用于治疗运动障碍的PDE7抑制剂的用途的制作方法
用于治疗运动障碍的pde7抑制剂的用途
1.本技术为分案申请,原申请的申请日为2010年5月3日,申请号为201080020136.9(pct/us2010/001305)(分案号201610458075.3),发明名称为“用于治疗运动障碍的pde7抑制剂的用途”。
发明领域
2.本发明涉及治疗与运动障碍病理有关的运动异常(movement abnormality)的方法,该方法包括给予有需要的患者一定量的有效抑制pde7酶活性的pde7抑制剂。
3.发明背景
4.帕金森病(parkinson's disease,pd)是影响中脑中的一小群神经元(称为黑质)的进行性疾病。pd与多巴胺耗竭有关,多巴胺对于在纹状体中通过与细胞相互作用以维持运动控制十分重要。每1,000人中大约有1人染上此病,65岁以上人群中大约1%患有pd。pd的常见症状包括静止性震颤、肌肉僵直(或强直)、动作迟缓(运动过慢)和平衡缺失(姿势功能障碍(postural dysfunction))。
5.帕金森病是可被一起归类为帕金森综合征(parkinsonism)的三种截然不同的病况之一。帕金森病或震颤麻痹,是帕金森综合征最常见的形式,累及大约75%的病例,且起因或病因未知。帕金森综合征的第二种类型由药物和毒素引起,包括一氧化碳、锰和称为mptp(甲基苯基四氢吡啶)的化合物。帕金森综合征的第三种类型,亦称血管性帕金森综合征,可由多次损伤产多巴胺的脑细胞的小中风引起。
6.自james parkinson于1817年命名并描述了该病症以来,已经尝试了许多疗法。大多数疗法是对症疗法,例如采用药物疗法(例如左旋多巴(levodopa)、多巴胺受体激动剂、mao-b抑制剂、comt抑制剂)或脑深部电刺激疗法,以减轻该病的症状。最近,神经保护疗法已成为深入的研发工作的主题。
7.多巴胺前体左旋多巴(l-dopa)和多巴脱羧酶抑制剂(卡比多巴(carbidopa))的治疗性组合,被认为是帕金森病症状最有效的疗法之一(themedicalletter 35:31-34,1993)。然而,该组合的某些局限在开始该组合疗法的2-5年内变得明显起来。随着该病的发展,从各剂量得到的益处越来越少(“疗效减退效应”),一些患者不可预测地在动与不动之间波动(“开/关效应”)。“开”周期通常与血浆左旋多巴浓度高有关,常常包括异常的不自主动作(即反常运动(dyskinesias))。“关”周期与血浆左旋多巴浓度低和运动过慢发作有关。因此,存在另外的有效治疗帕金森病的需要。
8.帕金森病的主要病理特征是投射到纹状体的黑质密部(snc)中的多巴胺能神经元变性(forno l.s.,j.neuropatholexp neurol 55:259-272,1996)。研究认为纹状体和其它基底核中,相对选择性多巴胺耗竭导致基底核-丘脑皮质运动原回路(motor loops)运动原区的冲动发放和同步化出现增加和失调(wichmann和delong,neuropsychopharmacology:the fifth generation of progress,第122章,“neurocircuitry of parkinson's disease(帕金森病的神经回路)”,2002)。除帕金森病以外,基底核功能异常也涉及多种具有运动异常的神经障碍。这类神经障碍包括腿多动综合征(restless leg syndrome)
(hening,w.等,sleep22:970-999,1999)和亨廷顿舞蹈病(huntington's disease)(vonsattel,j.p.等,j.neuropathol.exp.neurol.44:559-577,1985)。用mptp处理的灵长类和啮齿动物发生十分酷似人类帕金森病特征的行为和解剖变化,该发现促进了对由基底核中多巴胺能传递丧失所引起的基底核病理生理变化结果的研究。参见例如bankiewicz,k.s.等,life sci.39:7-16,1986;burns,r.s.等,pnas80:4546-4550,1983。
9.环核苷酸磷酸二酯酶(pdes)代表着将普遍存在的胞内第二信使腺苷3',5'-一磷酸(camp)和鸟苷3',5'-一磷酸(cgmp)水解成其相应的无活性5'-一磷酸代谢物的酶家族。一般认为存在至少11种不同类别的pde同工酶(pde1-11),各种酶具有独特的物理学特征和动力学特征,并代表独特的基因家族。在各个不同类别的pde内,可有多达4个独特的亚型(crocker,i.等,drugs today 35(7):519-535,1999;fawcett,l.等,pnas 97(7):3702-3703,2000;以及yuasa,k.等,j.biol.chem.275(40):31496-31479,2000)。
10.实际上,所有的磷酸二酯酶都在中枢神经系统(“cns”)中表达,使得该基因家族成为用于治疗精神疾病和神经变性性疾病特别有吸引力的新靶标的来源。然而,所有神经元均表达多种磷酸二酯酶,这类酶在环核苷酸特异性、亲和力、调控和亚细胞区室化方面各不相同,使得难以将特定疾病的靶标与疾病治疗联系起来。因此,有需要鉴定出治疗特定cns疾病(例如伴有运动异常的帕金森病和其它神经障碍)的磷酸二酯酶家族的靶标。
11.尽管帕金森病研究和治疗中有所进展,但是仍然存在对用于该疾病和伴有运动异常的其它神经障碍的新疗法的需要。本发明试图满足这一需要并提供更多相关的优势。
12.发明概述
13.如上所述,一方面,本发明提供治疗与神经性运动障碍(neurological movement disorder)病理有关的运动异常的方法。本发明这一方面的方法包括给予有需要的患者一定量的有效抑制pde7酶活性的pde7抑制剂,其中对pde7酶活性的这种抑制是pde7抑制剂在治疗运动异常中的主要治疗作用方式。
14.如上所述,一方面,本发明提供治疗与神经障碍病理有关的运动异常的方法。本发明这一方面的方法包括给予有需要的患者一定量的有效抑制pde7酶活性的pde7抑制剂,其中对pde7酶活性的这种抑制是pde7抑制剂在治疗运动异常中的主要治疗作用方式。
15.另一方面,本发明提供鉴定抑制pde7活性的药物的方法,该药物用于治疗有需要的哺乳动物受治疗者的与神经性运动障碍病理有关的运动异常。本发明这一方面的方法包括(a)测定抑制pde7a和/或pde7b活性的多种药物每一种的ic
50
;(b)从抑制pde7a和/或pde7b活性的ic
50
小于约1μm的多种药物中选出药物;(c)测定抑制pde7活性的ic
50
小于约1μm的药物抑制pde4活性的ic
50
;(d)通过选出抑制pde4活性的ic
50
是抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者10倍以上的化合物,鉴定出用于治疗运动障碍的药物;(e)在神经性运动障碍模型实验中对已鉴定出的化合物的活性进行评价,其中抑制pde7a和/或pde7b活性的ic
50
小于约1μm、且抑制pde4活性的ic
50
是抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者10倍以上、且在模型实验中被确定为有效治疗至少一种运动异常的药物,表明是用于治疗哺乳动物受治疗者的与神经性运动障碍病理有关的运动异常的pde7抑制剂。
16.另一方面,本发明提供治疗与神经性运动障碍病理有关的运动异常的方法。本发明这一方面的方法包括给予有需要的患者治疗有效量的作为pde7抑制剂的化合物,该化合
物的特征是:(i)该化合物抑制pde7a和/或pde7b活性的ic
50
小于约1μm;(ii)该化合物抑制pde3的ic
50
是抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中较小者的10倍以上。
17.本发明各个方面的方法用于治疗与神经障碍有关的运动异常。本发明各个方面的方法还用于治疗神经性运动障碍。本发明各个方面的方法进一步用于治疗与神经性运动障碍有关的运动异常。
18.在本发明各方面的一些实施方案中,该方法用于治疗可用多巴胺受体激动剂或多巴胺受体激动剂前体治疗的神经性运动障碍、与神经障碍有关的运动异常和/或与神经性运动障碍有关的运动异常。在一些实施方案中,该方法用于治疗选自以下的神经性运动障碍:帕金森病、脑炎后帕金森综合征、多巴胺反应性肌张力障碍(dopamine-responsive dystonia)、夏-德雷格综合征(shy-drager syndrome)、周期性肢体运动障碍(plmd)、睡眠中周期性肢体运动(periodic limb movements in sleep,plms)、图雷特综合征(tourette's syndrome)和腿多动综合征(rls)。
19.在本发明各方面的一些实施方案中,该方法用于治疗与神经性运动障碍病理和/或神经障碍病理有关的运动异常。在本发明各方面的一些实施方案中,该方法用于治疗与可用多巴胺受体激动剂或多巴胺受体激动剂前体治疗的神经性运动障碍病理有关的运动异常。在一些实施方案中,该方法用于治疗选自以下的与神经性运动障碍病理有关的运动异常:帕金森病、脑炎后帕金森综合征、多巴胺反应性肌张力障碍、夏-德雷格综合征、周期性肢体运动障碍(plmd)、睡眠中周期性肢体运动(plms)、图雷特综合征和腿多动综合征(rls)。
20.附图描述
21.连同附图一起参考下面的详述,在更好地理解本发明的同时,将更容易理解上述方面和本发明的许多随附的优势。在某些附图中,统计显著性用记号表示,其中“*”是指p值<0.05,“**”是指p值<0.01,“***”是指p值<0.005。附图中:
22.图1是健康哺乳动物受治疗者的中脑的基底核神经传递途径的流程图,其中“+”与阴影箭头表示刺激途径,
“‑”
与空心箭头表示抑制途径;
23.图2a表示健康受治疗者体内多巴胺受体活化途径的假设模型,图中显示出多巴胺受体激活的胞内信号转导途径被将camp水解成为其5'一磷酸(5'amp)的pde7下调或拮抗的这一新的发现;
24.图2b表示本发明的发明人提出的未接受治疗的患帕金森病(pd)受治疗者体内多巴胺受体活化途径的模型,图中显示出减量的多巴胺受体激活的胞内信号转导途径进一步被将camp水解成其5’一磷酸(5’amp)的pde7下调或拮抗,导致与健康受治疗者相比,活化pka水平低下,神经元活化减少;
25.图2c表示本发明的发明人假设的用pde7抑制剂治疗的患帕金森病(pd)受治疗者体内多巴胺受体活化途径的模型,图中显示出有效抑制pde7酶活性的pde7抑制剂的存在阻断camp的水解,有效提高胞内camp水平,激活在胞内信号转导途径中调节下游元件磷酸化的蛋白激酶a(“pka”),导致神经元活化增加,与本发明方法的各种实施方案一致;
26.图3a是说明用于本发明方法的代表性pde7抑制剂(om69)的pde7a抑制活性(ic
50
)曲线图,用每分钟计数(“cpm”)表示;
27.图3b是说明用于本发明方法的代表性pde7抑制剂(om69)的pde7b抑制活性(ic
50
)
曲线图,用cpm表示;
28.图4a是说明用于本发明方法的代表性pde7抑制剂(om955)的pde7a抑制活性(ic
50
)曲线图,用cpm表示;
29.图4b是说明用于本发明方法的代表性pde7抑制剂(om955)的pde7b抑制活性(ic
50
)曲线图,用cpm表示;
30.图5a是说明用于本发明方法的代表性pde7抑制剂(om956)的pde7a抑制活性(ic
50
)曲线图,用cpm表示;
31.图5b是说明用于本发明方法的代表性pde7抑制剂(om956)的pde7b抑制活性(ic
50
)曲线图,用cpm表示;
32.图6a是说明用于本发明方法的代表性pde7抑制剂(om056)的pde7a抑制活性(ic
50
)曲线图,用cpm表示;
33.图6b是说明用于本发明方法的代表性pde7抑制剂(om056)的pde7b抑制活性(ic
50
)曲线图,用cpm表示;
34.图7曲线图对用于本发明方法的代表性pde7抑制剂(om69)在血浆和脑组织中随时间变化的浓度(ng/g)进行了比较;
35.图8是说明在甲基苯基四氢吡啶(“mptp”)帕金森病小鼠模型中进行实验的流程图,与仅左旋多巴的作用相比较,对单独给予或者与左旋多巴组合给予用于本发明方法的代表性pde7抑制剂(om69)进行了初步评价,见实施例5;
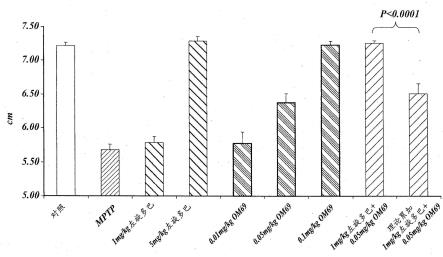
36.图9柱状图说明按照图8所示方案对mptp小鼠模型墨水涂染爪步长进行的试验,试验表明在mptp治疗小鼠中,用于本发明方法的代表性pde7抑制剂(om69)在单独给予或者与左旋多巴组合给予时加大步长,并对该抑制剂的疗效与仅左旋多巴以及与盐水对照进行了比较,见实施例5;
37.图10是表示图9中所显示的数据子集的柱状图,对mptp小鼠模型中各种剂量的用于本发明方法的代表性pde7抑制剂(om69(化合物1))、各种剂量的左旋多巴和om69与左旋多巴的组合对步长的作用进行了比较,见实施例5;
38.图11是表示图9所显示的数据子集的柱状图,图中与盐水对照(即非mptp处理)小鼠相比,对mptp小鼠模型中用于本发明方法的代表性pde7抑制剂(om69)、左旋多巴及其组合对步长的作用进行了比较,见实施例5;
39.图12流程图说明在mptp帕金森病小鼠模型进行的实验,以证实代表性pde7抑制剂(om69)加大mptp处理小鼠的步长,见实施例6;
40.图13a柱状图说明溶媒对照二甲基乙酰胺:聚乙二醇:甲磺酸(dma:peg:msa)当单独给予时不改变mptp处理小鼠的步长,见实施例7;
