;来自宿主的rna或dna;fk506;rs-61443;苯丁酸氮芥;脱氧精胍菌素;雷帕霉素;t-细胞受体(cohen等,美国专利号5,114,721);t细胞受体片段(offner等,science,251:430-432(1991);wo 90/11294;ianeway,nature,341:482(1989);和wo 91/01133);baff拮抗剂,如baff或br3抗体或免疫粘附素和ztnf4拮抗剂(综述见mackay和mackay,trends immunol.,23:113-5(2002),也参见下文的定义);干扰t细胞辅助信号的生物活性剂,如抗-cd40受体或抗-cd40配体(cd154),包括cd40-cd40配体的封闭抗体(例如durie等,science,261:1328-30(1993);mohan等,j.immunol.,154:1470-80(1995))和ctla4-ig(finck等,science,265:1225-7(1994));及t-细胞受体抗体(ep 340,109),如t10b9。
[0251]
本文所用的术语“改善”指病症、疾病、障碍或表型(包括异常或症状)的减少、降低或消除。
[0252]
疾病或障碍(例如炎性肠病,例如溃疡性结肠炎或克隆病)的“症状”是个体经历并指示疾病的任意病态现象或结构、功能或感觉偏离正常。
[0253]
表述“治疗有效量”指对预防、改善或治疗疾病或障碍(例如炎性肠病,例如溃疡性结肠炎或克隆病)有效的量。例如,抗体的“治疗有效量”指对预防、改善或治疗指定的疾病或障碍有效的抗体的量。类似地,抗体和第二化合物的组合的“治疗有效量”指组合起来对预防、改善或治疗指定的疾病或障碍有效的抗体的量和第二化合物的量。
[0254]
应理解,术语两种化合物的“组合”不意味着该化合物必须在相互的混合物中施用。因此,这种组合的治疗或用途涵盖化合物的混合物或化合物的分开施用,且包括在同一天或不同天施用。因此,术语“组合”意指将两种或多种化合物单独或在相互的混合物中用于治疗。在例如对个体组合施用抗体和第二化合物时,无论该抗体和第二化合物是单独对个体施用还是在混合物中对个体施用,在该第二化合物存在于该个体中时,该抗体也存在于该个体中。在某些实施方案中,在该抗体之前施用抗体以外的化合物。在某些实施方案中,在该抗体之后施用抗体以外的化合物。
[0255]
为了本文的目的,“肿瘤坏死因子-α(tnf-α)”指包含pennica等,nature,312:721(1984)或aggarwal等,jbc,260:2345(1985)中所述的氨基酸序列的人tnf-α分子。
[0256]
本文的“tnf-α抑制剂”是一般通过与tnf-α结合并中和其活性来以某种程度抑制tnf-α的生物学功能的物质。本文特别考虑的tnf抑制剂的实例是依那西普英利昔单抗阿达木单抗戈利木单抗(simponitm)和赛妥珠单抗
[0257]“皮质类固醇”指模拟或扩大天然存在的皮质类固醇的作用的具有类固醇的一般化学结构的几种合成或天然存在的物质中的任意一种。合成的皮质类固醇的实例包括强的松、强的松龙(包括甲基强的松龙)、地塞米松、曲安西龙和倍他米松。
[0258]“拮抗剂”指能够中和、阻断、抑制、废止、降低或干扰具体的或指定的蛋白质的活性(包括其在配体的情况下与一种或多种受体的结合,或在受体的情况下与一种或多种配
distribution center,san diego,calif.usa获得的衍生自mopc-21和mpc-11小鼠肿瘤的那些,及可从american type culture collection,manassas,va.usa获得的sp-2和衍生物,如x63-ag8-653细胞。还针对人单克隆抗体的产生描述了人骨髓瘤和小鼠-人杂骨髓瘤(heteromyeloma)细胞系(kozbor,j.immunol.,133:3001(1984);和brodeur等,monoclonal antibody production techniques and applications,51-63页(marcel dekker,inc.,new york,1987))。
[0280]
针对抗该抗原的单克隆抗体的产生测定杂交瘤细胞在其中生长的培养基。在某些实施方案中,通过免疫沉淀或通过诸如放射免疫测定(ria)或酶联免疫吸附测定(elisa)的体外结合测定来测定杂交瘤产生的单克隆抗体的结合特异性。
[0281]
可以例如通过描述于munson等,anal.biochem.,107:220(1980)中的scatchard分析来测定单克隆抗体的结合亲和力。一旦鉴定出产生具有希望的特异性、亲和力和/或活性的抗体的杂交瘤细胞,即可以通过有限稀释法亚克隆该克隆,并通过标准方法培养(goding,monoclonal antibodies:principles and practice,59-103页(academic press,1986))。适合用于此目的的培养基包括例如d-mem或rpmi-1640培养基。此外,可以例如通过将细胞腹膜内注射入小鼠,在动物中作为腹水肿瘤体内培养杂交瘤细胞。通过常规抗体纯化方法(例如亲和层析(例如使用a蛋白或g蛋白sepharose)或离子交换层析、羟基磷灰石层析、凝胶电泳、透析等)从培养基、腹水或血清适宜地分离由亚克隆分泌的单克隆抗体。
[0282]
编码该单克隆抗体的dna易于用常规方法分离和测序(例如,通过使用能够特异性结合编码鼠抗体的重链和轻链的基因的寡核苷酸探针)。杂交瘤细胞可作为这种dna的来源。一旦分离,即可以将该dna放入表达载体中,然后将该表达载体转染入诸如大肠杆菌(e.coli)细胞、猿猴cos细胞、中国仓鼠卵巢(cho)细胞或不以其他方式产生免疫球蛋白蛋白质的骨髓瘤细胞的宿主细胞中,以获得单克隆抗体在该重组宿主细胞中的合成。关于编码抗体的dna在细菌中的重组表达的综述文章包括skerra等,curr.opinion in immunol.,5:256-262(1993)和pluckthun,immunol.revs.130:151-188(1992)。
[0283]
在另一实施方案中,可以从用例如描述于mccafferty等,nature,348:552-554(1990)中的技术产生的抗体噬菌体文库分离单克隆抗体或抗体片段。clackson等,nature,352:624-628(1991)和marks等,j.mol.biol.,222:581-597(1991)分别描述了用噬菌体文库分离鼠抗体和人抗体。随后的公开描述了通过链改组产生高亲和力(nm范围)人抗体(marks等,bio/technology,10:779-783(1992)),以及作为构建非常大的噬菌体文库的策略的组合感染和体内重组(waterhouse等,nuc.acids.res.21:2265-2266(1993))。因此,这些技术是除传统单克隆抗体杂交瘤技术之外用于分离单克隆抗体的可行选择。
[0284]
可以修饰编码抗体的dna来产生嵌合或融合抗体多肽,例如,通过用人重链和轻链恒定结构域(ch和cl)序列取代同源鼠序列(美国专利号4,816,567;和morrison等,proc.natl.acad.sci.usa,81:6851(1984)),或通过将免疫球蛋白编码序列与非免疫球蛋白多肽(异源多肽)的编码序列的全部或部分融合。非免疫球蛋白多肽序列可以取代抗体的恒定结构域,或者它们可以取代抗体的一个抗原结合部位的可变结构域以产生嵌合二价抗体,该嵌合二价抗体包含对一种抗原具有特异性的一个抗原结合部位和对不同抗原具有特异性的另一抗原结合部位。
[0285]
示例性抗-β7抗体是fib504、fib 21、22、27、30(tidswell,m.j immunol.1997年8月1日;159(3):1497-505)或其人源化衍生物。fib504的人源化抗体详细公开于美国专利公开号20060093601(作为美国专利号7,528,236授权)中,其内容以其整体引入作为参考(还见下文的讨论)。
[0286]
3.人和人源化抗体
[0287]
本发明的抗-β7整联蛋白抗体可以进一步包含人源化抗体或人抗体。非人(例如鼠)抗体的人源化形式是嵌合免疫球蛋白、免疫球蛋白链或其片段(如fv、fab、fab'、f(ab')2或抗体的其他抗原结合子序列),其含有衍生自非人免疫球蛋白的最小序列。人源化抗体包括人免疫球蛋白(受体抗体),其中用来自非人物种(如小鼠、大鼠或兔)的具有希望的特异性、亲和力和能力的cdr(供体抗体)的残基取代来自受体的互补决定区(cdr)的残基。在一些情况下,用对应的非人残基取代人免疫球蛋白的构架残基。人源化抗体还可以包含不见于受体抗体中或输入的cdr或构架序列中的残基。通常,人源化抗体将包含至少一个、通常两个可变结构域的基本上全部,其中全部或基本上全部cdr区对应于非人免疫球蛋白的那些,且全部或基本上全部fr区是人免疫球蛋白共有序列的那些。人源化抗体还将可选地包含至少部分免疫球蛋白恒定区(fc),通常是人免疫球蛋白的恒定区[jones等,nature,321:522-525(1986);riechmann等,nature 332:323-329(1988);和presta,curr.op.struct.biol.,2:593-596(1992)]。
[0288]
用于人源化非人抗体的方法为本领域公知。通常,人源化抗体具有一个或多个从非人来源引入其中的氨基酸残基。这些非人氨基酸残基常称为“输入”残基,其通常取自“输入”可变结构域。基本上可以按照winter及其同事的方法[jones等,nature,321:522-525(1986);riechmann等,nature,332:323-327(1988);verhoeyen等,science,239:1534-1536(1988)],通过用啮齿动物cdr或cdr序列取代对应的人抗体序列来进行人源化。因此,这类“人源化”抗体是嵌合抗体(美国专利号4,816,567),其中用对应的来自非人物种的序列取代了基本上不再完整的人可变结构域。实际上,人源化抗体通常是人抗体,其中用来自啮齿动物抗体中类似位点的残基取代了一些cdr残基,且可能取代一些fr残基。在抗体预期用于人治疗用途时,用于制备人源化抗体的人可变结构域(轻链和重链二者)的选择对降低抗原性和hama反应(人抗-小鼠抗体)非常重要。按照所谓的“最佳适合”法,针对已知人可变结构域序列的整个文库筛选啮齿动物抗体的可变结构域的序列。然后鉴定出最接近于啮齿动物的v结构域序列的人v结构域序列,并接受其内的人构架区(fr)用于人源化抗体(sims等,j.immunol.151:2296(1993);chothia等,j.mol.biol.,196:901(1987))。另一种方法使用衍生自特定亚型的轻链或重链的所有人抗体的共有序列的特定构架区。同一构架可以用于几种不同的人源化抗体(carter等,proc.natl.acad.sci.usa,89:4285(1992);presta等,j.immunol.151:2623(1993))。保留对抗原的高亲和力和其他有利的生物学特性来人源化抗体也很重要。为了达到此目的,根据某些实施方案,通过用亲本序列和人源化序列的三维模型分析亲本序列和多种概念性人源化产物的方法来制备人源化抗体。三维免疫球蛋白模型通常可得,并为本领域技术人员所熟悉。说明和显示所选择的候选免疫球蛋白序列的可能的三维构象结构的计算机程序也可得。这些显示的检查允许分析残基在候选免疫球蛋白序列的功能发挥中可能的作用,即分析影响候选免疫球蛋白结合其抗原的能力的残基。这样,可以从受体序列和输入序列选择和组合fr残基,使得达到希望的抗体特征,如提高的对
一种或多种靶抗原的亲和力。通常,高变区残基直接且最实质性地涉及影响抗原结合。
[0289]
考虑多种形式的人源化抗-β7整联蛋白抗体。例如,人源化抗体可以是抗体片段,如fab,其可选地与一种或多种细胞毒剂缀合,以便产生免疫缀合物。备选地,人源化抗体可以是完整抗体,如完整的iggl抗体。
[0290]
示例性人源化抗β7抗体包括但不限于rhumabβ7,其是抗整联蛋白亚基β7的人源化单克隆抗体,且衍生自大鼠抗-小鼠/人单克隆抗体fib504(andrew等,1994j immunol 1994;153:3847-61)。已将它改造为包括人免疫球蛋白igg1重链和κ1轻链构架,并通过中国仓鼠卵巢细胞来产生。此抗体结合两种整联蛋白α4β7(holzmann等1989cell,1989;56:37-46;hu等,1992,proc natl acad sci usa 1992;89:8254-8)和αeβ7(cepek等,1993j immunol 1993;150:3459-70),这两种整联蛋白调节胃肠道中的淋巴细胞亚群的运输和保留,且涉及炎性肠病(ibd),如溃疡性结肠炎(uc)和克隆病(cd)。rhumabβ7是α4β7和其配体(粘膜地址素细胞黏附分子-l[madcam]-l、血管细胞黏附分子[vcam]-1和纤连蛋白)之间的细胞相互作用以及αeβ7和其配体(e-钙黏着蛋白)之间的相互作用的有效体外阻断剂。rhumabβ7以相似的高亲和力可逆地结合来自兔、食蟹猕猴和人类的淋巴细胞上的β7。它还以高亲和力结合小鼠β7。rhumabβ7及其变体的氨基酸序列及制备和使用详细公开于美国专利申请公开号20060093601(作为美国专利号7,528,236授权)中,该申请的内容以其整体引入。
[0291]
图6a和6b针对以下显示可变轻链和重链的序列的比对:轻链人亚型κi共有序列(图6a,seq id no:12);重链人亚型iii共有序列(图6b,seq id no:13);大鼠抗-小鼠β7抗体(fib504)可变轻链(图6a,seq id no:10);大鼠抗-小鼠β7抗体(fib504)可变重链(图6b,seq id no:11);及人源化抗体变体:人源化hu504k移植可变轻链(图6a,seq id no:14),人源化hu504k移植可变重链(图6b,seq id no:15),变体hu504-5、hu504-16和hu504-32(对于变体hu504-5、hu504-16和hu504-32,来自人源化hu504k移植的氨基酸变异显示在图6a(轻链)(按出现的顺序分别为seq id no:22-24)和图6b(重链)(seq id no:25)中)。