41.图13b柱状图说明溶媒对照酒石酸(ta)当单独给予时不改变mptp处理小鼠的步长,见实施例7;
42.图14是说明mptp小鼠模型中墨水涂染爪步长试验的柱状图,试验表明代表性pde7抑制剂om955(化合物2)加大mptp小鼠的步长,在0.5mg/kg剂量后20分钟基线步长完全恢复,见实施例7;
43.图15a是说明mptp小鼠模型中墨水涂染爪步长试验的柱状图,试验表明在给药后20分钟,1mg/kg左旋多巴不会加大mptp小鼠的步长至显著水平,见实施例7;
44.图15b是说明mptp小鼠模型中墨水涂染爪步长试验的柱状图,试验表明在给药后20分钟,0.1mg/kg om955(化合物2)不会加大mptp小鼠的步长至显著水平,见实施例7;
45.图15c是说明mptp小鼠模型中墨水涂染爪步长试验的柱状图,试验表明在给药后20分钟,给予0.1mg/kg om955(化合物2)和1mg/kg左旋多巴组合的小鼠显示mptp处理小鼠的步长完全恢复至显著水平,从而证实该组合的协同结果,见实施例7;
46.图16是说明mptp小鼠模型中墨水涂染爪步长试验的柱状图,试验表明代表性pde7抑制剂om956(化合物3)加大mptp处理小鼠的步长,在0.5mg/kg剂量后20分钟基线步长完全恢复,见实施例7;
47.图17是说明mptp小鼠模型中墨水涂染爪步长试验的柱状图,试验表明代表性pde7抑制剂om056(化合物4)加大mptp处理小鼠的步长,0.05mg/kg剂量后20分钟基线步长完全恢复,见实施例7;
48.图18是说明mptp小鼠模型中墨水涂染爪步长试验的柱状图,试验表明代表性pde7抑制剂om69(化合物1)以剂量依赖性方式加大mptp处理小鼠的步长,进一步证实了om69和左旋多巴的组合使mptp处理小鼠的步长超过累加性(即协同)增加,见实施例6。
49.发明详述
50.本发明是基于本发明的发明人预料不到的发现,即在1-甲基,4-苯基,1,2,3,6-四氢吡啶(mptp)帕金森病(pd)小鼠模型中,环核苷酸磷酸二酯酶7型(pde7)的选择性抑制剂使运动功能显著改善。通过使用mptp动物模型,本发明的发明人证实在mptp损伤小鼠中,以相当于用左旋多巴治疗的方式,但却以与要达到同等反应水平所需的左旋多巴剂量相比是预料不到的低的剂量,给予选择性pde7抑制剂可有效地恢复这些动物的步长。此外,本发明的发明人证实,当同时给予低于最佳剂量的左旋多巴和选择性pde7抑制剂的组合时,提供超出累加(即协同)效应,也恢复mptp损伤小鼠的步长至正常值。
51.i.定义
52.除非本文特别规定,否则本文所使用的所有术语具有与本发明领域普通技术人员所理解的相同含义。本发明提供下列定义,以便当该术语用于说明书和权利要求书以说明本发明时提供有关该术语的明确含义。
53.本文所用术语“神经性运动障碍”是指以多巴胺信号转导缺乏或缺陷为特征的运动障碍,临床表现为一种或多种与运动障碍病理有关的运动异常,例如异常的不自主动作、静止性震颤、肌张力改变、动作难以启动(运动过慢)和/或姿势稳定性障碍。
54.本文所用术语“帕金森病”是指以4种主要体征为特征的临床综合征:(1)静止性震颤;(2)强直,(3)运动过慢,和(4)缺乏姿势反射。
55.本文所用术语“脑炎后帕金森综合征”是指在脑炎之后发生并假定是脑炎造成的帕金森综合征。
56.本文所用术语“帕金森综合征”是指与帕金森病类似的任何类型的神经障碍,以帕金森病的4种主要体征为特征:静止性震颤、肌肉强直、运动过慢和缺乏姿势反射。
57.本文所用术语“运动过慢”或“运动不能”是指缺乏自动或自发运动。
58.本文所用术语“运动过度”或“反常运动”是指不自主动作过度或异常。
59.本文所用术语“震颤”是指相对节律性的摆动性动作,它可以是由例如相对肌群的交替收缩产生(例如帕金森震颤)。
60.本文所用术语“张力障碍”是指具有在动作结束时持续性收缩的不自主动作。
61.本文所用术语“多巴胺反应性肌张力障碍”是指其中持续性肌肉收缩引起颤搐和重复性动作或姿势异常的神经性运动障碍,可通过增加多巴胺水平或提高经多巴胺能途径的信号转导的药物予以减轻。这种障碍可能与帕金森病、青少年型帕金森综合征、进行性核上性麻痹、皮质基底神经节变性(cortical basal ganglionic degeneration)、某些类型的多系统萎缩或dyt3 x连锁隐性张力障碍-帕金森综合征有关。
62.本文所用术语“睡眠中周期性肢体运动”(plms)是指在睡眠期间患者腿部不自主地摆动或颤搐的障碍。如果这些动作导致睡眠障碍,则该综合征亦称周期性肢体运动障碍(plmd)。
63.本文所用术语“腿多动综合征”(rls)是指一种病理生理不确定的神经障碍,其特征是通常当平躺时(像入睡前),尤其是在夜间发生的腿部疼痛、烧灼感、蠕动感或爬行感,急切地迫使腿部摆动,常常伴有难以入睡或保持安睡以及睡眠期间腿部不自主的颤搐。
64.本文所用术语“夏-德雷格综合征”是指变性性神经障碍,其特征是直立性低血压、自主功能障碍(autonomic dysfunction)、膀胱功能障碍和帕金森样运动缺失(parkinson's-like deficits inmovement)。
65.本文所用术语“多巴胺能药物”是指其功能是在中枢神经系统中提高或重复由多巴胺介导的作用的药物,包括多巴胺(如果可开发出临床上有效的递药方法)、多巴胺前体(例如左旋多巴)、多巴胺辅因子、多巴胺代谢酶的抑制剂、其它多巴胺受体激动剂和经代谢转化成多巴胺受体激动剂的前体化合物以及多巴胺重摄取抑制剂。
66.本文所用术语“多巴胺受体激动剂”是指引起一个或多个多巴胺受体蛋白家族亚型活化的任何分子。
67.本文所用术语“已知涉及帕金森病病理的分子靶标”包括儿茶酚-o-甲基转移酶(comt)、单胺氧化酶b(mao-b)、多巴胺转运蛋白(dat)、酪氨酸羟化酶、多巴胺受体、腺苷a2a受体和加巴喷丁(gabapentin)受体。
68.本文所用术语“已知与多巴胺信号转导途径有关的分子靶标”包括儿茶酚-o-甲基转移酶(comt)、单胺氧化酶b(mao-b)、多巴胺转运蛋白(dat)、酪氨酸羟化酶、多巴脱羧酶、多巴胺受体、n-甲基d-天冬氨酸(nmda)受体、毒蕈碱性乙酰胆碱受体、γ氨基丁酸(gaba)受体、腺苷酸环化酶、蛋白激酶a(pka)、多巴胺和环腺苷酸调节的分子量为32,000的磷蛋白(darpp32)及蛋白质磷酸酶-1。
69.本文所用术语“治疗”包括减轻、缓解或掩蔽疾病或障碍的症状的对症疗法,以及用于防止、降低、停止或逆转待治疗病症或症状严重性发展的疗法。因此,术语“治疗”包括已确诊病症或症状的医药治疗性治疗和/或适当时的医药预防性给药。
70.本文所用术语“治疗与神经性运动障碍病理有关的运动异常”是指逆转、缓解、改善或抑制一种或多种与神经性运动障碍有关的运动异常。
71.本文所用术语“治疗神经性运动障碍”包括:(1)治疗与神经性运动障碍病理有关的运动异常;和/或(2)治疗神经性运动障碍。
72.本文所用术语“治疗神经障碍”包括:(1)治疗与神经障碍病理有关的运动异常;和/或(2)治疗神经障碍。
73.本文所用术语“治疗”还包括根据有需要的受治疗者的情况,预防神经性运动障碍
或预防与神经性运动障碍病理有关的运动异常或者预防神经障碍或预防与神经障碍病理有关的运动异常,包括运动异常的发作或与之有关的任何症状,以及降低运动异常的严重程度或防止运动异常的复发。
74.本文所用术语“pde7”一般是用来指由这两个基因(pde7a和/或pde7b)中的任一个或两个的转录物编码的所有翻译产物。
75.本文所用术语“pde7抑制剂”是指直接或间接抑制或阻断pde7a、pde7b或pde7a与pde7b的磷酸二酯酶活性的物质,例如化合物、肽或核酸分子。在一些情况下,所述物质可直接与pde7蛋白结合或相互作用。与pde7结合的该物质可通过任何合适的方法,例如通过抑制camp或底物配体与pde7的结合,起抑制或阻断pde7活化的作用。在其它情况下,pde7抑制剂可间接地抑制pde7活性,例如通过降低pde7蛋白的表达。在某些情况下,pde7抑制剂可通过改变pde7的细胞分布来抑制pde7活性,例如通过干扰pde7和细胞内锚定蛋白(anchoring protein)之间的缔合(association)。
76.本文所用术语“哺乳动物受治疗者”包括所有哺乳动物,包括而不限于人、非人类灵长类、狗、猫、马、绵羊、山羊、牛、兔子、猪和啮齿动物。
77.ii.治疗与神经性运动障碍病理有关的运动异常的pde7抑制剂的用途
78.多巴胺能系统通常与自主活动能力(locomotor activity)和动作的调节密切相关。参见例如tran,a.h.等,pnas 102:2117-2122,2005;tran,a.h.等,pnas99:8986-8991,2002。例如,有证据表明多巴胺能功能障碍在以下疾病中起决定性作用:帕金森病、帕金森综合征、腿多动综合征(“rls”)、周期性肢体运动障碍(plmd)、睡眠中周期性肢体运动(“plms”)和其它运动障碍。在帕金森病中,纹状体中缺乏多巴胺,这是由于黑质和蓝斑中色素性神经元丧失与其多巴胺和去甲肾上腺素神经递质相应的丧失所导致的。在脑炎后帕金森综合征中,随着黑质神经元的丧失,中脑受到累及(wyngaarden和smith,cecil textbook ofmedicine,第17版,“neurological and behavioral disease:第5节:the extrapyramidal disorders:parkinsonism:(锥体束外障碍:帕金森综合征)”,1985)。
79.一般认为纹状体和其它基底核中的相对选择性多巴胺耗竭导致基底核-丘脑皮质运动原回路运动原区中冲动发放和同步化出现增加和失调(wichmann和delong,neuropsychopharmacology:the fifth generation ofprogress,第122章,“neurocircuitry ofparkinson's disease(帕金森病的神经回路)”,2002)。
80.基底核起通向锥体束运动系统的主要输入的作用。基底核包含5对核,包括:尾状核、豆状核、苍白球、底丘脑核和黑质。底丘脑核在间脑内。黑质位于中脑。尾状核、豆状核和苍白球位于大脑半球内,统称纹状体。尾状核和豆状核合起来被视为纹状体,用作通向基底核的神经输入的主要部位。纹状体接收来自大脑皮质所有部分和来自丘脑中央中核的输入。纹状体的主要输出是通向苍白球和黑质的网状带部分。黑质的背侧部分向纹状体发送输出(多巴胺能黑质纹状体途径),黑质的腹侧部分接收来自纹状体的纤维。
81.图1表示健康哺乳动物受治疗者中脑基底核内的神经传递途径,其中“+”与阴影箭头表示刺激途径,
“‑”
与空心箭头表示抑制途径。如图1所示,神经途径将基底核的输出途径与纹状体连接,所述基底核是一组功能上相关的皮质下核,包括苍白球外部(“gpe”)、苍白球内部(“gpi”)、黑质密部(“snc”)和黑质网状部(“snr”)。图1还表示将底丘脑核(“stn”)与gpe、gpi和snr连接的途径。如图1所示,在健康受治疗者中,来自snc内的产多巴胺细胞的多
巴胺(“da”)向多巴胺d1受体(“d1”)发出刺激信号,该受体一旦被激活,便向snr发出抑制信号,并向gpi发出抑制信号。再如图1所示,来自snc内产多巴胺细胞的da还向多巴胺d2受体(“d2”)发出抑制信号,这就抑制了d2受体向gpe发送抑制信号。
82.帕金森病突出的病理特征(“pd”)是投射到纹状体黑质密部(snc)的多巴胺能神经元发生变性(forno,l.s.,j.neuropathol exp neurol55:259-272,1996)。现已确定,在帕金森病早期,在纹状体感觉运动区中多巴胺消耗最大,这与运动功能障碍的早期表现一致(kish,s.j.等,n.engl.j.med.318:876-880,1988)。
83.在pd和帕金森综合征疾病中,snc内产多巴胺的细胞丧失,导致通向纹状体的多巴胺能信号转导缺失。因为在健康受治疗者中,da通常通过d1受体激活通向snr的抑制性纹状体输出(见图1),所以该途径在pd中减弱。相反,因为在健康受治疗者中,da通过d2受体抑制通往gpe的抑制性纹状体输出(见图1),所以该途径在pd中增强。因此,在pd中通向纹状体的多巴胺能信号转导缺失具有引起自丘脑到皮质的刺激途径的净抑制的净效应(net effect)。
84.环腺苷酸一磷酸(camp)是介导细胞对各种胞外刺激产生的生物应答的第二信使。当合适的激动剂与特定的细胞表面受体结合时,腺苷酸环化酶被激活,使腺苷三磷酸(atp)转化成camp。据推测,激动剂诱导的细胞内camp的作用主要是由camp依赖性蛋白激酶的作用介导的。通过将核苷酸转运到细胞外,或者通过环核苷酸磷酸二酯酶(pdes)的酶促切割,来终止camp的胞内作用,该酶将3'-磷酸二酯键水解形成5'-腺苷一磷酸(5'-amp),这是一种无活性的代谢物。因此,pdes的胞内酶家族在细胞中调节camp的水平。
85.图2a表示提出的健康受治疗者体内多巴胺受体活化途径的模型。如图2a所示,在健康受治疗者中,黑质密部(snc)中多巴胺能神经元产生的多巴胺(da)(用3个箭头表示)结合并激活多巴胺d1受体,这就导致腺苷酸环化酶被激活并增加camp水平。camp激活蛋白激酶a(“pka”),该酶调节胞内信号转导途径中下游元件的磷酸化,导致神经元活化。如图2a所示,据推测,多巴胺受体激活的胞内信号转导途径受使camp水解成其5’一磷酸(5’amp)的pde7下调或拮抗。