[0292]
4.人抗体
[0293]
作为人源化之外的选择,可以产生人抗体。例如,现在可能产生转基因动物(例如小鼠),其在免疫时能够在缺乏内源免疫球蛋白产生的情况下产生人抗体的所有组成成分。例如,已描述了嵌合和种系突变体小鼠中抗体重链连接区(jh)基因的纯合缺失导致内源抗体产生的完全抑制。将人种系免疫球蛋白基因阵列转入这类种系突变体小鼠将导致在抗原攻击时产生人抗体。见例如jakobovits等,proc.natl.acad.sci.usa,90:2551(1993);jakobovits等,nature,362:255-258(1993);bruggemann等,year in immuno.7:33(1993);美国专利号5,545,806、5,569,825、5,591,669(全都属于genpharm);美国专利号5,545,807;和wo 97/17852。
[0294][0295]
备选地,可以用噬菌体展示技术(mccafferty等,nature 348:552-553[1990])来在体外从来自未免疫供体的免疫球蛋白可变(v)结构域基因库产生人抗体和抗体片段。根据此技术,将抗体v结构域基因符合读框地克隆入丝状噬菌体(如m13或fd)的主要或次要外被蛋白质基因中,并作为功能性抗体片段展示在噬菌体颗粒的表面上。因为丝状颗粒包含噬菌体基因组的单链dna拷贝,基于抗体功能特性的选择还导致编码显示那些特性的抗体
的基因的选择。因此,噬菌体模拟了b细胞的一些特性。噬菌体展示可以以综述于例如johnson,kevin s.和chiswell,david j.,current opinion in structural biology 3:564-571(1993)中的多种形式进行。可以将v基因区段的几个来源用于噬菌体展示。clackson等,nature,352:624-628(1991)从衍生自免疫小鼠的脾的v基因的小的随机组合文库分离了一系列多样化的抗-恶唑酮抗体。可以构建来自未免疫的人供体的v基因库,且可以基本上按照marks等,j.mol.biol.222:581-597(1991)或griffith等,embo j.12:725-734(1993)所述的技术分离抗一系列多样化抗原(包括自身抗原)的抗体。还见美国专利号5,565,332和5,573,905。
[0296]
如上文所讨论,还可以通过体外活化b细胞来产生人抗体(见美国专利号5,567,610和5,229,275)。
[0297]
5.抗体片段
[0298]
在某些情况下,使用抗体片段而不是完整抗体是有利的。片段的较小尺寸允许快速清除,且可以导致更易接近实体瘤。
[0299]
已发展了多种技术来产生抗体片段。通常,通过完整抗体的蛋白水解消化来衍生这些片段(见例如morimoto等,journal of biochemical and biophysical methods 24:107-117(1992)和brennan等,science,229:81(1985))。但是,现在可以通过重组宿主细胞直接产生这些片段。例如,可以从上文讨论的抗体噬菌体文库分离抗体片段。备选地,可以从大肠杆菌直接回收fab
’‑
sh片段,并化学偶联来形成f(ab’)2片段(carter等,bio/technology 10:163-167(1992))。根据另一种方法,可以从重组宿主细胞培养物直接分离f(ab’)2片段。用于产生抗体片段的其他技术对熟练的从业者而言显而易见。在其他实施方案中,所选择的抗体是单链fv片段(scfv)。见wo 93/16185;美国专利号5,571,894;及美国专利号5,587,458。抗体片段还可以是描述于例如美国专利号5,641,870中的“线性抗体”。这类线性抗体片段可以是单特异性的或双特异性的。
[0300]
6.双特异性抗体
[0301]
双特异性抗体是对至少两个不同表位具有结合特异性的抗体。示例性双特异性抗体可以结合本文所述的β7整联蛋白的两个不同表位。其他这类抗体可以将tat结合部位与用于另一蛋白质的结合部位组合。备选地,抗-β7整联蛋白臂可以与结合白细胞上的触发分子(如t细胞受体分子(例如cd3),或igg的fc受体(fcγr),如fcγri(cd64)、fcγrii(cd32)和fcγriii(cd16))的臂组合,以将细胞防御机制集中和定位在表达tat的细胞。双特异性抗体还可以用来将细胞毒剂定位至表达tat的细胞。这些抗体具有tat结合臂和结合细胞毒剂(例如肥皂草毒蛋白、抗干扰素-α、长春花生物碱、蓖麻毒蛋白a链、氨甲喋呤或放射性同位素半抗原)的臂。双特异性抗体可以作为全长抗体或抗体片段(例如f(ab’)2双特异性抗体)制备。
[0302]
用于制备双特异性抗体的方法为本领域已知。全长双特异性抗体的传统产生基于两个免疫球蛋白重链-轻链对的共表达,其中两条链具有不同的特异性(millstein等,nature 305:537-539(1983))。由于免疫球蛋白重链和轻链的随机分配,这些杂交瘤(quadromas)产生10种不同抗体分子的可能的混合物,其中只有一种具有正确的双特异性结构。正确分子的纯化(通常通过亲和层析步骤进行)非常麻烦,且产物产率低。wo 93/08829和traunecker等,embo j.10:3655-3659(1991)中公开了类似的方法。
[0303]
根据不同的方法,将具有希望的结合特异性(抗体-抗原结合部位)的抗体可变结构域与免疫球蛋白恒定结构域序列融合。在某些实施方案中,该融合是与包含至少部分铰链区、ch2区和ch3区的ig重链恒定结构域融合。在某些实施方案中,包含轻链结合所必需的部位的第一重链恒定区(c
h1
)存在于该融合的至少一个中。将编码免疫球蛋白重链融合物和(如果希望)免疫球蛋白轻链的dna插入分开的表达载体,并共转染入适宜的宿主生物。在用于构建的不等比例的三条多肽链提供希望的双特异性抗体的最佳产率时,这为在实施方案中调整三个多肽片段的相互比例提供了极大的灵活性。但是,在表达等比例的至少两种多肽链导致高产率时,或在该比例对希望的链组合的产率无显著影响时,可能将两种或全部三种多肽链的编码序列插入单个表达载体中。
[0304]
在某些实施方案中,该双特异性抗体由一条臂中具有第一结合特异性的杂合免疫球蛋白重链和另一条臂中的杂合免疫球蛋白重链-轻链对(提供第二结合特异性)组成。发现此不对称结构便于从不想要的免疫球蛋白链组合分离希望的双特异性化合物,因为免疫球蛋白轻链仅存在于该双特异性分子的一半中提供了容易的分离方式。此方法公开于wo 94/04690中。产生双特异性抗体的进一步细节见例如suresh等,methods in enzymology121:210(1986)
[0305]
根据美国专利号5,731,168中所述的另一种方法,可以改造抗体分子对间的界面来最大化从重组细胞培养物回收的异二聚体的百分比。在某些实施方案中,该界面至少包含部分c
h3
结构域。在此方法中,用更大的侧链(例如酪氨酸或色氨酸)取代来自第一抗体分子的界面的一个或多个小的氨基酸侧链。通过用更小的氨基酸侧链(例如丙氨酸或苏氨酸)取代大的氨基酸侧链来在第二抗体分子的界面上产生与一个或多个大的侧链相同或相似大小的补偿“空洞”。这提供了增加异二聚体的产率超过其他不想要的终产物(如同二聚体)的机制。
[0306]
双特异性抗体包括交联抗体或“异缀合物”抗体。例如,异缀合物中的抗体之一可以与抗生物素蛋白偶联,另一种与生物素偶联。例如,已提出这类抗体将免疫系统细胞靶向至不需要的细胞(美国专利号4,676,980),并用于治疗hiv感染(wo 91/00360、wo 92/200373和ep 03089)。可以用任意方便的交联方法产生异缀合物抗体。适宜的交联剂为本领域公知,并连同许多交联技术一起公开于美国专利号4,676,980中。
[0307]
还已在文献中描述了用于从抗体片段产生双特异性抗体的技术。例如,可以用化学连接制备双特异性抗体。brennan等,science 229:81(1985)描述了蛋白酶解切割完整抗体来产生f(ab')2片段的方法。在二巯基络合剂亚砷酸钠的存在下还原这些片段,以稳定邻位二巯基并防止分子间二硫化物形成。然后将产生的fab’片段转化为硫代硝基苯甲酸盐(tnb)衍生物:然后通过用巯基乙胺还原来将fab
’‑
tnb衍生物之一再转化为fab
’‑
巯基,并与等摩尔量的其他fab
’‑
tnb衍生物混合来形成双特异性抗体。可以将产生的双特异性抗体用作用于选择性固定酶的物质。
[0308]
最近的进展已便于从大肠杆菌直接回收fab'-sh片段,该片段可以化学偶联来形成双特异性抗体。shalaby等,j.exp.med.175:217-225(1992)描述了充分人源化的双特异性抗体f(ab')2分子的产生。每个fab'片段分别从大肠杆菌分泌,并在体外进行定向化学偶联来形成双特异性抗体。这样形成的双特异性抗体能够结合过量表达erbb2受体的细胞和正常的人t细胞,以及触发人细胞毒性淋巴细胞对人乳腺肿瘤靶标的裂解活性。还已描述了
用于直接从重组细胞培养物制备和分离双特异性抗体片段的多种技术。例如,已用亮氨酸拉链产生了双特异性抗体。kostelny等,j.immunol.148(5):1547-1553(1992)。通过基因融合将来自fos和jun蛋白质的亮氨酸拉链肽与两种不同抗体的fab’部分连接。在铰链区还原抗体同二聚体形成单体,然后再氧化形成抗体异二聚体。还可以利用此方法来产生抗体同二聚体。hollinger等,proc.natl.acad.sci.usa 90:6444-6448(1993)所述的“双抗体”技术提供了用于制备双特异性抗体片段的备选机制。片段包含通过接头与v
l
连接的vh,该接头太短而不允许同一条链上的两个结构域间配对。因此,迫使一个片段的vh和v
l
结构域与另一片段的互补的v
l
和vh结构域配对,从而形成两个抗原结合部位。还已报道了通过使用单链fv(sfv)二聚体来制备双特异性抗体片段的另一策略。见gruber等,j.immunol.152:5368(1994)。
[0309]
考虑超过二价的抗体。例如,可以制备三特异性抗体。tutt等,j.immunol.147:60(1991)。
[0310]
7.异缀合物抗体
[0311]
异缀合物抗体也在本发明的范围之内。异缀合物抗体由两个共价结合的抗体组成。例如,已提出这类抗体将免疫系统细胞靶向至不需要的细胞[美国专利号4,676,980],并用于治疗hiv感染[wo 91/00360;wo92/200373;ep 03089]。考虑用合成蛋白质化学中已知的方法(包括涉及交联剂的那些)在体外制备该抗体。例如,可以使用二硫键交换反应或通过形成硫醚键来构建免疫毒素。适合用于此目的的试剂的实例包括亚氨基硫醇盐和甲基-4-巯基butyrimidate和例如美国专利号4,676,980中公开的那些。
[0312]
8.多价抗体
[0313]
多价抗体可以比二价抗体更快地被表达该抗体所结合的抗原的细胞内化(和/或分解代谢)。本发明的抗体可以是(除igm种类外的)具有三个或多个抗原结合部位的多价抗体(例如四价抗体),其可以容易地通过重组表达编码该抗体的多肽链的核酸来制备。该多价抗体可以包含二聚化结构域和三个或多个抗原结合部位。在某些实施方案中,该二聚化结构域包含fc区或铰链区(或由fc区或铰链区组成)。在这种情况下,该抗体将包含fc区及fc区氨基端的三个或多个抗原结合部位。在某些实施方案中,本文的多价抗体包含三个至约八个,但通常四个抗原结合部位(或由三个至约八个,但通常四个抗原结合部位组成)。该多价抗体包含至少一条多肽链(且通常两条多肽链),其中该一条或多条多肽链包含两个或多个可变结构域。例如,该一条或多条多肽链可以包含vd1-(x1)n-vd2-(x2)n-fc,其中vd1是第一可变结构域,vd2是第二可变结构域,fc是fc区的一条多肽连,x1和x2代表氨基酸或多肽,n是0或1。例如,该一条或多条多肽链可以包含:vh-ch1-柔性接头-vh-ch1-fc区链;或vh-ch1-vh-ch1-fc区链。本文的多价抗体可以进一步包含至少两条(且通常四条)轻链可变结构域多肽。例如,本文的多价抗体可以包含约两条至约八条轻链可变结构域多肽。此处考虑的轻链可变结构域多肽包含轻链可变结构域,且可选地进一步包含cl结构域。
[0314]
9.效应子功能改造
[0315]
可以希望就效应子功能修饰本发明的抗体,例如,以增强该抗体的依赖抗原的细胞毒性(adcc)和/或依赖补体的细胞毒性(cdc)。这可以通过在该抗体的fc区中引入一个或多个氨基酸取代来达到。备选地或此外,可以在fc区中引入一个或多个半胱氨酸残基,从而允许在此区域中形成链间二硫键。这样产生的同二聚体抗体可以具有改善的内化能力和/
或提高的补体介导的细胞杀伤和抗体依赖性细胞毒作用(adcc)。见caron等,j.exp med.176:1191-1195(1992)和shopes,b.j.,immunol.148:2918-2922(1992)。还可以用wolff等,cancer research 53:2560-2565(1993)中所述的异双功能交联剂来制备具有增强的抗肿瘤活性的同二聚体抗体。备选地,可以改造抗体,使其具有双fc区,从而可以具有增强的补体裂解和adcc能力。见stevenson等,anti-cancer drug design3:219-230(1989)。为了增加抗体的血清半衰期,可以按例如美国专利号5,739,277中所述在抗体(尤其是抗体片段)中掺入挽救受体结合表位。本文所用的术语“挽救受体结合表位”指igg分子(例如igg1、igg2、igg3或igg4)的fc区的表位,其负责增加igg分子的体内血清半衰期。
[0316]
10.免疫缀合物
[0317]
用于本文的方法的拮抗剂或抗体可选地缀合至另一物质,如细胞毒剂或细胞因子。