86.图2b表示提出的未接受治疗的患帕金森病(pd)受治疗者体内多巴胺受体活化途径的模型。如图2b所示,在pd患者中,含量减少的多巴胺(da)(与健康受治疗者的3个箭头相比,用1个箭头表示)可用来与多巴胺受体(d1)结合,因为snc内产多巴胺的细胞丧失,导致通向纹状体的多巴胺能信号转导缺失(参照图1中所示)。在pd患者中,低水平的da结合并激活多巴胺d1受体至较低程度,这就导致腺苷酸环化酶的活化最小,camp水平的增加减缓。因此,蛋白激酶a(“pka”)的活化程度较低,进而又导致胞内信号转导途径的下游元件较少磷酸化,神经元活化程度降低。如图2b所示,据推测,多巴胺受体激活的胞内信号转导途径量的降低受使camp水解成其5’一磷酸(5’amp)的pde7的进一步下调或拮抗,导致与健康受治疗者相比,活化pka水平低下,神经元活化降低。
87.图2c表示提出的用pde7抑制剂治疗的患帕金森病(pd)的受治疗者体内多巴胺受体活化途径的模型。如图2c所示,在pd受治疗者中,含量减少的多巴胺(da)(与健康受治疗者的3个箭头相比,用1个箭头表示)可用来与多巴胺受体(d1)结合,因为snc内产多巴胺的细胞丧失,导致通向纹状体的多巴胺能信号转导缺失(参照图1中所示)。在pd受治疗者中,水平降低的da结合并激活多巴胺d1受体至较低程度,导致了腺苷酸环化酶的活化最小,
camp水平的增加减缓。然而,再如图2c中所示,有效抑制pde7酶活性的pde7抑制剂的存在阻断camp的水解,从而增加胞内camp水平,允许蛋白激酶a(“pka”)活化至更正常的程度,这就调节了胞内信号转导途径下游元件的磷酸化,引起神经元活化增加。
88.为了支持图2a-2c中所示的多巴胺信号转导模型,本发明的发明人发现给予抑制pde7酶活性的pde7抑制剂导致与运动障碍病理有关的运动异常(例如帕金森病)得到改善。本文所提供的数据证实pde7抑制剂有效恢复肢体运动,正如在mptp处理小鼠中通过前爪步长所测量的一样,而且在mptp小鼠模型中,当pde7抑制剂与左旋多巴组合时,观察到协同效应。根据本发明的发明人预料不到的发现,相信pde7在脑部,尤其是在已知与运动有关的区域的突触后多巴胺信号转导中起作用。
89.除帕金森病以外,基底核功能异常还涉及多种具有运动异常的神经障碍。这类神经障碍包括腿多动综合征(hening,w.等,sleep22:970-999,1999)。因此,根据本文上述研究,我们认为pde7抑制剂对这类神经性运动障碍可具有治疗效果。
90.因此,虽然不希望受理论的束缚,但是我们认为pde7抑制剂可用于通过抑制pde7活性,从而防止基底核中camp的降解,来治疗以缺乏多巴胺受体信号转导等基底核功能异常为特征的神经障碍,例如帕金森病、脑炎后帕金森综合征、药物诱发的帕金森综合征、多巴胺反应性肌张力障碍、夏-德雷格综合征、周期性肢体运动障碍(plmd)、睡眠中周期性肢体运动(plms)和腿多动综合征(rls)。因此我们认为pde7抑制剂可用于治疗以运动异常为特征的这些和其它神经性运动障碍和神经障碍,这些障碍目前用左旋多巴、其它多巴胺激动剂或前体或其它多巴胺能药物治疗。
91.在本发明的一些方面,pde7抑制剂用于治疗与神经障碍病理有关的运动异常,不论这类障碍是否与多巴胺信号转导缺陷缺乏有关,其中对pde7酶活性的这种抑制是pde7抑制剂在治疗运动异常中的主要治疗作用方式。
92.在一些实施方案中,本发明提供治疗与神经性运动障碍病理有关的运动异常的方法,该方法包括给予有需要的患者一定量的有效抑制pde7酶活性的pde7抑制剂,其中对pde7酶活性的这种抑制是pde7抑制剂在治疗运动异常中的主要治疗作用方式。在一些实施方案中,本发明提供改善运动障碍症状的方法,包括但不限于多巴胺受体胞内信号转导途径障碍,该方法包括给予抑制pde7酶活性的pde7抑制剂。在一些实施方案中,神经性运动障碍可用多巴胺受体激动剂或多巴胺受体激动剂前体治疗。
93.帕金森病
94.帕金森综合征是由4种主要体征组成的临床综合征:(1)静止性震颤;(2)强直,(3)运动过慢,(4)缺乏姿势反射。运动过慢占帕金森症状和体征的大部分。帕金森综合征可分成下列病因组:原发性障碍亦称帕金森病,继发性获得性帕金森综合征(由于暴露于药物或毒素、之前的中风或脑炎所致)和“帕金森综合征叠加(parkinsonism-plus)”综合征(帕金森患者的眼部运动受损、直立性低血压、小脑共济失调或痴呆)。
95.黑质受损以及随之产生的纹状体多巴胺丧失导致帕金森综合征的运动过慢综合征。在帕金森病中,黑质和蓝斑存在色素性神经元丧失,其多巴胺和去甲肾上腺素神经递质也随之丧失。
96.pd动物模型十分依赖于以下这一偶然发现:即全身性给予mptp(1-甲基-4-苯基-1,2,3,6-四氢吡啶)引起人、猴和啮齿动物黑质内特定神经元细胞死亡(jakowec,m.w.等,
comp.med.54(5):497-513,2004)。细胞死亡的模式使人联想到尸体解剖时在pd患者中观察到的模式。常用的帕金森病动物模型包括猴mptp模型、大鼠6-ohda模型和小鼠mptp模型。如本文实施例5-7中所示,可以使用mptp损伤小鼠pd模型来评价用于本发明方法的pde7抑制剂在以下方面的功效:减小或减少由mptp诱导的其步长(stride length)、网格步长(grid step length)和网格足过错(grid foot faults)方面的变化(tillerson,j.l.等,exp.neurol.178(1):;80-90,2002)。
97.如实施例5-7中所示,在mptp处理小鼠中,pde7抑制剂有效恢复肢体运动。尽管治疗帕金森病的现有方法一般包括用多巴胺受体激动剂治疗,在多巴胺信号转导减少的受治疗者体内,本发明的方法针对对pde7磷酸二酯酶活性的抑制以便提高camp水平,从而引起pka活性提高。据推测,pde7的抑制剂可能具有优于现有pd药物或降低这类药物的需求水平的优势。例如,长期使用最常见的pd药物左旋多巴会引起严重的反常运动(bezard,e.等,nat.rev.neurosci.2(8):577-88,2001)。替代左旋多巴的任何pd药物可避免这种严重的副作用。
98.再如实施例5-7中所示,pde7抑制剂和左旋多巴(一种多巴胺受体激动剂)的组合提供协同效应,在mptp处理小鼠中导致肢体运动甚至更大的改善。可供降低左旋多巴剂量的药物(例如pde7抑制剂)与左旋多巴联用,可延迟反常运动的发作。此外,由于由左旋多巴疗法引起的多巴胺水平提高可能对黑质密部神经元造成氧化性损伤,可供降低左旋多巴剂量的药物(例如pde7抑制剂)可延迟该病的进程。因此,本发明的pde7抑制剂可与左旋多巴、其它多巴胺受体激动剂、多巴胺受体激动剂前体或其它多巴胺能药物联合给予、以组合剂型给予、同时(即同一时间)给予或序贯(例如依次)给予。
99.腿多动综合征(rls)