[0318]
缀合通常将通过共价连接实现,其确切的性质将由靶向分子和整联蛋白β7拮抗剂或抗体多肽上的连接位点决定。通常,通过加入接头来修饰非肽试剂,该接头允许通过抗-β7整联蛋白抗体的氨基酸侧链、碳水化合物链或者通过化学修饰引入抗体上的反应基而缀合到该抗体。例如,药物可以通过赖氨酸残基的ε氨基、通过自由α氨基、通过与半胱氨酸残基的二硫键交换、或者通过用高碘酸氧化碳水化合物链中的1,2-二醇以允许通过希夫碱连接附着含有多种亲核试剂的药物来进行附着。见例如美国专利号4,256,833。蛋白质修饰剂包括胺反应性试剂(例如,反应性酯、异硫氰酸酯、醛和磺酰卤)、硫醇反应性试剂(例如,卤代乙酰基衍生物和马来酰亚胺)及羧酸和醛反应性试剂。整联蛋白β7拮抗剂或抗体多肽可以通过使用双功能交联剂来共价连接到肽试剂。异双功能试剂更常用,且允许通过使用两种不同的反应性部分(例如胺反应性加上硫醇、碘乙酰胺或马来酰亚胺)来控制两种不同蛋白质的偶联。这类连接试剂的用途为本领域公知。见例如brinkley,上文和美国专利号4,671,958。也可以使用肽接头。在备选方案中,抗β7整联蛋白抗体多肽可以通过融合多肽的制备连接到肽部分。
[0319]
其他双功能蛋白质偶联剂的实例包括n-琥珀酰亚胺基-3-(2-吡啶基二硫基)丙酸酯(spdp)、琥珀酰亚胺基-4-(ν-马来酰亚胺基甲基)环己烷-1-羧酸酯、亚氨基硫烷(it)、亚胺酯的双功能衍生物(如二甲基己二酸hcl)、活性酯(如辛二酸二琥珀酰亚胺酯)、醛(如戊二醛)、二-叠氮基化合物(如二(对-叠氮基苯甲酰基)己二胺)、二-重氮衍生物(如二-(对-重氮基苯甲酰基)-乙二胺)、二异氰酸酯(如2,6-二异氰酸甲苯酯)和双活性氟化合物(如1,5-二氟-2,4-二硝基苯)。
[0320]
11.免疫脂质体
[0321]
本文公开的抗β7整联蛋白抗体也可以配制为免疫脂质体。“脂质体”是由多种类型的脂质、磷脂和/或表面活性剂组成的小囊,其用于将药物递送至哺乳动物。脂质体的成分通常以双层形式排列,类似于生物膜的脂质排列。通过本领域已知的方法来制备含有抗体的脂质体,如epstein等,proc.natl.acad.sci.usa 82:3688(1985);hwang等,proc.natl acad.sci.usa77:4030(1980);美国专利号4,485,045和4,544,545;及1997年10月23日公开的wo97/38731中所述。美国专利号5,013,556中公开了具有增强的循环时间的脂质体。
[0322]
可以用包含磷脂酰胆碱、胆固醇和peg-衍生的磷脂酰乙醇胺(peg-pe)的脂质组合物,通过反相蒸发法来产生尤其有用的脂质体。将脂质体通过确定孔径的滤器挤出以产生
具有所希望直径的脂质体。
[0323]
可以按martin等,j.biol.chem.257:286-288(1982)中所述通过二硫键交换反应来将本发明抗体的fab'片段缀合至脂质体。化疗剂可选地包含在脂质体内。见gabizon等,j.national cancer inst.81(19):1484(1989)。
[0324]
12.用于抗体产生的载体、宿主细胞和重组方法
[0325]
还提供分离的编码本文所述抗-β7抗体或多肽试剂的核酸、包含该核酸的载体和宿主细胞及用于产生该抗体的重组技术。
[0326]
为了重组产生抗体,可以分离编码它的核酸,并插入可复制载体用于进一步克隆(dna的扩增)或用于表达。在另一实施方案中,可以通过例如美国专利号5,204,244(在此明确引入作为参考)中所述的同源重组来产生抗体。编码单克隆抗体的dna易于用常规方法分离和测序(例如,通过使用能够特异性结合编码抗体的重链和轻链的基因的寡核苷酸探针)。许多载体可用。载体成分一般包括但不限于以下一种或多种:信号序列、复制起点、一种或多种标记基因、增强子元件、启动子和转录终止序列,例如,如1996年7月9日授权的美国专利号5,534,615中所述,其在此明确引入作为参考。
[0327]
适合用于克隆或表达本文的载体中的dna的宿主细胞是上文所述的原核生物、酵母或高级真核生物细胞。适合用于此目的的原核生物包括真细菌,如革兰氏阴性或革兰氏阳性生物,例如肠杆菌科(enterobacteriaceae),如埃希氏菌属(escherichia),例如大肠杆菌,肠杆菌属(enterobacter),欧文氏菌属(erwinia),克雷伯氏菌属(klebsiella),变形杆菌属(proteus),沙门氏菌属(salmonella),例如,鼠伤寒沙门氏菌(salmonella typhimurium),沙雷氏菌属(serratia),例如serratia marcescans,志贺氏菌属(shigella),以及芽抱杆菌属(bacilli),如枯草芽孢杆菌(b.subtilis)和地衣芽孢杆菌(b.iicheniformis)(例如,1989年4月12日公开的dd 266,710中公开的地衣芽孢杆菌41p),假单胞菌属(pseudomonas),如铜绿假单胞菌(p.aeruginosa),和链霉菌属(streptomyces)。一个大肠杆菌克隆宿主是大肠杆菌294(atcc 31,446),虽然诸如大肠杆菌b、大肠杆菌x1776(atcc 31,537)和大肠杆菌w3110(atcc 27,325)的其他菌株也适宜。这些实例是说明性的而非限制性的。
[0328]
除原核生物外,诸如丝状真菌或酵母的真核微生物也是适合用于编码抗-β7整联蛋白抗体的载体的克隆或表达宿主。酿酒酵母(saccharomyces cerevisiae)或常见的面包酵母是低级真核宿主微生物中最常用的。但是,许多其他属、种和菌株是通常可得到的,并在本文中使用,如粟酒裂殖酵母(schizosaccharomyces pombe);克鲁维酵母属(kluyveromyces)宿主,例如乳酸克鲁维酵母(k.iactis)、脆壁克鲁维酵母(k.fragilis)(atcc 12,424)、保加利亚克鲁维酵母(k.bulgaricus)(atcc 16,045)、威克克鲁维酵母(k.wickeramii)(atcc 24,178)、瓦尔提克鲁维酵母(k.waltii)(atcc 56,500)、果蝇克鲁维酵母(k.drosophilarum)(atcc 36,906)、耐热克鲁维酵母(k.thermotolerans)和马克思克鲁维酵母(k.marxianus);耶氏酵母属(yarrowia)(ep 402,226);巴斯德毕赤酵母(pichia pastoris)(ep 183,070);假丝酵母属(candida);里氏木霉(trichoderma reesia)(ep 244,234);粗糙链孢霉(neurospora crassa);许旺酵母属(schwanniomyces),如许旺酵母(schwanniomyces occidentalis);和丝状真菌,例如脉孢霉属(neurospora)、青霉属(penicillium)、弯颈霉属(tolypocladium)和曲霉属(aspergillus)宿主,如构巢曲
霉(a.nidulans)和黑曲霉(a.niger)。
[0329]
适合用于表达糖基化的抗-β7抗体的宿主细胞衍生自多细胞生物。无脊椎动物细胞的实例包括植物细胞和昆虫细胞。已鉴定了来自诸如草地夜蛾(spodoptera frugiperda)(毛虫)、埃及伊蚊(aedes aegypti)(蚊子)、白纹伊蚊(aedes albopictus)(蚊子)、黑腹果蝇(drosophila melanogaster)(果蝇)和家蚕(bombyx mori)的宿主的许多杆状病毒株和变体及对应的受纳昆虫宿主细胞。多种用于转染的病毒株是公开可得的,例如苜蓿银纹夜蛾(autographa californica)npv的l-1变体和家蚕npv的bm-5病毒株,且可以将这类病毒用作本发明的病毒,尤其是用于转染草地夜蛾细胞。棉花、玉米、马铃薯、大豆、矮牵牛、西红柿和烟草的植物细胞培养物也可以用作宿主。
[0330]
但是,在脊椎动物细胞中的兴趣最大,且在培养物(组织培养物)中繁殖脊椎动物细胞已成为常规方法。有用的哺乳动物宿主细胞系的实例是sv40转化的猴肾cv1细胞系(cos-7,atcc crl 1651);人胚肾细胞系(293或为在悬浮培养物中生长而亚克隆的293细胞,graham等,j.gen virol.36:59(1977));幼仓鼠肾细胞(bhk,atcc ccl 10);中国仓鼠卵巢细胞/-dhfr(cho,urlaub等,proc.natl.acad.sci.usa 77:4216(1980));小鼠支持细胞(tm4,mather,biol.reprod.23:243-251(1980));猴肾细胞(cvl atcc ccl 70);非洲绿猴肾细胞(vero-76,atcc crl-1587);人宫颈癌细胞(hela,atcc ccl 2);犬肾细胞(mdck,atcc ccl 34);buffalo大鼠肝细胞(brl 3a,atcc crl 1442);人肺细胞(w138,atcc ccl 75);人肝细胞(hep g2,hb 8065);小鼠乳腺肿瘤(mmt 060562,atcc ccl51);tri细胞(mather等,annals n.y.acad.sci.383:44-68(1982));mrc 5细胞;fs4细胞;和人肝癌细胞系(hep g2)。
[0331]
用上述用于抗-β7整联蛋白抗体产生的表达或克隆载体转化宿主细胞,并在根据诱导启动子、选择转化体或扩增编码希望的序列的基因的需要改变的常规营养培养基中培养。
[0332]
可以在多种培养基中培养用于产生本发明的抗-β7整联蛋白抗体的宿主细胞。诸如ham's f10(sigma)、minimal essential medium(mem)(sigma)、rpmi-1640(sigma)和dulbecco's modified eagle's medium(dmem,sigma)的市售培养基适合用于培养宿主细胞。此外,可以将ham等,meth.enz.58:44(1979);barnes等,anal.biochem.1 02:255(1980);美国专利号4,767,704;4,657,866;4,927,762;4,560,655;或5,122,469;wo 90/03430;wo 87/00195;或美国专利公开30,985中所述的培养基中的任意种用作宿主细胞的培养基。可以根据需要向任意这些培养基中补充激素和/或其他生长因子(如胰岛素、转铁蛋白或表皮生长因子)、盐(如氯化钠、钙、镁和磷酸盐)、缓冲液(如hepes)、核苷酸(如腺苷和胸苷)、抗生素(如gentamycin.tm.药物)、痕量元素(定义为通常以微摩尔范围内的终浓度存在的无机化合物)和葡萄糖或等同的能量来源。还可以按本领域技术人员已知的适当浓度包含任意其他必需的补充物。诸如温度、ph等的培养条件是之前用于所选择的表达宿主细胞的那些,且对普通技术人员而言显而易见。
[0333]
在使用重组技术时,抗体可以在胞内产生,产生在周质空间中,或者直接分泌进入培养基。如果抗体在胞内产生,作为第一步骤,例如通过离心或超滤去除颗粒碎片(宿主细胞或裂解片段)。carter等,bio/technology10:163-167(1992)描述了用于分离分泌至大肠杆菌的周质空间的抗体的方法。简言之,在乙酸钠(ph 3.5)、edta和苯甲基磺酰氟(pmsf)的
存在下融化细胞糊约30分钟。通过离心去除细胞碎片。在抗体分泌进入培养基时,通常首先用市售蛋白质浓缩滤器(例如amicon或millipore pellicon超滤单元)浓缩来自这类表达系统的上清。可以在任意前述步骤中包含蛋白酶抑制剂(如pmsf)来抑制蛋白水解,且可以包含抗生素来防止外来污染物的生长。
[0334]
可以用例如羟基磷灰石层析、凝胶电泳、透析和亲和层析纯化从细胞制备的抗体组合物,亲和层析是典型的纯化技术。a蛋白作为亲和配体的适合性取决于抗体中存在的任意免疫球蛋白fc结构域的种类和同种型。可以用a蛋白来纯化基于人γ1、γ2或γ4重链的抗体(lindmark等,j.immunol.meth.62:1-13(1983))。建议g蛋白用于所有小鼠同种型和人γ3(guss等,embo j.5:15671575(1986))。亲和配体所附着的基质最常是琼脂糖,但其他基质也可用。与用琼脂糖可以达到的流速和处理时间相比,机械稳定的基质(如可控孔度玻璃或聚(苯乙烯二乙烯)苯)允许更快的流速和更短的处理时间。在抗体包含c
h3
结构域时,用bakerbond abx
tm
树脂(j.t.baker,phillipsburg,nj)进行纯化。取决于待回收的抗体,用于蛋白质纯化的其他技术(如离子交换柱上的分级分离、乙醇沉淀、反相hplc、二氧化硅上的层析、肝素sepharose
tm
上的层析、阴离子或阳离子交换树脂(如聚天冬氨酸柱)上的层析、层析聚焦、sds-page和硫酸铵沉淀)也可用。任意一个或多个初步纯化步骤后,可以对包含目的抗体和污染物的混合物进行低ph疏水作用层析,该层析使用ph在约2.5-4.5之间的洗脱缓冲液,通常在低盐浓度(例如约0-0.25m盐)下进行。
[0335]
c.药物制剂
[0336]
通过将具有希望的纯度的抗体与可选的生理可用载体、赋形剂或稳定剂(remington's pharmaceutical sciences第16版,osol,a.编辑(1980))混合,以水溶液、冻干或其他干燥制剂的形式制备包含本发明的治疗剂、拮抗剂或抗体的治疗制剂用于保存。可用载体、赋形剂或稳定剂在所用的剂量和浓度下对受体无毒性,且包括缓冲剂,如磷酸盐、柠檬酸盐、组氨酸和其他有机酸;抗氧化剂,包括抗坏血酸和甲硫氨酸;防腐剂(如十八烷基二甲基苄基氯化铵;氯化六甲双铵;苯扎氯铵、苄索氯铵;苯酚、丁醇或苯甲醇;对羟基苯甲酸烷基酯,如对羟基苯甲酸甲酯或丙酯;儿茶酚;间苯二酚;环己醇;3-戊醇;和间甲酚);低分子量(小于约10个残基)多肽;蛋白质,如血清白蛋白、明胶或免疫球蛋白;亲水聚合物,如聚乙烯吡咯烷酮;氨基酸,如甘氨酸、谷氨酰胺、天冬酰胺、组氨酸、精氨酸或赖氨酸;单糖、二糖和其他糖类,包括葡萄糖、甘露糖或糊精;螯合剂,如edta;糖,如蔗糖、甘露醇、海藻糖或山梨醇;成盐抗衡离子,如钠;金属络合物(例如,zn-蛋白质络合物);和/或非离子型表面活性剂,如tween.tm.、pluronics.tm.或聚乙二醇(peg)。
[0337]
本文的制剂还可以包含所治疗的具体适应症必需的一种以上活性化合物,通常是相互无不利影响的具有互补活性的那些。这类分子适宜地以对预期目的有效的量组合存在。
[0338]
活性成分还可以包载在例如通过凝聚技术或通过界面聚合制备的微囊(例如,分别为羟甲基纤维素微囊或明胶微囊和聚-(甲基丙烯酸甲酯)微囊)中、胶体药物递送系统(例如脂质体、白蛋白微球、微乳、纳米颗粒和纳米囊(nanocapsule))中或粗滴乳状液中。这类技术公开于remington's pharmaceutical sciences第16版,osol,a.编辑(1980)中。
[0339]
待用于体内施用的制剂必须无菌。这易于通过滤过无菌滤膜来实现。
[0340]
可以制备缓释制剂。缓释制剂的适宜的实例包括含有本发明的免疫球蛋白的固体
疏水聚合物的半透性基质,该基质是成形物品的形式,例如膜或微囊。缓释基质的实例包括聚酯、水凝胶(例如聚(甲基丙烯酸-2-羟乙酯)或聚(乙烯醇))、聚交酯(美国专利号3,773,919)、l-谷氨酸和γ乙基-l-谷氨酸的共聚物、不可降解的乙烯-乙酸乙烯酯、可降解的乳酸-乙醇酸共聚物如lupron depot.tm.(由乳酸-乙醇酸共聚物和醋酸亮丙瑞林组成的可注射微球)和聚-d-(-)-3-羟基丁酸。诸如乙烯-乙酸乙烯酯和乳酸-乙醇酸的聚合物使得能够释放分子超过100天,而某些水凝胶释放蛋白质较短时期。包封的免疫球蛋白长时间存留在体内时,它们可由于在37℃暴露于湿度而变性或聚集,导致生物学活性的丧失和可能的免疫原性改变。取决于所涉及的机制,可以设计合理的策略来稳定。例如,如果发现聚集机制是通过硫代二硫化物(thio-disulfide)交换形成分子间s-s键,那么可以通过修饰硫氢残基、从酸性溶液冻干、控制含水量、使用适当的添加剂和发展特异性聚合物基质组合物来达到稳定。
[0341]
d.施用
[0342]
实施治疗的医生将能够针对基于体重的剂量或针对固定剂量确定单个个体的适当剂量,将遵循标签上的说明。与整联蛋白β7拮抗剂组合施用的市售第二治疗剂和其他化合物的制备和施用方案可以按厂家的说明使用或由熟练的从业人员按经验确定。
[0343]
对于疾病的预防或治疗,整联蛋白β7拮抗剂及与非排除抗体组合施用的任意第二治疗剂或其他化合物的适当剂量将取决于待治疗的胃肠炎性障碍的类型(例如ibd、uc、cd)、疾病的严重度和病程、为了预防还是治疗的目的而施用该整联蛋白β7拮抗剂或组合、之前的治疗、患者的临床病史和对整联蛋白β7拮抗剂或组合的反应及主治医生的判断。在某些实施方案中,每周一次、或每两周一次、或每四周一次、或每六周一次、或每八周一次施用整联蛋白β7拮抗剂,施用一个月(4周)、或两个月、三个月、或六个月、或12个月、或18个月、或24个月、或在患者一生中长期施用。在某些实施方案中,治疗由患者自己施用。
[0344]
取决于疾病的类型和严重度,约0.5mg/kg至4.0mg/kg抗-β7抗体是对患者施用的初始候选剂量,例如,通过一次或多次分开施用,或通过连续输注。对于在几天或更长时间内反复施用,取决于病症,治疗持续至出现希望的疾病症状的抑制。但是,可以使用其他给药方案。
[0345]
例如,在某些实施方案中,对患者施用固定剂量的抗-β7抗体。固定剂量是不考虑体重而对每名患者施用的抗-β7抗体的具体量。取决于疾病的类型和严重度,对患者施用约50mg和450mg之间的抗-β7抗体的固定剂量,其可以是一次或多次分开的注射或输注或施用。这种固定剂量可以静脉内或皮下或通过本文所述的其他途径施用。在某些实施方案中,该固定剂量是50mg、或100mg、或150mg、或200mg、或300mg、或400mg、或420mg、或450mg。
[0346]
在某些实施方案中,抗-β7抗体的起始统一负荷剂量后跟随抗-β7抗体的一个或多个维持剂量。该负荷剂量是比维持剂量更大量的抗-β7抗体。在某些实施方案中,该负荷剂量在400mg和450mg之间,该维持剂量在50mg和350mg之间。在某些实施方案中,该负荷剂量是400mg、或420mg、或430mg、或450mg。在某些实施方案中,该维持剂量是50mg、或100mg、或150mg、或200mg、或300mg、或350mg。在某些实施方案中,该负荷剂量是400mg,该维持剂量是50mg。在某些实施方案中,该负荷剂量是400mg,该维持剂量是100mg。在某些实施方案中,该负荷剂量是400mg,该维持剂量是150mg。在某些实施方案中,该负荷剂量是400mg,该维持剂量是200mg。在某些实施方案中,该负荷剂量是400mg,该维持剂量是300mg。在某些实施方案
中,该负荷剂量是400mg,该维持剂量是350mg。在某些实施方案中,该负荷剂量是420mg,该维持剂量是50mg。在某些实施方案中,该负荷剂量是420mg,该维持剂量是100mg。在某些实施方案中,该负荷剂量是420mg,该维持剂量是150mg。在某些实施方案中,该负荷剂量是420mg,该维持剂量是200mg。在某些实施方案中,该负荷剂量是420mg,该维持剂量是300mg。在某些实施方案中,该负荷剂量是420mg,该维持剂量是350mg。在某些实施方案中,该负荷剂量是430mg,该维持剂量是50mg。在某些实施方案中,该负荷剂量是430mg,该维持剂量是100mg。在某些实施方案中,该负荷剂量是430mg,该维持剂量是150mg。在某些实施方案中,该负荷剂量是430mg,该维持剂量是200mg。在某些实施方案中,该负荷剂量是430mg,该维持剂量是300mg。在某些实施方案中,该负荷剂量是430mg,该维持剂量是350mg。在某些实施方案中,该负荷剂量是450mg,该维持剂量是50mg。在某些实施方案中,该负荷剂量是450mg,该维持剂量是100mg。在某些实施方案中,该负荷剂量是450mg,该维持剂量是150mg。在某些实施方案中,该负荷剂量是450mg,该维持剂量是200mg。在某些实施方案中,该负荷剂量是450mg,该维持剂量是300mg。在某些实施方案中,该负荷剂量是450mg,该维持剂量是350mg。
[0347]
在某些实施方案中,与该负荷剂量同一天施用第一维持剂量。在某些实施方案中,在该负荷剂量后一天施用第一维持剂量。在某些实施方案中,在该负荷剂量后一周施用第一维持剂量。在某些实施方案中,在该负荷剂量后两周施用第一维持剂量。在某些实施方案中,在该负荷剂量后四周施用第一维持剂量。在某些实施方案中,每两周施用第二及每个后续维持剂量。在某些实施方案中,每四周施用第二及每个后续维持剂量。在某些实施方案中,每八周施用第二及每个后续维持剂量。在某些实施方案中,在该负荷剂量后两周施用第一维持剂量,在该第一维持剂量后两周施用第二维持剂量,每四周施用每个后续维持剂量。在某些实施方案中,施用该抗-β7抗体2个月的时间。在某些实施方案中,施用该抗-β7抗体3个月的时间。在某些实施方案中,施用该抗-β7抗体6个月的时间。在某些实施方案中,施用该抗-β7抗体12个月的时间。在某些实施方案中,施用该抗-β7抗体18个月的时间。在某些实施方案中,施用该抗-β7抗体24个月的时间。在某些实施方案中,该个体终生施用该抗-β7抗体。
[0348]
通常,临床医生将施用本发明的抗体(单独或与第二化合物组合),直至达到提供所需生物学作用的一个或多个剂量。容易地通过常规技术和测定来监测本发明的疗法的进展。
[0349]
可以通过任意适宜的手段来施用整联蛋白β7拮抗剂,包括胃肠外、局部、静脉内、皮下、腹膜内、肺内、鼻内和/或病灶内施用。胃肠外输注包括肌内、静脉内、动脉内、腹膜内或皮下施用。还考虑鞘内施用(见例如grillo-lopez的美国专利公开号2002/0009444)。此外,可以通过例如用剂量逐渐降低的抗体脉冲输注来适宜地施用整联蛋白β7拮抗剂。在某些实施方案中,静脉内或皮下给药。可以用相同或不同的施用手段来提供每个暴露。在一个实施方案中,通过皮下施用来提供抗-β7抗体的每个暴露。在一个实施方案中,通过静脉内施用来提供抗-β7抗体的第一暴露(例如负荷剂量),通过皮下施用来提供每个后续暴露。
[0350]
在某些实施方案中,用例如自注射装置、自动注射装置或设计用于自我施用的其他装置来施用抗-β7抗体。包括自动注射装置的多种自注射装置为本领域已知,且是市售的。示例性装置包括但不限于预装注射器(如来自becton dickinson的bd hypakreadyfilltm和sterifill scftm;来自baxter的clearshottm共聚物预装注射器;及可从
west pharmaceutical service获得的daikyo seiko crystal预装注射器);一次性笔注射装置,如来自becton dickinson的bd pen;超锋利(ultra-sharp)和微针装置(如来自becton dickinson的inject-easetm和微输注器;可从valeritas获得的h-patchtm);以及无针注射装置(如可从bioject获得的和可从medtronic获得的和贴剂装置)。在某些实施方案中,rhumabβ7是包含含有2ml(150mg)rhumabβ7的预装注射器的制品。在某些实施方案中,rhumabβ7是包含含有1ml(180mg)rhumabβ7的预装注射器的制品。
[0351]
如所指出,整联蛋白β7拮抗剂可以单独施用或与至少第二治疗化合物组合施用。这些第二治疗化合物通常以之前所用的剂量和给药途径相同的剂量和给药途径使用,或之前所用剂量的约1%至99%。如果使用这类第二化合物,则它们在某些实施方案中以低于不存在整联蛋白β7拮抗剂时的量使用,以消除或降低因此引起的副作用。
[0352]
同样如所指出(例如,见下文),多种适合用于治疗ibd(例如溃疡性结肠炎和克隆病)的第二治疗化合物为本领域已知,同样描述了这类第二治疗化合物的剂量和施用方法。
[0353]
整联蛋白β7拮抗剂和任意第二治疗化合物的施用可以同时进行,例如作为单种组合物或作为使用相同或不同给药途径的两种或多种不同组合物。备选地,或此外,施用可以以任意顺序顺次进行。在某些实施方案中,两种或多种组合物的施用之间可以存在从几分钟至几天、几周至几个月的间隔。例如,可以先施用整联蛋白β7拮抗剂,然后施用第二治疗化合物。但是,也考虑同时施用或在整联蛋白β7拮抗剂之前施用第二治疗化合物。
[0354]
活性中度至严重活性uc个体的治疗标准涉及用以下的标准剂量治疗:氨基水杨酸、口服皮质类固醇、6-巯基嘌呤(6-mp)和/或硫唑嘌呤。整联蛋白β7拮抗剂(如本文公开的抗-β7整联蛋白抗体)治疗将导致疾病缓解的改善(疾病的快速控制和/或延长的缓解),和/或优于用这类个体的治疗标准达到的临床反应的临床反应。
[0355]
在一个实施方案中,本发明的患有ibd的人个体中的炎性肠病(ibd)治疗包括对该个体施用有效量的治疗剂,如抗-β7整联蛋白抗体,且进一步包括对该个体施用有效量的第二药物,该药物是免疫抑制剂、疼痛控制剂、抗腹泻剂、抗生素或其组合。
[0356]
在示例性实施方案中,该第二药物选自氨基水杨酸、口服皮质类固醇、6-巯基嘌呤(6-mp)和硫唑嘌呤。在另一示例性实施方案中,该第二药物是另一整联蛋白β7拮抗剂,如另一抗-β7整联蛋白抗体或抗细胞因子的抗体。
[0357]
所有这些第二药物可以分别相互组合或它们自身与第一药物组合使用,使得本文所用的表述“第二药物”并非意味着它是除第一药物外唯一的药物。因此,第二药物无需是一种药物,但可以组成或包含一种以上这种药物。
[0358]
本文的组合施用包括用分开的制剂或单种药物制剂共同施用,及以任一顺序连续施用,其中通常存在两种(或所有)活性剂同时发挥其生物学活性的时期。
[0359]
第二药物的组合施用包括用分开的制剂或单种药物制剂共同施用(同时施用),及以任一顺序连续施用,其中通常存在两种(或所有)活性剂(药物)同时发挥其生物学活性的时期。
[0360]
e.设计治疗方案
[0361]
药物开发是复杂并且昂贵的过程。估计新药上市的费用在8亿到10亿美元之间。i
期临床试验中不到10%的药物进入批准期。药物在晚期失败的两个关键原因是缺乏对剂量-浓度反应和非预期的安全性事件之间的关系的理解。考虑到此情况,关键是有工具帮助预测药物在体内表现如何及协助临床治疗候选者的成功(lakshmi kamath,drug discovery and development;modeling success in pk/pd testing drug discovery&development(2006))。
[0362]
药物动力学(pk)表征药物的吸收、分布、代谢和清除特性。药效学(pd)定义对所施用的药物的生理学和生物学反应。pk/pd建模建立了这两种方法之间的数学和理论联系,并帮助更好地预测药物作用。通过模拟进行的集成pk/pd建模和计算机辅助试验设计正被整合到许多药物开发项目中,并具有不断增长的影响(lakshmi kamath,drug discovery and development;modeling success in pk/pd testing drug discovery&development(2006))。
[0363]
pk/pd测试通常在药物开发过程的每个阶段进行。因为开发正变得越来越复杂、费时和昂贵,许多公司正寻求更好地利用pk/pd数据,以在开始时消除有缺陷的候选物,并鉴定临床成功机会最高的那些候选物(lakshmi kamath,上文)。
[0364]