100.腿多动综合征(rls)是同样涉及多巴胺系统的常见神经病症。rls是一种感觉运动障碍,用于其诊断的主要强制性标准为:(1)迫使腿部摆动,通常伴有肢体不适感,(2)在休息或不活动期间症状加重,(3)通过运动症状得到改善,(4)在傍晚或夜晚症状出现或加重(allen,r.p.等,sleep med 4:101-119,2003)。对于rls诊断是常用但非必不可少的支持性标准,包括存在睡眠中周期性肢体运动(plms),这是在睡眠期间下肢的不自主动作,常常连续发生至少4次,动作间的间隔为5-90秒钟(baier等,j.neurological sciences 198:71-77,2002)。诊断rls的其它支持性标准是易对低剂量多巴胺能治疗起反应(allen,r.p.等,同上)。在受累于帕金森病和其它形式的帕金森综合征的患者中,出现严重的rls和plms(poewe,w.等,neurology 63:s12-s16,2004)。
101.已经确定rls的发病机制以多巴胺能系统的神经功能障碍为特征。通过功能性成像研究(turjanski,n.等,neurology 52:932-37,1999)及人rls和plms的强效多巴胺激动剂治疗(montplaisier,j.等,neurology 52:938-43,1999;trenkwalder,c.等,neurology 62:1391-97,2004;及walters,a.s.等,mov.disord.19:1414-23,2004),表明多巴胺能系统涉及rls。例如,用下列用来治疗帕金森病的药物的临床研究表明对rls具有功效:(1)da激动剂:sinemet
tm
(左旋多巴、卡比多巴)、stalevo
tm
(左旋多巴、卡比多巴、恩他卡朋(entacapone))、permax
tm
(培高利特(pergolide))、parlodel
tm
(溴隐亭(bromocryptine));(2)d2、d3、d4激动剂:mirapex
tm
(普拉克索(pramipexole))、requip
tm
(罗匹尼罗(ropinirole));(3)mach拮抗剂:cogentin
tm
(本扎托品(benztropine))、artane
tm
(苯海索
(trihexyphenidyl));(4)mao抑制剂:eldepryl
tm
(司来吉兰(selegiline));(5)comt抑制剂tasmar
tm
(托卡朋(tolcapone))。参见例如hentz j.g.等,mov disord.15(2):324-7(2000);walters a.s.等,ann neurol 24(3):455-8(1988);trenkwalder c.等,neurology 62(8):1391-7(2004);polo o.等,clin neuropharmacol31(1):61(2007);kohnen r.sleep 22(8):1073-81(1999);以及shapiro c.mov disord 17(2):398-401(2002)。
102.本文所述mptp小鼠模型被普遍公认为帕金森病模型,但它还可代表特征是多巴胺不足的障碍或响应多巴胺受体激动剂的障碍(例如腿多动综合征)。因此,如同上的实施例5-7中所证实的一样,在mptp处理动物中所观察到的反应,可适度地视作可转换成腿多动综合征及特征是多巴胺不足的其它运动障碍,例如多巴胺反应性肌张力障碍、夏-德雷格综合征、睡眠中周期性肢体运动(plms)和图雷特综合征。
103.周期性肢体运动障碍(plmd)/睡眠中周期性肢体运动(plms)
104.周期性肢体运动障碍(plmd)是以睡眠期间周期性肢体运动(plms)继发的睡眠障碍为特征的综合征。虽然常与rls有关(manconi m.等,sleep med.8(5):491-7(2007);haba-rubio j.等,neurophysiol clin.33(4):180-4(2003)),但是在下面的情况下也可观察到plmd:脊髓损伤(de mello m.t.等,spinal cord.42(4):218-21(2004))、嗜眠症(hornyak m.等,sleep med rev.10(3):169-77(2006))、其它睡眠障碍(horyak,2006supra,saletu m.等,humpsychopharmacol.16(2):177-187(2001))或尿毒症(walker s.l.,等,sleep 19(3):214-8(1996))。
105.在不存在可识别的主要病理时可发生plmd(vetrugno r.等,neurol sci.28增刊1:s9-s14(2007);horyak,2006,同上)。在所有这些情况下,用左旋多巴(wolkove n.等,cmaj.176(10):1449-54(2007);de mello m.t.等,2004,同上)或多巴胺能激动剂(manconi m.等,sleepmed.8(5):491-7(2007);haba-rubio j.等,neurophysiol clin.33(4):180-4(2003);saletum.等,hum psychopharmacol.16(2):177-187(2001))观察到的临床改善表明多巴胺信号转导是主要功能障碍。因此,由于plmd和plms以多巴胺信号转导功能障碍为特征并可用左旋多巴治疗,所以我们认为当单独或与左旋多巴或其它多巴胺受体激动剂联合同时或序贯给予有需要的受治疗者时,pde7抑制剂可用于治疗plmd和/或plms。老年大鼠动物模型(参见baier p.c.等,j neurol sci.15;198(1-2):71-7(2002))可用来评价pde7抑制剂治疗plms的功效。
106.包括夏-德雷格综合征在内的多系统萎缩
107.多系统萎缩是一组进行性神经变性性疾病,包括夏-德雷格综合征、橄榄体脑桥小脑萎缩和纹状体黑质变性。特征性症状包括帕金森样运动异常、直立性低血压、膀胱功能障碍和小脑功能障碍(vanacore n.,jneural transm.112(12):1605-12(2005)。α突触核蛋白沉积于这两种疾病的尸体解剖样品中的发现,表明了与帕金森病的病理相似性(yoshida m.,neuropathology 27(5):484-93(2007);wenning g.k.等,acta neuropathol.109(2):129-40(2005);moore d.j.等,annu rev neurosci.28:57-87(2005)。左旋多巴通常用于减轻帕金森症状的疗法中,估计其反应速度介于33%和60%之间(gilman s.等,jneural transm.112(12):1687-94(2005);colosimo c.等,j neural transm.112(12):1695-704(2005))。由于一些多系统萎缩疾病(包括夏-德雷格综合征)可用左旋多巴治疗,因此我们认为当单独给予或与左旋多巴、多巴胺受体激动剂或其它多巴胺能药物联合同时或序贯给
予有需要的受治疗者时,pde7抑制剂可用于治疗对用多巴胺能药物治疗有治疗反应的多系统萎缩疾病的这些类型,例如夏-德雷格综合征。已知mptp模型是预测多系统萎缩(包括夏-德雷格综合征)的模型(stefanova n.等,trends neurosci.28(9):501-6(2005))。多系统萎缩的动物模型(参见stefanovan.等,trendsneurosci.28(9):501-6(2005))还可用来评价pde7抑制剂治疗多系统萎缩疾病(例如夏-德雷格综合征)的功效。
108.因此,根据本文上述研究,我们认为当单独给予或与多巴胺受体激动剂联合同时或序贯给予有需要的受治疗者时,pde7抑制剂可用于治疗对用多巴胺能药物治疗有治疗反应的多系统萎缩疾病,包括夏-德雷格综合征。
109.图雷特综合征
110.图雷特综合征是其中主要症状为刻板动作和秽语症(vocalizations)或抽搐(tics)的神经发育障碍(m
ü
ller n.dialogues clin neurosci.9(2):161-71(2007);leckman jf,等,j child neurol.21(8):642-9(2006))。解剖和神经成像证据表明该病涉及基底核的多巴胺能系统(m
ü
ller n.dialogues clin neurosci.9(2):161-71(2007))。虽然阻断d2多巴胺受体的抗精神病药是用于治疗图雷特综合征致残抽搐的药物类别之一,但是用多巴胺受体激动剂培高利特的双盲交叉临床研究表明该药显著改善抽搐(gilbert dl等,neurology.28;54(6):1310-5(2000))。
111.由于图雷特综合征的特征是多巴胺信号转导功能障碍,并可用多巴胺激动剂培高利特治疗,因此,我们认为当单独给予或与多巴胺受体激动剂或其它多巴胺能药物结合同时或序贯给予有需要的受治疗者时,pde7抑制剂可用于治疗图雷特综合征。
112.亨廷顿舞蹈病
113.亨廷顿舞蹈病是进行性遗传决定的致命性神经疾病,其特征是急动(舞蹈病),严重程度增加并伴有认知减退,最终导致完全无法运动,日常生活的活动性功能丧失。纹状体内中等棘状神经元的选择性丧失是主要的病理特征,一般被认为是舞蹈病动作(choreic movements)的首先要病因(standaert dg和young ab,goodman and gilman’s pharmacological basis of therapeutics,第10版,mcgraw-hill new york 2001;第22章,第562-564页)。没有药物可用于减缓亨廷顿舞蹈病的进程速度,且极少数药物可连续用于改善症状。最新综述将抗精神病药例如氟哌啶醇(haloperidol)引用为“可能用于”治疗舞蹈病动作。同一综述指出左旋多巴和多巴胺激动剂普拉克索“可能用于”治疗强直(bonelli rm等,currpharm des.12(21):2701-20.(2006))。少数报道提出左旋多巴或普拉克索可用于其中以帕金森症状为主的亨廷顿舞蹈病的特定变型(韦斯特法尔变型(westphal variant))(bonelli rm等,clin neuropharmacol.25(1):58-60(2002);reuter i,j neurol neurosurgpsychiatry.68(2):238-41(2000))。然而,没有进行对照试验。因此,有可能的是,pde7抑制化合物可用于对左旋多巴、其它多巴胺激动剂或前体或者其它多巴胺能药物起反应的亨廷顿舞蹈病患者。
114.多巴胺反应性肌张力障碍:
115.多巴胺反应性肌张力障碍(drd)是早期发作性进行性且主要由遗传决定的神经疾病,其特征是弥散性强直(diffuse rigidity)和其它帕金森样症状(segawa m等,advneurol.14:215-33(1976))。在纹状体中观察到多巴胺耗竭,但神经末梢却完整无缺。drd的主要病因是遗传性缺乏gtp环化水解酶,该酶是四氢生物蝶呤合成(segawa病)中的限
速酶,四氢生物蝶呤接着又是酪氨酸羟化酶的重要辅因子(ichinose h等,jbiol chem.380(12):1355-64(1999))。这种缺乏导致黑质纹状体末梢中的多巴和多巴胺耗竭。用左旋多巴/卡比多巴组合(例如息宁(sinemet))治疗取得重大成功,并成为该病的治疗标准(jeon b,jkorean medsci.12(4):269-79(1997))。因为该病易对左旋多巴和中等棘状神经元中多巴胺信号转导途径的完整性起反应,所以我们认为也将证实本发明的pde7抑制化合物可有效治疗drd。
116.iii.pde7抑制剂
117.根据环核苷酸磷酸二酯酶7型(pde7)的主要氨基酸序列和截然不同的酶促活性,pde7被鉴定为独特家族。被鉴定为pde7(pde7a和pde7b)的pde基因编码camp特异性pdes。pde7的生化和药理特性具有高亲和力camp特异性pde(km=0.2μm),它不受cgmp影响也不受其它pdes选择性抑制剂影响。pde7酶选择性分解camp,并被表征为不受咯利普兰(rolipram)抑制的酶,咯利普兰是独特的camp特异性pde家族的pde4的选择性抑制剂。在pde7家族内鉴定出两个亚型,即pde7a(michael,t.等,jbiol.chem.268(17):12925-12932,1993;han,p.等,jbiol.chem.272(26):16152-16157,1997)和pde7b(美国专利第6,146,876号;gardner,c.等,biochem.biophys.res.commun.272(1):186-192,2000;saski,t.等,biochem.biophys.res.commun.271(3):575-583,2000)。两个基因产物的c端催化结构域具有70%同一性(hetman j.m.等,pnas97(1):472-476(2000))。
118.pde7a具有3个剪切变异体(pde7a1、pde7a2和pde7a3);通过n端和c端两端的可变剪接产生这些变异体(bloom,t.j.和j.a.beavo,proc.natl.acad.sci.usa.93:14188-14192,1996)。pde7a的核苷酸序列,即转录物变异体1,容易通过登录号nm_002603在公共数据库中获取。人pde7a1蛋白(seq id no:2,由seq id no:1编码)具有456个氨基酸,沿还原sds-page迁移的表观分子量为53-55kda。