pk/pd建模方法在确定生物标记应答、药物水平和给药方案之间的关系中被证明是有用的。药物候选者的pk/pd谱和预测患者对它的应答的能力对于临床试验的成功是很关键。分子生物学技术中的最近进展和多种疾病靶标的更好理解已将生物标记验证为药物治疗功效的良好临床指标。生物标记测定法帮助鉴定对药物候选者的生物应答。一旦在临床上验证了生物标记,就可以对试验模拟有效建模。生物标记具有达到替代品状态的潜力,其可以在某天代替药物开发中的临床结果(lakshmi kamath,上文)。
[0365]
外周血中生物标记的量可用于鉴定对整联蛋白β7拮抗剂治疗的生物学反应,并因此可以作为候选物治疗的治疗功效的良好临床指标发挥功能。
[0366]
药物开发中的传统pk/pd建模定义诸如药物剂量浓度、药物暴露作用、药物半衰期、药物浓度对时间及药物作用对时间的参数。在更宽泛地使用时,诸如药物建模、疾病建模、实验建模和市场建模的定量技术可以支持整个开发过程,其通过风险的明确考虑和知识的更好利用导致更好的决策。对药物开发研究员而言,多种pk/pd建模工具可用,例如由pharsight,inc.mountain view,california开发的winnonlin and the knowledgebase server(pks)。
[0367]
认为前述书面说明和以下实施例足以使本领域技术人员能够实施本发明。除本文所显示和描述的那些之外,对本领域技术人员而言,本发明的多种修改将从前述描述和以下实施例变得显而易见,并落在所附权利要求的范围之内。
[0368]
应理解,按照本文所含的教导下,本发明的教导在具体问题或情况中的应用将在本领域普通技术人员的能力范围之内。
[0369]
通过以下非限制性实施例来阐明本发明的其他细节。说明书中所有引文的公开内容在此明确引入作为参考。
实施例
[0370]
实施例1
[0371]
在溃疡性结肠炎患者中评估在单剂量、剂量递增阶段后接着多剂量、平行治疗阶
段中施用的静脉内和皮下rhumabβ7的安全性、药物动力学、药效学和免疫原性的i期、多中心、随机化、安慰剂对照、双盲研究
[0372]
研究描述
[0373]
rhumabβ7(etrolizumab)的描述
[0374]
rhumabβ7(etrolizumab)是人源化单克隆抗体,其基于人igg1亚型iii vh、κ亚型-i v
l
共有序列,且特异性抗整联蛋白异二聚体的β7亚基。已显示它以高亲和力结合α4β7(约116pm的kd)和αeβ7(约1800pm的kd)。
[0375]
此重组抗体具有通过igg1抗体典型的链间和链内二硫键共价连接的两条重链(446个残基)和两条轻链(214个残基)。对于本文所述的研究,它是在中国仓鼠卵巢(cho)细胞中产生的。完整的非糖基化rhumabβ7分子的分子量约为144kda。rhumabβ7的每条重链在asn297处具有一个保守的n连接糖基化位点。存在于此位点上的寡糖是在cho细胞中表达的重组抗体中观察到的那些典型寡糖,主要的糖型是无唾液酸的双分枝g0和g1聚糖。包含两个g0聚糖且无c端赖氨酸残基的最普遍的rhumabβ7形式的质量约为147kda。
[0376]
rhumabβ7由genentech作为生理可用缓冲液中的澄清至轻微乳白、无色至微黄的水溶液提供。
[0377]
研究的总体设计
[0378]
人类中的首次i期研究由随机化(剂量群组内)、双盲、安慰剂对照、单剂量、剂量递增阶段后接着随机化(跨剂量群组)、双盲、安慰剂对照、多剂量、平行治疗阶段组成。接受rhumabβ7的参与研究的单剂量阶段的患者不允许参加多剂量阶段。但是,在研究的单剂量阶段接受安慰剂的患者允许参加多剂量阶段。48名溃疡性结肠炎患者参加此研究。患者需已诊断溃疡性结肠炎12周。研究在18-70岁的患者中进行,该患者已诊断为中度至严重溃疡性结肠炎,是门诊患者,且在基线时具有≥5点的mayo临床评分(mcs)。mcs的范围为0至12,越高的评分指示越严重的疾病活性(schroeder等,n.engl.j.med.317:1625-1629(1987);还见下文表1)。此研究中不包括由于其溃疡性结肠炎而全身不舒服且需要住院或手术的患者。此外,由研究人员考虑符合条件的患者成为生物制品疗法的候选者,因为他们患有不响应5-asa、免疫抑制剂(硫唑嘌呤或6-巯基嘌呤)或类固醇的标准疗法的中度至严重疾病。
[0379]
表1.用于评估溃疡性结肠炎活性的mayo临床评分系统
[0380][0381]a每名患者作为他或她自己的对照来建立大便频率的异常程度。
[0382]b每日出血评分代表该天最严重的出血。
[0383]c医生的总体评估承认三个其他标准,患者每天的腹部不适的记忆和健康的一般感觉,及其他观察结果,如体检发现和患者的体力状态。
[0384]
研究的单剂量剂量递增阶段的目的是在uc患者中评估rhumabβ7施用的安全性和耐受性。在此阶段,依次招募患者进入5个递增剂量群组(群组a-f;注意,省去了最初计划的接受1.0mg/kg sc剂量的群组e,但此处保留a-f的命名),并按四个剂量水平(0.3、1.0、3.0或10.0mg/kg)之一用单个静脉内(iv)或皮下(sc)剂量治疗(见表2)。
[0385]
计划每个群组招募5名患者。在每个群组内,随机分配患者来接受rhumab beta7或安慰剂。对于单剂量阶段,剂量递增决定基于通过iv途径接受研究药物的群组的临床安全性评价(群组a、b、c和d)。根据之前在人类中sc施用其他单克隆抗体的经验,预期sc施用后的暴露低于iv施用后所达到的暴露。
[0386]
预期在14天的安全性随访期内表征和可能解决任何潜在的急性毒性(例如过敏反应、输注反应或血清病型反应)。完成单剂量阶段中的给药后,在开始多剂量阶段之前评价安全性和药物动力学学(pk)数据。
[0387]
完成单剂量阶段群组f(3.0mg/kg sc)中治疗的最后一名患者的开始于给药后约10周的多剂量平行治疗阶段(群组g-j)。在这10周中,审查研究的单剂量阶段的pk和临床安全性数据。多剂量阶段的平行设计及所提出的剂量范围有来自单剂量阶段的最大耐受剂量(mtd)>10mg/kg的条件,并通过审查单剂量阶段中的pk数据来确认,以确保支持基于食蟹猕猴研究的预期的人药物动力学(见下文)。多剂量阶段中每4周施用的4.0mg/kg iv的最大剂量三个循环处于从食蟹猕猴毒理学数据确定的安全系数之内(国际专利公开号wo2009/140684;stefanich等,br.j.pharmacol.,已接收文章,doi:10.1111/j.1476-5381.2011.01205.x),基于人等同剂量(hed)具有16倍的安全系数,基于通过曲线下面积(auc)测量的暴露的具有21倍的安全系数(见下文)。多剂量阶段的主要目的是表征在所提出的剂量范围(0.5-4mg/kg)内多次给药的安全性,并评价在溃疡性结肠炎患者组中反复给药潜在的免疫原性。在研究的此部分中,在四个剂量群组之一中随机分配患者接受rhumabβ7或安慰剂(见表2)。在第1、29和57天每4周皮下(0.5mg/kg、1.5mg/kg或3.0mg/kg)或静脉内(4.0mg/kg)施用rhumabβ7或安慰剂三个循环(见表2)。4.0mg/kg水平下的暴露安全性的评估对考虑按固定剂量施用rhumabβ7很重要。
[0388]
评估不良事件、严重不良事件和实验室异常的发生率和性质。通过按national cancer institute common toxicity criteria for adverse events(nci ctcae),v3.0分级的不良事件的发生率来评估安全性。在研究的单剂量阶段中追踪患者的安全性18周(通过第127天),在多剂量阶段中追踪患者的安全性28周(通过第197天)。采集血样用于pk、研究性pd和抗治疗剂抗体(ata)评估。
[0389]
表2.研究群组
[0390]
[0391][0392]
iv=静脉内;sc=皮下。
[0393]
在第1、29和57天每4周一次施用研究药物三个循环。
[0394]
剂量限制毒性(dlt)
[0395]
dlt定义为在研究的单剂量阶段期间在研究药物施用的14天内发生的任何nci ctcae分级≥3的不良事件,其由研究人员或发起人确定为与rhumabβ7具有任何合理的可能的关系。在此范畴内,将输注期间或完成输注后24小时内发生的3级输注相关毒性(例如变态反应/过敏反应、发热、疼痛、支气管痉挛、喘鸣或缺氧)认为是dlt。溃疡性结肠炎的恶化不认为是dlt。
[0396]
mtd定义为低于rhumabβ7治疗的2名或多名患者经历上文定义的dlt的剂量水平的剂量水平。如果六个单剂量群组的每一个中不超过1名患者经历dlt,则未定义mtd,最高测试剂量为10mg/kg。
[0397]
研究设计的原理
[0398]
此研究在短期和中期内评价rhumabβ7的安全性、耐受性、pk谱、免疫原性和探究性pd标记。
[0399]
两阶段设计的原理
[0400]
单剂量阶段主要设计用于评价急性毒性。这种类型的潜在毒性的实例包括sc施用后的过敏反应、血清病和局部组织刺激。这种类型的毒性可以随任何蛋白质治疗发生而不考虑其给药途径。虽然rhumabβ7的靶标是淋巴细胞的β7
高
肠归巢亚群,其占人类中总淋巴细胞群体的小比例(约2%
–
10%)(rott等,j immunol 156:3727
–
36,1996),但密切监测任何输注反应。
[0401]
多剂量施用允许以28周的随访来评价反复sc和iv剂量的rhumabβ7的毒性。为了更好地在ibd群体中评价潜在的免疫原性,在多剂量阶段中每4周一次对患者施用研究药物三个周期。此外,研究的多剂量期期间使得能够评价多剂量药物动力学、探究性pd标记和生物学活性的证据,便于选择用于ii期研究的剂量和剂量间隔(见下文)。
[0402]
研究群体的原理
[0403]
此研究中的患者需在基线时患有mcs≥5的溃疡性结肠炎,并由研究人员考虑符合生物活性剂疗法的条件,因为他们患有已证明对使用5-asa药物、免疫抑制剂(硫唑嘌呤或6-巯基嘌呤)或类固醇的标准疗法反应不足的中度至严重疾病。排除具有临床上显著的机会性感染、慢性或潜在性感染、活性全身性感染或wbc计数抑制的历史的任意患者参与研究。选择此患者群体来最好地逼近此治疗最终的靶群体,同时最小化用高剂量皮质类固醇和免疫抑制药物共同治疗引入的提高的感染易感性的潜在风险。
[0404]
起始剂量、剂量范围和给药方案的原理
[0405]
i期剂量的选择基于非临床安全系数、预期人药物动力学和探究性非临床pd数据的组合。将多次给药并入此研究中来确定反复给药的潜在免疫原性,并评估rhumabβ7的累积暴露的安全性。所提出的剂量水平和频率提供机会来评价pk谱、探究性pd谱和溃疡性结肠炎患者中的生物学活性的任意证据之间的关系。这便于为ii期概念证明研究(见下文)选择适当的剂量范围和剂量频率。
[0406]
用来自食蟹猕猴的rhumabβ7pk数据来确定β7受体的饱和(国际专利公开号o2009/140684;stefanich等,br.j.pharmacol.,已接收文章,doi:10.1111/j.1476-5381.2011.01205.x)。1、3和10mg/kg humabβ7的单剂量施用后收集的数据暗示3mg/kg以下的非线性药物动力学,表明需要3mg/kg或以上的剂量来充分饱和β7受体介导的清除。因此,食蟹猕猴中1mg/kg的剂量似乎是非饱和剂量;此剂量下对应的平均观测最大血清浓度是25.1μg/ml。食蟹猕猴中1mg/kg非饱和剂量的人等同剂量(hed)是约0.3mg/kg;预期这个提出的0.3mg/kg的起始单剂量将不产生>10μg/ml的血清浓度,因此应不是饱和剂量。
[0407]
选择提出的人类中的起始剂量(通过iv输注施用的0.3mg/kg rhumabβ7)来评价研究药物施用引起的任何急性反应,并为食蟹猕猴中的非临床毒理学研究所支持(国际专利公开号wo2009/140684;stefanich等,br.j.pharmacol.,已接收文章,doi:10.1111/j.1476-5381.2011.01205.x)。以≥50mg/kg的noael(每周给药一次,给药12周)(国际专利公开号wo2009140684)、预期的人清除(cl)和预期的人中央区室体积(v1)为基础来估计起始剂量的单剂量安全系数。安全系数以hed为基础是53.7,以暴露(auc)为基础是76.2,以剂量为基础是167,以预期的最大浓度(cmax)为基础是175(见表3)。在提出的4mg/kg iv总计三个剂量的最高多给药方案下,安全系数以hed为基础是16.1,以auc为基础是21.1,以剂量为基础是50,以cmax为基础是34.4(见表3)。
[0408]
用noael而不是mabel选择0.3mg/kg的起始剂量的原理基于几个因素。首先,靶向整联蛋白并不新颖,因为在之前的临床中已通过natalizumab和mln02(也称为mln0002或vedolizumab)靶向α4β7而无急性严重不良事件(feagan等,n engl j med 352:2499
–
507(2005);sandborn等,n engl j med 353:1912
–
25(2005))。第二,提出的rhumabβ7的作用机制是阻断淋巴细胞向肠运输,且可能阻断淋巴细胞在上皮中的停留。已显示此作用机制与之前的整联蛋白拮抗剂的急性不良事件相关。此外,rhumabβ7未显示任何明显的拮抗特性,
也不在体外诱导细胞因子释放。最后,rhumabβ7未在高达10μg/ml的浓度下在体外显示任何依赖抗体的细胞毒性或依赖补体的细胞毒性。在小鼠和食蟹猕猴中完成的非临床研究中也没有证据证明外周细胞亚型的变化。因此,认为0.3mg/kg是在溃疡性结肠炎中进行的这项首次人试验的合理的起始剂量。
[0409]
表3.rhumabβ7的单剂量和重复剂量安全系数估计值。
[0410][0411]
auc=血清浓度-时间曲线下面积;auc
inf
=从时间0至无穷的血清浓度-时间曲线下面积;cl=清除;c
max
=观测到的最大浓度;hed=人等同剂量;iv=静脉内;noael=未观察到不良作用的水平。
[0412]a最低单剂量是0.3mg/kg iv。最高单剂量是10mg/kg iv。最高重复剂量是每4周4mg/kg iv,持续三个总剂量iv(平行招募)。.
[0413]b安全系数=hed/剂量h,其中h是人。食蟹猕猴noael向hed的转化基于体重,且通过将noael除以3.1来计算。假设noael≥50mg/kg
×
12,hed对于单剂量安全系数是16.1mg/kg,对于重复剂量安全系数是193mg/kg(16.1mg/kg
×
12)。
[0414]c安全系数=剂量c/剂量h,其中c是食蟹猕猴,h是人。假设noael≥50mg/kg
×
12,剂量c对于单剂量安全系数是50mg/kg,对于重复剂量安全系数是600mg/kg(50mg/kg
×
12)。
[0415]d安全系数=aucc/auch,其中c是食蟹猕猴,h是人。对于单剂量安全系数,用关系aucc=剂量c/clc计算aucc。通过在食蟹猕猴中25mg/kg的四个周剂量后计算平均cl来确定2.93ml/天/kg的clc。假设noael≥50mg/kg,对于单剂量安全系数,aucc为17065天
·
μg/ml。对于重复剂量安全系数,通过计算50mg/kg
×
12iv剂量组的几何平均值auc
inf
来确定188846天
·
μg/ml的aucc。用关系auch=剂量h/(clh)估计auch。用clh的最保守的估计值来计算基于暴露的安全系数。用物种不变-时间法(dedrick 1973)估计clh。针对iv施用的0.3mg/kg单剂量、10mg/kg单剂量和三个4mg/kg剂量估计的auch分别224、7463和8955天
·
μg/ml。
[0416]e安全系数=c
max-c
/c
max-h
,其中c是食蟹猕猴,h是人。c
max-c
是食蟹猕猴中的最大观测浓度的几何平均值。假设noael≥50mg/kg,单次iv施用50mg/kg后观察到的几何平均值c
0-c
是1315μg/ml。50mg/kg的12个iv剂量后观察到的几何平均值c
max-c
是3444μg/ml。将c
max-h
估计和确定为类似于在人类中针对其他igg1分子观察到的c
max-h
。针对iv施用的0.3mg/kg单
剂量、10mg/kg单剂量和三个4mg/kg剂量估计的c
max-h
分别为7.5、250和100μg/ml。
[0417]f安全系数基于估计为剂量/v1的预测c
max-h
,其假设每4周施用4mg/kg、总计三个剂量的重复剂量后无累积。
[0418]
以预期维持稳态波谷水平高于1-10μg/ml的靶浓度的模拟血清rhumabβ7浓度-时间谱为基础来估计靶剂量范围和给药方案的选择。此靶浓度基于来自小鼠和猕猴中的rhumabβ7的体内研究的结果,其显示1
–
10μg/ml的波谷浓度是维持循环外周血t细胞上的β7受体的占据所必需的。循环外周血t细胞上的β7受体的占据是在i期研究中评估的探究性pd标记。在获知这种标记将如何与临床活性相关之前,1
–
10μg/ml靶浓度与临床有效性的相关性也未知。
[0419]
预期所计划的剂量递增方案优化评估rhumabβ7在宽范围的剂量和暴露内施用时的安全性的能力。不预期每4周iv施用后研究药物的过度或意外的累积。
[0420]
结果测量
[0421]
此研究的主要结果测量如下:(1)不良事件(按3.0版nci ctcae分级)的发生率、性质和严重度;和(2)基于血液学、临床化学和尿液分析测试结果的实验室异常的发生率和性质。
[0422]
此研究的次要结果测量是源自施用rhumabβ7后的血清浓度-时间谱的pk谱和参数,包括所列出的那些参数:cmax;皮下施用的药物的cl或表观cl(cl/f);皮下施用的药物的分布体积(v)或表观分布体积(v/f);auc;清除半衰期(t1/2);剂量比例性;及sc生物利用率。次要结果测量还包括抗rhumabβ7的抗体的发生率,其基于在每名患者给药之前和之后的多个时间点获得的样品。
[0423]
还考查了某些实验室结果测量。这些包括通过淋巴细胞亚群的流式细胞术测量的pd标记从基线的变化(其可以包括rhumabβ7的受体占据,相对于细胞表面β7受体水平,和/或表达β7整联蛋白的t细胞亚群);及改变的mcs从基线的变化。
[0424]
研究群体
[0425]
选择患有中度至严重溃疡性结肠炎且mcs≥5的门诊患者参与研究。如果患者已为了其疾病的急性治疗而接受高剂量皮质类固醇,则在研究药物施用之前,将剂量降低至≤20mg/天强的松或强的松等同物,持续最少2周。
[0426]
在研究期间的任意时间,允许其溃疡性结肠炎发作的患者接受类固醇(iv、口服和/或局部)的挽救疗法。还允许5-asa(口服或局部)和/或免疫抑制剂(即硫唑嘌呤、6-巯基嘌呤或氨甲喋呤)作为挽救疗法。在研究过程中不允许用抗-tnf剂(例如英利昔单抗、阿达木单抗给、塞妥珠单抗)、(natalizumab)、环孢菌素、他克莫司或任意研究性药物作为挽救疗法。
[0427]
在研究的多剂量阶段接受溃疡性结肠炎发作的挽救疗法的患者不接受其他剂量的研究药物,但继续进行安全性、pk、探究性pd和ata评估的随访。
[0428]
不将筛查时具有疑似临床上显著的免疫抑制(基于之前的机会性感染的频率和严重度)及外周血中wbc计数减少的迹象的任意患者纳入研究。由于一部分患者可以接受过之前的英利昔单抗治疗,患者需要在启动此研究中的治疗之前至少12周停用抗-tnf疗法。英利昔单抗具有8-9天的半衰期;因此,认为12周(约10个半衰期)的间歇期对生物制品的药物清除是足够的,确保患者安全性及任意rhumabβ7相关不良事件的公平评价。
[0429]
在参与研究之前,专门针对任何感染迹象,尤其是结肠的jc病毒感染(通过血浆jcv dna测量)、巨细胞病毒(cmv)感染(通过结肠活组织检查)、呼吸系统、蠕虫或寄生虫感染筛查患者。如果发现任何活性感染,则排除患者。
[0430]
纳入标准
[0431]
为了符合进入研究的条件,患者必须满足以下标准:(1)能且愿意提供书面知情同意书;(2)18-70岁;(3)具有生育能力的男性和女性:愿意从研究开始至最后一剂研究药物后最少4个月(约5个半衰期)使用可靠的避孕方法(例如激素避孕、patch、阴道环、宫内节育器或物理障碍);(4)在筛查时诊断为mcs≥5分的中度至严重溃疡性结肠炎(见上文);(5)筛查前6个月内胸部x光检查无临床上显著的异常;(6)在研究人员看来符合接受生物制品疗法的条件,因为它们患有已证明对使用5-asa药物、免疫抑制剂(硫唑嘌呤、6-巯基嘌呤或氨甲喋呤)或类固醇的标准疗法反应不足的中度至严重疾病。在美国,参与md阶段的患者必须已证明对免疫抑制剂和抗-tnf疗法二者都反应不足或不耐受;(7)筛查时疾病持续时间≥12周(医生首次诊断后);(8)处于高剂量皮质类固醇的患者必须在第一天用研究药物给药前将剂量降低至≤20mg/天强的松或强的松等同物至少2周。局部皮质类固醇和局部5-asa制剂必须在第1天用研究药物给药前停用至少1周;(9)处于口服5-asa的患者必须在第1天用研究药物给药前处于稳定剂量或停药至少4周;(10)之前接受抗-tnf疗法的患者必须在第1天用研究药物给药前中断治疗至少12周;(11)单剂量阶段的免疫抑制剂疗法:在美国,患者必须在第1天用研究药物给药时中断免疫抑制剂疗法(即硫唑嘌呤、6-巯基嘌呤或氨甲喋呤)。在美国之外,根据研究人员的判断,患者可以在第1天用研究药物给药时中断免疫抑制剂疗法(即即硫唑嘌呤、6-巯基嘌呤或氨甲喋呤),或者可以在研究期间继续按稳定剂量接受这些疗法;(12)多剂量阶段的免疫抑制剂疗法:在美国,根据研究人员的判断,患者可以在第1天首次用研究药物给药时中断免疫抑制剂疗法(即即硫唑嘌呤、6-巯基嘌呤或氨甲喋呤),或者可以在研究期间继续按稳定剂量接受这些疗法至多6周(第43天)。但是,在第43天,所有患者必须在研究的md阶段的剩余时间(直至第197天)中断其免疫抑制剂疗法。此外,对于正在接受类固醇的患者,第57天必须在已达到临床反应的患者中开始减量,或在随后达到临床反应时的就诊时。临床反应将由研究人员判断,且考虑第43天的可屈性乙状结肠镜检查。在美国之外,根据研究人员的判断,患者可以在第1天首次用研究药物给药时中断免疫抑制剂疗法(硫唑嘌呤、6-巯基嘌呤或氨甲喋呤),或者可以在研究期间继续按稳定剂量接受这些疗法。
[0432]
排除标准
[0433]
从研究准入排除满足任意以下标准的患者。与uc相关的排除标准包括:(1)由于溃疡性结肠炎的严重性而需要住院;(2)中度至严重贫血(血红蛋白<10g/dl);(3)按研究人员的判断,溃疡性结肠炎的任意表现可能需要在研究过程中用>20mg/天强的松或强的松等同物治疗。
[0434]
与一般健康相关的排除标准包括:(1)怀孕或哺乳;(2)缺乏外周静脉入口;(3)在研究人员看来不能顺从研究流程;(4)筛查时粪便的卵或寄生虫测试为阳性、粪便的病原体培养为阳性或针对难辨梭菌(clostridium difficile)的粪便毒素测定为阳性;(5)显著的不受控疾病,如神经、心脏、肺、肾、肝、内分泌或gi障碍;(6)显著的筛查ecg异常,包括急性心肌梗塞、完全性左束支传导阻滞、第二度心脏传导阻滞或完全性心脏传导阻滞的迹象;
(7)控制不佳的糖尿病(糖基化血红蛋白[hba1c]>8.0%);(8)需要在研究过程中用>20mg/天强的松或强的松等同物治疗的除溃疡性结肠炎之外的病症(例如哮喘);(9)除完全切除的皮肤基底细胞癌或鳞状细胞癌之外的恶性肿瘤病史;(10)肾功能受损(肌酐>1.5x正常上限[uln]);(11)肝功能受损(转氨酶>2.5x uln,或研究人员判断合成功能测试中的异常在临床上显著)。
[0435]
与感染的风险因子相关的排除标准包括:(1)先天性免疫缺陷;(2)血清免疫球蛋白iga和igg(包括igg亚型2和3)低于实验室正常下限;(3)血清jcv dna测试为阳性;(4)指示hiv或乙肝或丙肝的活性感染或之前的感染的抗体的测试为阳性;(5)在抗eb病毒(ebv)的阴性igg效价的存在下,igm抗体效价为阳性;(6)肠黏膜活组织检查为cmv阳性;(7)潜伏性结核分枝杆菌(mycobacterium tuberculosis)感染筛查测试为阳性,纯蛋白衍生物(ppd)皮肤测试或诸如gold(qft-g,或等同的血液测试)的实验室测定是可接受的筛查测定。qft-g(或等同的血液测试)应用于之前接种过卡介苗(bcg)的患者。如果在筛查前3个月内记录过阴性ppd测试,则无需重复;(8)严重全身性细菌、真菌或寄生虫感染的病史(每年超过两次住院或每年超过两次iv抗生素的疗程);(9)启动研究治疗之前6个月内诸如假丝酵母属或曲霉属的侵袭性真菌感染(排除鹅口疮或其他浅表真菌感染)的病史;(10)启动研究治疗之前12周内严重疱疹(1型单纯疱疹、2型单纯疱疹或带状疱疹)感染或复活的病史,或疱疹频繁复发(每年超过2次);(11)启动研究治疗之前12周内任意其他机会性感染的病史;(12)pml病史;(13)任意当前或最近(启动研究治疗之前4周内)的感染病征或症状;(14)启动研究治疗之前4周内接受过口服抗生素,或启动研究治疗之前8周内接受过iv抗生素;(15)筛查前4周内接受过活减毒疫苗;(16)筛查前4周内住院。
[0436]
与药物相关的排除标准包括:(1)筛查前12周内(或研究性产品的5个半衰期内,无论哪一个时间更长)接受过任意研究性治疗;(2)完成此研究的sd阶段但未接受rhumabβ7(即接受安慰剂)的患者将不排除参与md阶段;(3)筛查前12周内接受过任意生物制品疗法;(4)血小板减少(血小板计数<75,000/μl)、中性粒细胞减少(绝对中性粒细胞计数<1500/μl)或淋巴细胞减少(绝对淋巴细胞计数<800/μl);(5)启动研究治疗之前3个月内接受过环孢菌素或他克莫司(fk506)。
[0437]
测定方法
[0438]
jcv dna评估
[0439]
患者在基线时及然后在研究期间每4周用jcv dna detectr qualitative assay(#8145)测试jcv dna,阳性测试分类为80拷贝/ml或更高。对在研究期间变为jcv dna阳性的患者进行定量jcv dna测定(jcv dna ultraquant
tm quantitative assay[#8147])。此测定具有80拷贝/ml的检测下限。
[0440]
药物动力学和抗-治疗剂抗体(ata)评估
[0441]
从所有患者获得血清样品来进行rhumabβ7的药物动力学的测定和表征及ata的评估。通过免疫测定法检查临床样品的血清rhumabβ7和ata水平作为下文所述测试项目的部分。
[0442]
ata的测试项目遵循ata测试的总体指导(mire-sluis等j immunol methods 289:1
–
16(2004))。ata的测试项目具有两层(tier):筛查层和表征层。筛查层具有血清样品的起始筛查测试、在最初的筛查中测试为阳性的那些的验证性测试及确认的阳性样品的滴定。
保存阳性血清用于将来可能的用途。验证筛查层测定。由于预期高血清水平的rhumabβ7干扰ata的检测,在施用最后一剂rhumabβ7后获得洗出样品,此时预期药物水平降至预期干扰测定的水平以下。
[0443]
药物动力学测定
[0444]
验证具有比色检测系统的夹心法elisa来定量人血清中的rhumabβ7(pro145223)。按1.0μg/ml用抗-rhumabβ7包被微量滴定板来捕获rhumabβ7。将稀释的样品、标准品和对照加至平板,并孵育。随后,加入生物素化的抗-rhumabβ7和缀合辣根过氧化物酶(hrp)的链霉抗生物素蛋白进行检测,并孵育。加入过氧化物酶底物(四甲基联苯胺)来显色,并通过加入1m磷酸来终止反应。在450nm读板来检测吸光度,在620或630nm读板来检测参考吸光度。此测定的最小可定量浓度为12.5ng/ml。
[0445]
抗-治疗剂抗体测定
[0446]
验证抗体桥接电化学发光测定(ecla)来检测人血清中抗pro145223(rhumabβ7)的抗体。该测定用缀合生物素的pro145223和缀合sulfo-tag的pro145223来捕获抗pro145223的抗体。将2μg/ml的生物素化rhumabβ7缀合物和2μg/ml的sulfo-tag rhumabβ7缀合物加至96孔聚丙烯微孔板(corning)。随后,将稀释的样品和对照加至微孔板,并与缀合物过夜共孵育。还用测定稀释液在2℃-8℃过夜封闭链霉抗生物素蛋白包被的meso scale(msd;gaithersburg,md)平板。然后将来自聚丙烯平板的样品转移至封闭的msd平板,并在室温孵育2小时。然后用洗涤缓冲液洗涤msd平板三次。向msd平板中加入msd 2x read buffer t,并在msd sector imager 6000酶标仪上读平板。用对数效价数据还原程序来确定抗体效价值。
[0447]
在测定验证期间,确定最小可报告效价为:log
10
(最小稀释度)=1.30效价单位。
[0448]
药效学评估
[0449]
从患者抽取外周血样品,并用标准流式细胞术法通过外周血淋巴细胞亚型的荧光激活细胞分选(faca)分析来实时测量探究性pd标记。测量所有样品后针对每个群组分析数据。
[0450]
设计β7测定来评估β7占据和表达,用pe(藻红蛋白)缀合的rhumabβ7(竞争性抗体)来检测给药前和给药后可用的自由位点,以及pe缀合的抗-β7抗体(非竞争性抗体),其不受rhumabβ7的存在的抑制,且提供给药期间总β7表达的评估。
[0451]
简言之,在冰上将100ml肝素钠抗凝的全血加至适当标记的染色管,并孵育30分钟。孵育后,向适当的管中加入含有适当的抗体混合物的混合物,并在冰上避光孵育30分钟。然后用含2%正常牛血清的磷酸缓冲液溶液洗涤细胞,最后重悬在1%多聚甲醛中,并保存在2-8℃,直至在becton dickinson facscalibur流式细胞仪上获取。以50,000总事件的终止计数对管进行获取。
[0452]
结果
[0453]
单剂量阶段和多剂量阶段两个阶段的患者基线特征显示在以下两表格中。
[0454]
表4.sad阶段的患者基线特征。
[0455][0456]
表5.mad阶段的患者基线特征。
[0457][0458]
如上文所示,此i期研究的主要结果测量是安全性和耐受性。对于单剂量阶段,我们未观察到任何剂量限制毒性,且无输注或注射部位反应。在单剂量阶段,七名患者经历了严重不良事件(六名接受rhumabβ7,一名接受安慰剂)。在因uc恶化而进行了紧急结肠切除的两名接受活性治疗的患者中存在两例与受损的创伤愈合相关的严重不良事件,但是,存在显著的混杂因子。研究的多剂量阶段不存在临床上显著的注射部位反应。研究的多剂量阶段存在一例严重不良事件。
[0459]
在i期研究中所测试的剂量(单剂量和多剂量)下未针对rhumabβ7鉴定出相关的安全性信号。在任意剂量群组中都未满足流程定义的dlt标准。ae的总体发生率一般在群组之间相当,不存在ae发生率的剂量依赖性提高的趋势。大部分ae为nci ctcae1级或2级。在i期研究中接受rhumabβ7的七名患者报告sae:六名在sd阶段,一名在md阶段。研究的sd阶段中不存在感染相关事件的发生率的明显的剂量相关提高。在研究的任一阶段中都未报告严重
的输注或注射相关ae。与安慰剂(sd和md两个阶段都是20%的患者)相比,在rhumabβ7治疗的患者(sd阶段为45%的患者,md阶段为55.6%的患者)中,在研究药物施用后24小时内报告的ae的发生率似乎提高。但是,研究药物施用后24小时内出现的所有ae都是nci ctcae 1级或2级。
[0460]
在整个研究的某些时间点采集来自每名患者的血液样品,并测量血清rhumabβ7浓度来评估pk。通过使用标准非房室(nca)pk法和计算机软件winnonlin pro version 5.1.1(pharsight)来针对可用的血清浓度-时间数据进行pk评估。使用实际的采血时间。结果显示在图1a-1c中。图1a显示研究的sad阶段的结果,图1b显示按3.0mg/kg iv施用的单剂rhumabβ7的pk谱,与3.0mg/kg sc相比较,图1c显示研究的mad阶段的结果。
[0461]
图1a中所呈现的数据的分析显示,血清rhumabβ7浓度显示初始快速分布期后跟随缓慢清除期的两阶段倾向。iv施用时,c
max
以与剂量成比例的方式提高,0.3-mg/kg剂量组平均为9.33μg/ml,10-mg/kg剂量组平均为239μg/ml。类似地,如整个剂量范围内相似的剂量归一化auc值所证明,组浓度-时间曲线下平均面积(auc
0-inf
)在从76.9至2500天
×
μg/ml的范围内,且大致与剂量成比例。终点清除半衰期约为2周(~14天)。低剂量(0.3mg/kg和1mg/kg)的pk谱表明非线性pk的轻微趋势,可能是由于低浓度下靶标介导的清除的贡献。
[0462]
图1b中所呈现的数据的分析显示,sc群组的清除一般与iv群组一致。将单个3mg/kg sc剂量群组的auc
0-inf
值与单个3mg/kg iv剂量群组的auc
0-inf
相比较的初步评估估计出66-67%的大致生物利用率。
[0463]
图1c中所呈现的数据的分析显示,与单剂量pk数据一致,rhumabβ7血清暴露一般与剂量成比例。每4周一次的给药方案导致最小或中等的药物随时间累积。4mg/kg iv的最高剂量群组在第一剂和最后一剂后分别具有117
±
31和136
±
24μg/ml的平均(
±
标准差)c
max
。类似于单剂量动力学,多剂量群组中的终点清除半衰期约为2周。
[0464]
用nonmem vi(icon)对来自所有单剂量群组的数据进行了初步的群体pk评估。通过将清除(cl)或中央分布体积(v1)的个体间变异性(iiv)作为协变量对体重(bw)作图来进行单变量探究性分析。rhumabβ7的cl或v1未在此评估中显示与bw相关,表明可以用固定剂量取代基于bw的剂量。
[0465]
总的来说,上述分析显示在《3mg/kg的剂量下具有非线性pk的轻微趋势的总体线性药物动力学。3和10mg/kg的单个iv剂量后的清除半衰期约为2周(~14天),sd生物利用率约为66-67%。
[0466]
抗-治疗剂抗体(ata)分析的结果揭示,接受0.3mg/kg iv的sad群组中的一名患者在第29天后为ata阳性,其余患者在第127天后为阳性。此ata阳性对pk无影响(数据未显示)。
[0467]
在多种时间在来自每名患者的样品中测量了整联蛋白β7的占据。对于研究的sad阶段,占据保持在1-10μg/ml(数据未显示)。图2a-2d显示来自研究的mad阶段的占据分析的结果。数据显示低至0.5mg/kg sc的每月给药保持β7受体饱和的持续时间与完全占据的剂量关系。
[0468]
我们还针对可能的临床活性的迹象分析了数据。所考察的一个参数是mayo临床评分(mcs)的个体变化。这些结果显示在图3a-3e中。对于多剂量阶段的所有患者,平均(
±
标准差)基线mcs为9.3
±
1.9。临床反应定义为mcs减少3分加上从基线降低30%及直肠出血减
少≥1分或绝对出血得分为0/1。如图3a-3e中所示,在第71天,用rhumabβ7治疗的12/18患者和用安慰剂治疗的4/5患者显示符合这些标准的临床反应。此外,临床缓解定义为绝对mcs≤2加上没有单个小分》1。图3a-3e显示,用rhumabβ7治疗的3/18患者和用安慰剂治疗的1/5患者显示符合这些标准的临床缓解。值得注意的是,在未使用类固醇的rhumabβ7治疗的患者中观察到了临床活性,且临床活性在群组h和i中更一致。
[0469]
实施例2
[0470]
在中度至严重溃疡性结肠炎患者中评价rhumabβ7的有效性和安全性的ii期、随机化、双盲、安慰剂对照研究及开放标签延长研究
[0471]
研究设计
[0472]
研究描述
[0473]
此ii期研究是在中度至严重uc患者中与安慰剂比较评价两个rhumabβ7剂量水平的有效性和安全性的随机化、双盲、安慰剂对照多中心研究。此目标患者群体与在上述i期研究中研究的群体相同。将在第10周(施用最后一剂研究药物后2周)评价主要有效性终点,在第6周评价次要有效性终点。
[0474]
在第0、4和8周rhumabβ7 100mg sc(固定剂量)及第0周420mg sc(统一负荷剂量)后跟随第2、4和8周300mg sc或匹配的安慰剂sc的剂量范围内按1:1:1的比例随机化患者。研究方案显示在图7中。研究将划分为0-35天的筛查期、10周的双盲治疗期、18周的安全性随访期和17个月(随机化后2年)的进行性多灶性白质脑病(pml)随访期。
[0475]
为符合条件,患者必须具有按照美国胃肠病学会(acg)实践指南诊断的最少12周的uc持续时间;即,组织病理学报告所确证的临床和内窥镜检查证据,具有在某些情况下由mcs≥5或在某些情况下由mcs≥6(包括内窥镜检查小分≥2)证明的中度至严重疾病的证据;直肠出血小分≥1(见表1);距离肛外缘最小25cm的疾病活性的内窥镜检查证据。
[0476]
在随机化之前,患者必须处于共同施用的uc药物的稳定剂量。在第1天的随机化之前,口服5-氨基水杨酸(5-asa)和免疫抑制剂(硫唑嘌呤[aza]、6-巯基嘌呤[6-mp]或氨甲喋呤)剂量必须保持稳定至少4周。在第1天的随机化之前,接受局部5-asa或皮质类固醇的患者必须停药2周。在第1天的随机化之前,口服皮质类固醇剂量必须保持稳定2周。在第1天的随机化之前,接受高剂量类固醇的患者必须将剂量降低至≤20mg/天2周。对于在研究治疗期内接受口服皮质类固醇的患者,必须在第10周按每周5-mg强的松或强的松等同物的速率开始减少类固醇2周,然后按每周2.5mg强的松或强的松等同物的速率减少,直至停药。对于接受口服免疫抑制剂(口服皮质类固醇以外)的患者,免疫抑制剂的减量必须在第8周开始,且患者必须在第10周后完全停用免疫抑制剂。之前接受抗-tnf疗法的患者必须在第1天随机化来接受研究药物之前最少8周中断该疗法。如果患者在此研究期间的任意时间经历持久或增加的疾病活性,可以根据研究人员的临床判断来增加或起始提高类固醇和/或免疫抑制剂剂量的形式的挽救疗法。将允许需要挽救疗法的患者保留在研究中,但将中断研究治疗,且在数据分析期间将分类为经历了治疗失败。
[0477]
将评估患者来确定它们是否未响应常规疗法(包括至少一种抗-tnf剂)。本文所用的对抗-tnf剂和免疫抑制剂的反应丧失和/或不耐受意指以下。就抗-tnf剂而言,反应丧失和/或不耐受意指尽管之前用以下的一种或多种治疗,但活性疾病的症状持续:(a)英利昔单抗:5mg/kg iv,6周内3个剂量,在第8周评估;(b)阿达木单抗:第0周一个160mg sc剂量,
然后第2周一个80mg剂量,然后在第4和6周40mg,在第8周评估;或之前的反应后规律安排的维持给药期间复发的活性症状(已反应且未丧失反应的患者选择性中断治疗不合格);或对至少一种抗-tnf抗体不耐受的病史(包括但不排除或限于输注相关反应或注射部位反应、感染、充血性心力衰竭、脱髓鞘)。就免疫抑制剂而言,反应丧失和/或不耐受意指尽管之前每周用硫唑嘌呤(≥1.5mg/kg)或等同剂量的6-巯基嘌呤mg/kg(≥0.75mg/kg)或氨甲喋呤25mg sc/肌内(或如所示)治疗至少8周,但活性疾病的症状持续;或不耐受至少一种免疫抑制剂的病史(包括但不排除胰腺炎、药物热、皮疹、恶心/呕吐、肝功能测试标高、硫代嘌呤s-甲基转移酶遗传突变、感染)。
[0478]
向研究治疗的随机化将按与皮质类固醇并行治疗(是/否)、与免疫抑制剂并行治疗(是/否)、之前的抗-tnf暴露(是/否)(除在美国随机化的患者外)和研究地点分层。
[0479]
将在筛查时(且这将视为基线mcs)、第6周(第4周给药后2周)和第10周(最后一剂研究药物后2周)用mcs评估uc疾病活性。将在这些相同时间点进行的可屈性乙状结肠镜检查过程中获得结肠的活组织检查。还将在整个研究中收集部分mcs。还将用简短的炎性肠病问卷(sibdq)和mcs来评估患者报告的结果(pro),其由患者在第1天及第6和10周完成。此外,将在患者日记中收集疾病活性、每日症状和uc的影响,该日记由患者从筛查(第1天之前约7天至第1天)及第6和10周的研究就诊之前至少7天至第6和10周的研究就诊每天完成。还将获得血清和粪便样品进行生物标记分析。将在筛查时及第6、10和28周获得粪便进行生物标记的测量。将测量的示例性生物标记包括但不限于脂质运载蛋白、钙防卫蛋白和乳铁蛋白。将在筛查时、第1天及第4、6、10、16和28周获得血清和血浆进行探究性生物标记的测量。
[0480]
此研究的主要有效性终点是第10周时达到临床缓解的患者的比例,临床缓解定义为mcs绝对降低至≤2,没有单个小分超过1分。附加的次要终点在研究结果测量中列出。
[0481]
开放标签延长期
[0482]
此研究将是在招募入上述ii期研究且满足进入ole研究的合格标准的中度至严重uc患者中评价rhumabβ7的安全性和耐受性的标签开放延长(ole)试验。此研究将招募约120名患者。
[0483]
此ole研究中的所有患者将按每4周300mg sc的剂量接受rhumabβ7治疗。研究将划分为104周的开放标签治疗期、治疗后12周的安全性随访及最后一剂rhumabβ7后104周的延长的进行性多灶性白质脑病(pml)监测期。
[0484]
如果它们满足任意以下标准,则参与上述ii期研究的所有患者(安慰剂或活性)都可以进入此ole研究:
[0485]
在ii期研究中在第10周还未获得临床反应;患者可以在第10周后直至且包括第28周的任意时间进入;按ii期研究定义临床反应:mcs至少减少3分且从基线降低30%,及直肠出血小分减少≥1分或绝对直肠出血评分为0或1。
[0486]
在ii期研究中在第10周具有临床反应(如上文定义),但在第10周和第28周之间具有发作;按ii期研究中定义uc的发作:部分mcs减少2分,伴随3天的连续直肠出血,且可屈性乙状结肠镜检查上的内窥镜检查得分为2。
[0487]
已完成直至第28周的随访的ii期研究。
[0488]
美国的患者还必须在进入ole研究前中断并行的免疫抑制剂疗法,且必须已在招募至ole的24周内完成口服皮质类固醇的减量。
[0489]
研究设计的原理
[0490]
剂量原理
[0491]
下文总结所提出的方案的原理。
[0492]
对于80kg中位数体重的患者,所提出的待在ii期研究中sc施用的固定剂量(100、300和420mg)分别等同于1.25、3.75和5.25mg/kg。预期选择用于ii期研究的剂量提供显示ib期研究(上述多剂量阶段)中的初步临床活性的观测暴露水平。此外,如通过用初步的i期pk数据模拟所预测的血清rhumabβ7-时间谱中所示,在解释暴露的个体间变异性(iiv)后,所提出的100和420/300mg剂量臂提供约4.4倍的剂量范围和明显的暴露分开。此外,预期100mg剂量臂在所有患者中达到1-10μg/ml的稳态波谷浓度水平(受体占据所需的波谷浓度),预期420/300mg剂量臂在>90%的患者中提供高于10μg/ml的稳态波谷浓度水平。此剂量臂中420mg的第一负荷剂量和附加的第2周的300mg剂量旨在最大化疗程早期的rhumabβ7暴露,以探究早期更高的药物暴露是否将诱导临床反应的更快起始。与观察到的来自i期多剂量4mg/kg iv群组的组平均暴露数据相比,针对所提出的高剂量臂预测的前2或4周(第0-14天或第0-28天)内的暴露具有2.5(基于c
max
)或1.3-2.2(基于auc)的安全限度(表6)。
[0493]
表6.所提出的高剂量臂的安全限度与i期多剂量iv群组暴露相比。
[0494][0495]
提出固定剂量,并根据以下考虑来证明。首先,进行了单变量探究性分析,其中将清除(cl)或中央分布体积(v1)(获自初步的基本群体iv pk模型)的个体间变异性(iiv)作为协变量对体重作图。cl的iiv或v1的iiv对体重作图显示,cl或v1对体重的依赖性较低或无依赖性。第二,对于接受固定剂量(300mg)或等同的基于体重的剂量(3.75mg/kg)的患者,在群体pk模型预测的稳态最大血清浓度(c
max.ss
)及从第0天至第84天的血清浓度-时间曲线下面积(auc
0-84
)中比较变异性。如图5中所示,中位数c
max.ss
和auc
0-84
在固定剂量和基于体重的剂量之间相似。模拟并在图5中显示的给药方案是3.75mg/kg的基于体重的剂量和300mg的固定剂量。图5还显示,与基于体重的给药方案相比,在统一给药方案中,暴露的iiv相似或略小。
[0496]
总的来所,所提出的第0、4和8周100mg sc及第0周420mg sc后跟随第2、4和8周300mg sc的ii期给药方案基于综合的临床和pk/pd数据,平衡了满足安全性和剂量范围的需要,以及可能显示临床活性。rhumabβ7的已知pk特征及本文所述的暴露变异性评估的结果支持并证明固定剂量。
[0497]
结果测量
[0498]
主要有效性结果测量
[0499]
主要有效性结果测量是第10周的临床缓解。临床缓解定义为mcs≤2,没有单个小分超过1分(见表1)。
[0500]
次要有效性结果测量
[0501]
此研究的次要有效性结果测量是(1)第6周和第10周的临床反应,其中临床反应定
义为mcs减少至少3分,且从基线降低30%,直肠出血小分减少≥1分或绝对直肠出血得分为0或1;(2)第6周的临床缓解(上文定义);和(3)第6周和第10周的内窥镜评分和直肠出血评分的指示物。
[0502]
探究性结果测量
[0503]
此研究的探究性结果测量是已达到反应或缓解的患者中uc发作的时间。对于此结果测量,发作定义为部分mcs增加2分,伴随3天的连续直肠出血及可屈性乙状结肠镜检查上的内窥镜评分为2。
[0504]
安全性结果测量
[0505]
将用以下测量来评估rhumabβ7的安全性和耐受性:(1)按national cancer institute common terminology criteria for adverse events(nci ctcae)4.0版分级的不良事件和严重不良事件的发生率;(2)生命体征和安全性实验室测量的临床上显著的变化;(3)由于一个或多个不良事件而停药;(4)注射部位反应和过敏反应的发生率和性质;(5)感染并发症的发生率;及(6)通过ata的发生率测量的免疫原性。
[0506]
药物动力学结果测量
[0507]
药物动力学结果测量包括以下:(1)第一剂和最后一剂之后的c
max
;(2)第一剂和最后一剂之后达到最大浓度的时间(t
max
);(3)最后一剂之后的剂量间隔内的血清浓度-时间曲线下面积(auc);(4)从时间0至最后可检测的观察的时间(auc
last
)或至无穷(auc
inf
)的auc;(5)表观清除(cl/f);(6)表观分布体积(v/f);和(7)清除半衰期(t
1/2
)。
[0508]
纳入标准
[0509]
为了符合进入研究的条件,患者必须满足:(1)能且愿意提供书面知情同意书;(2)18-75岁;(3)具有生育能力的男性和女性必须愿意从研究开始至最后一剂研究药物后最少4个月(约5个半衰期)使用高效的避孕方法(例如激素避孕[口服或patch]、阴道环、宫内节育器、物理障碍或切除输精管的配偶);(4)中度至严重uc门诊患者的诊断,在某些情况下mcs≥5,或在某些情况下mcs≥6(包括内窥镜检查小分≥2);直肠出血小分≥1(见表1)及距离肛外缘最小25cm的疾病活性;(5)筛查时疾病持续时间≥12周(即首次由医生按照acg指导诊断后);(6)接受皮质类固醇的患者必须在第1天用研究药物给药前处于不超过20mg/天强的松或强的松等同物的稳定剂量至少2周;(7)在第1天用研究药物给药前必须已停用局部皮质类固醇和局部5asa制剂至少2周;(8)处于口服5-asa的患者必须在第1天用研究药物给药前处于稳定剂量至少4周;(9)以aza、6-mp或氨甲喋呤的形式接受免疫抑制剂的患者应在第1天用研究药物给药前处于稳定剂量4周。如果没有禁忌症,还应建议使用氨甲喋呤的患者使用1mg/天叶酸(或等同物)补充剂;(10)处于益生菌剂(例如culturelle、saccharomyces boulardii)的患者必须在第1天用研究药物给药前处于稳定剂量2周;(11)处于抗腹泻剂(例如氯苯哌酰胺、含阿托品的氰苯哌酯)的患者必须在第1天用研究药物给药前处于稳定剂量2周;(12)之前接受抗-tnf疗法的患者必须在第1天用研究药物给药前中断疗法最少8周;(13)之前用环孢菌素或他克莫司(fk506)治疗的患者必须在第1天起始研究药物前中断疗法最少4周;(14)之前用管饲、限定的配方饮食或肠外营养(alimentation/nutrition)治疗的患者必须在第1天起始研究药物前停用这些3周;(15)诊断为中度至严重uc的患者需对免疫抑制剂(例如aza、6-mp或氨甲喋呤)和至少一种抗-tnf剂无反应或不耐受。
[0510]
排除标准
[0511]
将从研究准入排除满足任意以下标准的患者。与uc相关的排除标准包括:(1)大量结肠切除或结肠次全切除或全结肠切除;(2)存在回肠造口或结肠造口;(3)中度至严重贫血(血红蛋白<9g/dl);(4)结肠黏膜发育异常的病史或迹象。
[0512]
与一般健康相关的排除标准包括:(1)怀孕或哺乳;(2)缺乏外周静脉入口;(3)在研究人员看来不能顺从研究流程;(4)显著的不受控的共发病,如神经、心脏、肺、肾、肝、内分泌或胃肠(gi)障碍;(5)显著的筛查ecg异常,包括急性心肌梗塞、完全性左束支传导阻滞、第二度心脏传导阻滞或完全性心脏传导阻滞的迹象;(6)控制不佳的糖尿病(糖基化血红蛋白[hba1c]>8.0%);(7)活性酗酒;(8)需要在研究过程中用>20mg/天强的松或强的松等同物治疗的除uc之外的病症(例如中度至严重哮喘);(9)除完全切除的皮肤基底细胞癌或鳞状细胞癌之外的恶性肿瘤病史;(10)肾功能受损(血清肌酐>1.5x正常上限[uln]);(11)缺乏原发性硬化性胆管炎的情况下的肝功能受损(血清转氨酶>2.5x uln,碱性磷酸酶>2.5x uln,总胆红素》1.5x uln,或研究人员判断合成肝功能测试中的异常在临床上显著)。如果患者已诊断出原发性硬化性胆管炎,则血清转氨酶》3uln,碱性磷酸酶》3x uln,总胆红素》2.5x uln,或研究人员判断合成肝功能测试中的异常在临床上显著;(12)筛查前4周内住院(除选择原因外);(13)血小板减少(血小板计数<75,000/μl);(14)基线神经检查上显著的异常(包括mini-mental state examination(mmse)和pml评估中的异常)。
[0513]
与感染的风险因子相关的排除标准包括:(1)先天性免疫缺陷;(2)指示hiv或乙肝(hbv)或丙肝(hcv)的活性感染或之前的感染的抗体的测试为阳性;(3)在抗eb病毒的阴性igg效价的存在下,igm抗体效价为阳性;(4)筛查时粪便的卵或寄生虫测试为阳性、粪便的病原体培养为阳性或针对难辨梭菌的粪便毒素测定为阳性;(5)筛查时肠黏膜活组织检查为巨细胞病毒(cmv)阳性;(6)潜伏性结核分枝杆菌感染筛查测试为阳性;(7)纯蛋白衍生物(ppd)皮肤测试或诸如gold(qft-g)和等同的血液测试的实验室测定是可接受的筛查测定;(8)qft-g(或等同的血液测试)应用于之前接种过卡介苗(bcg)的患者;(9)如果在筛查前3个月内记录过阴性ppd测试,则无需重复;(10)如果筛查检查和筛查前3个月内进行的胸部x光检查显示没有当前的活性感染的迹象,则可以包括已接受适当且有记录的疗程的具有潜伏性tb感染病史的患者。在这种情况下,无需进行tb筛查测试。(11)启动研究治疗之前12周内任意其他机会性感染的病史;(12)脱髓鞘疾病或pml的病史;(13)任意当前或最近(启动研究治疗之前4周内)的感染病征或症状;(14)启动研究治疗之前4周内接受过口服抗生素,或启动研究治疗之前8周内接受过iv抗生素;(15)启动研究治疗之前4周内接受过活减毒疫苗;(16)中性粒细胞减少(绝对中性粒细胞计数<1500/μl)或淋巴细胞减少(绝对淋巴细胞计数<500/μl)。
[0514]
与共同施用的uc药物相关的排除标准包括:(1)启动研究治疗之前12周内(或研究性产品的5个半衰期内,无论哪一个时间更长)接受过任意研究性治疗;(2)之前(前6个月内)暴露于利妥昔单抗(rituximab);(3)之前暴露于rhumabβ7;(4)对嵌合抗体、人抗体或人源化抗体或融合蛋白质的严重变态反应或过敏反应的病史。
[0515]
研究治疗
[0516]
试验药物和安慰剂
[0517]
rhumabβ7药物产品和安慰剂由genentech制备。它们是澄清至轻微乳白、无色至微
黄的水溶液。两种溶液都是预期用于iv和sc施用的无菌且不含防腐剂的液体。
[0518]
剂量和施用
[0519]
将随机分配患者(1:1:1)来:(1)在第0周接受100mg rhumabβ7,随后在第2周接受安慰剂,在第4和8周接受100mg rhumabβ7;或(2)在第0周接受420mg rhumabβ7,随后在第2、4和8周接受300mg;或(3)在第0、2、4和8周接受安慰剂。
[0520]
所有剂量都将sc递送。将对腹部实施注射。如果腹部不可用于注射,则患者可以在大腿中接受sc剂量。每名患者将根据待施用的具体剂量的需要接受一次或多次注射。
[0521]
测定方法
[0522]
抗-治疗抗体测定
[0523]
将从所有患者获得血清样品进行ata评估。将用经验证的抗体桥接电化学发光测定分析血清ata样品。根据需要,可以用pk样品来评估ata反应。测定将遵循ata测试的总体指导(mire-sluis等j immunol methods 289:1
–
16(2004))。上文详述了测定方法。
[0524]
药物动力学测定
[0525]
将从所有患者获得血清样品进行rhumabβ7的pk的测定和表征。将用经过验证的桥接elisa分析血清pk样品,最小可定量浓度为12.5ng/ml。上文详述了测定方法。
[0526]
药效学测定
[0527]
将用探究性pd生物标记分析的定量方法分析全血和/或血清样品,其包括但不限于流式细胞术、qrt pcr和/或elisa。上文描述了关于该测定的其他细节。但是,对于ii期研究,将用不同的竞争性抗-β7抗体fib504(其与rhumabβ7结合相同的表位)代替rhumabβ7。
[0528]
有效性分析
[0529]
主要有效性终点
[0530]
主要终点是缓解的维持。缓解的维持指在给定时间点(例如6个月、1年或2年时)处于临床缓解的患者的比例。将按与皮质类固醇并行治疗、与免疫抑制剂并行治疗及之前的抗-tnf暴露分层,用mantel-haenszel检验统计来评价每个rhumabβ7臂和安慰剂之间的差异。此外,将通过构建主要有效性终点的80%置信区间来评价治疗组之间的差异。将提供每个治疗组的描述性概要统计。
[0531]
还可以评估持续缓解。持续缓解是一旦他们诱导了缓解就随时间推移保持缓解的患者的数目。它是从诱导缓解的时间至下一次发作的个体患者经历。这意味着患者无需增加共同施用的uc药物来保持缓解。
[0532]
次要有效性终点
[0533]
此研究的次要有效性终点如下:(1)在第6周和第10周具有临床反应的患者的比例,其中临床反应定义为mcs减少至少3分,且从基线降低30%,直肠出血小分减少≥1分或绝对直肠出血得分为0或1;(2)第6周处于临床缓解(上文定义)的患者的比例;(3)达到内窥镜评分和直肠出血评分为0的患者的比例。。
[0534]
将以与主要有效性结果测量相同的方式(上述的)来分析次要有效性终点。
[0535]
安全性分析
[0536]
将通过记录不良事件、进行临床实验室评价及通过测量ata评估免疫原性来评估安全性。将按照所接受的实际治疗来分析患者。
[0537]
将从提供知情同意书时起,直至患者完成研究或提前中断记录不良事件。将为筛
查期、10周的治疗期和18周的随访期提供分开的不良事件摘要。将按治疗组总结不良事件。
[0538]
将用描述性统计按治疗组总结临床实验室数据(血清化学、血液学评价(包括具有分类计数和血小板计数的全血计数)和尿液分析值)。
[0539]
所有患者都将包括在安全性分析中。
[0540]
药物动力学和药效学分析
[0541]
将用非房室或群体pk方法针对所有患者估计衍生自rhumabβ7的血清浓度的pk参数,包括第一剂和最后一剂之后的c
max
,第一剂和最后一剂之后的t
max
,最后一个给药间隔内(auc
57-85
)及从时间0至最后一次可检测的观察的时间(auc
last
)或至无穷(auc
inf
)的auc,cl/f,v/f和t
1/2
。将制备单个及平均血清rhumabβ7浓度-时间谱的图。血清rhumabβ7浓度和pk参数将按患者和给药方案列出,并将通过描述性统计来总结。
[0542]
可以将来自此研究的pk数据与来自i期研究的数据组合来进行群体pk分析。将针对整个研究群体估计群体典型的pk参数值,连同患者内和患者间变异的估计及随机误差的估计。将用事后分析法计算单个患者参数估计。将准备前瞻性分析计划,将在与临床研究报告分开的报告中提供群体pk分析。群体pk分析可以包括探究性分析来鉴定在此患者群体中影响rhumabβ7的pk的基线协变量。待检查的基线协变量将包括人口统计数据及其他患者特征,如疾病严重度和所选择的实验室测量。将用适当的描述性统计来总结数据。
[0543]
暴露-临床反应的关系的评估将是探究性的,且可以评价rhumabβ7暴露(c
max
、c
min
或auc)和临床终点(如mcs)之间的相关性。
[0544]
与pd生物标记目标相关的所有分析都将是探究性的。结果将作为绝对值和/或从基线的变化总结。将根据需要进行附加的pk和pd分析。
[0545]
开放标签延长研究的初步结果
[0546]
用部分mayo临床评分(pmcs)评价开放标签延长研究的前16名患者(他们全都未响应抗-tnf剂)。pmcs并入了完全mayo临床评分的三个成分,它们是:患者每天排便的次数;患者每天经历的直肠出血的数量;医生对患者疾病的总体评价,其基于前两种成分,但也考虑患者所遭受的任何腹部疼痛及患者总体上与其uc相关的感觉有多好。16名患者中的每一名的初步结果显示在图8中。
[0547]
如图8中可见,16名患者中的13名显示pmcs至少提高1分。16名患者中的11名显示pmcs提高2分。此外,6名患者治疗12-16周,他们中的3-4名随时间推移显示改善。因此,虽然是初步的数据,但这些数据提供了rhumabβ7在患有抗-tnf疗法难治的中度至严重uc的患者中的临床活性的证据。
[0548]
总体而言,rhumabβ7结合在肠黏膜中分别调节淋巴细胞亚群的运输和停留的受体α4β7和αeβ7。临床研究已证明抗-α4抗体(natalizumab)对cd的治疗的有效性,在uc的治疗中已针对抗-α4β7抗体(ldp02/mln02/mln0002/vedolizumab)观察到了振奋人心的结果。这些发现将α4β7确认为治疗靶点,并支持α4β7和madcam-1之间的相互作用有助于ibd的发病机制的假说。除靶向αeβ7的潜在的附加疗效外,rhumabβ7对阻断淋巴细胞向肠黏膜运输的特异性是追求此抗体在ibd中的临床开发的引人入胜的原因。rhumabβ7潜在的益处可以包括在uc患者中降低疾病严重度,如通过mayo临床评分测量,且还可以包括诱导缓解,同样如通过mayo临床评分测量。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1