119.pde7a的核苷酸序列,即转录物变异体2,容易通过登录号nm_002604在公共数据库中获取。人pde7a2蛋白(seq id no:4,由seq id no:3编码)具有424个氨基酸。
120.pde7a蛋白在羧基端具有约270个氨基酸区域,与其它水解camp的pde的类似区域具有显著相似性(~23%同源性)。该区域起催化结构域的作用。该蛋白质的氨基端区域不同于其它pdes的此区域,并可能介导该酶家族所独有的特殊性质和调节性质。
121.人pde7b的核苷酸序列容易通过登录号nm_018945在公共数据库中获取,以seq id no:6提供,由seq id no:7编码。研究报道了pde7b的3个剪切变异体:pde7b1、pde7b2和pde7b3。pde7b公布于wo 01/62904、美国专利第6,146,876号。
122.pde7b2和pde7b3两者均具有独特的n端序列。人pde7b基因产物在还原sds-page上的表观分子量为53-55kda(sasaki,t.,kotera,j.,omori,k.,biochemicalj.361:211-220,2002)。同pde7a中一样,pde7b具有十分保守的大约270个氨基酸区域,这是所有pdes羧基端常见的区域,起催化结构域的作用。与pde7a蛋白相似,pde7b蛋白质的氨基端区域不同,可能解释pdes家族所独有的特殊性质和调节性质。pde7b蛋白在催化结构域内显示与其它camp依赖性pde同源(23%)。按照wo 2004/044196,pde7b多肽与pde7a有61%同源性。
123.比起其它pde家族,pde7也仅位于哺乳动物受治疗者体内。在大多数经分析的组织中检测出有pde7a表达,包括脑、心脏、肾、骨骼肌、脾和子宫(bloom等,pnas93:14188,1996)。在脑内,pde7a广泛分布在神经元细胞群和非神经元细胞群中(miro等,synapse40:
201,2001)。pde7a广泛表达于脑内(包括基底核和黑质),为pde7a在运动控制以及其它脑功能中所起的作用提供了理论基础。
124.虽然pde7a表达广泛分布于脑组织内,但是pde7b脑部表达受到更严格的限制,并高度富集于与运动控制有关的区域,例如纹状体(reyes-irisarri等,neuroscience 132:1173,2005)。然而,虽然脑组织中存在pde7,但是在本技术公开的数据之前,没有数据将pde7与任何特定cns疾病(例如帕金森病)联系起来。相反,根据表明用小干扰rnas(sirna)抑制pde7可调节t细胞增殖的研究,将pde7抑制剂的使用集中在免疫应用方面。参见rotella,d.p.,drug discovery2007,22-23。
125.与图2a-2c中所示的多巴胺信号转导模型一致,pde7a和pde7b的表达模式与多巴胺能系统重叠,支持了这样的理论,即pde7参与调节运动功能。因此,虽然不希望受理论的束缚,但是我们认为通过抑制pde7治疗pd起促进多巴胺信号转导的作用,与多巴胺受体激动剂相比,可以是用于治疗pd的选择性机制。我们还认为pde7抑制剂可用作与一种或多种多巴胺受体激动剂或其它多巴胺能药物联合(即在组合产品中,同时或序贯)给药的治疗剂。
126.在本发明方法的实施中,抑制pde7磷酸二酯酶活性的代表性pde7抑制剂包括:与pde7结合并抑制pde7酶活性的分子(例如与pde7结合并降低酶促活性的小分子抑制剂或封闭性肽(blocking peptide)),以及在转录和/或翻译水平上降低pde7表达的分子(例如pde7反义核酸分子、pde7特异性rnai分子和pde7核酶),从而防止pde7切割camp。pde7抑制剂可单独作为主要疗法或者可与其它治疗药(例如多巴胺受体激动剂)组合作为辅助疗法使用以提高治疗益处,正如上文所论述的一样。
127.pde7抑制的特征是由于按照本发明方法给予pde7抑制剂所产生的至少一种下列变化:抑制pde7依赖性酶促切割camp中的3'-磷酸二酯键形成5'-腺苷一磷酸(5'-amp)(例如按实施例1中所述方法测定)、降低pde7的基因或蛋白质表达水平,例如通过基因表达分析(例如rt-pcr分析)或蛋白质分析(例如蛋白质印迹)测定。
128.在一些实施方案中,pde7抑制剂是抑制pde7a、pde7b或pde7a与pde7b两者表达的分子或组合物,例如与对应于靶pde7基因的细胞mrna和/或基因组dna特异性杂交以抑制其转录和/或翻译的反义核苷酸或抑制性小核苷酸(例如sirna),或者特异性切割靶pde7的mrna的核酶。
129.pde7抑制剂的效价强度
130.在一个实施方案中,用于本发明方法的pde7抑制剂足以强效抑制pde7(pde7a、pde7b或pde7a与pde7b)的酶活性的化合物,其ic
50
≤1μm,优选小于或约为0.1μm。在一个实施方案中,pde7抑制剂足以强效抑制pde7(pde7a、pde7b或pde7a与pde7b)的酶活性,其ic
50
为约0.1nm至约500nm。在一个实施方案中,pde7抑制剂强效抑制pde7(pde7a、pde7b或pde7a与pde7b)的酶活性,ic
50
为约1nm至约100nm。
131.用于测定pde7(pde7a或pde7b)抑制剂的ic
50
的代表性方法如本文实施例1中提供,且是本领域众所周知的,例如披露于bardelle等人的闪烁亲近测定法(spa)(bardelle等,anal biochem15:275(2):148-55(1999))。
132.pde7a或pde7b选择性抑制剂
133.在一个实施方案中,用于本发明方法的pde7抑制剂是pde7a抑制剂。在一个实施方
案中,pde7a抑制剂强效抑制pde7a的酶活性,其ic
50
约为0.1nm至约500nm。在一个实施方案中,pde7a抑制剂的ic
50
约为1nm至约100nm。用于测定pde7a抑制剂的ic
50
的一个合适实验采用在杆状病毒系统中表达的重组人pde7a2酶。该实验方法是spa测定法(bardelle等,同上)的改动方法。用于测定pde7a抑制的一个示例性测定法见实施例1。
134.在一些实施方案中,pde7抑制剂具有针对pde7a的同工酶选择性活性。pde7a选择性抑制剂降低pde7a活性超过降低pde7b活性的至少2倍,更优选至少10倍、至少20倍、至少50倍或至少100倍。在一些实施方案中,pde7a抑制剂是抑制pde7a活性的选择性超出抑制任何其它pde(pde1-6、7b和8-11)酶活性的至少10倍(例如至少20倍或至少50倍或至少100倍)的抑制剂。
135.在另一个实施方案中,用于本发明方法的pde7抑制剂是pde7b抑制剂。由于因pde7b表达受限制以及在与运动控制有关的脑区(例如纹状体)中高水平表达可能降低副作用,因此pde7b的抑制剂可用于治疗神经性运动障碍,例如帕金森病。
136.在一个实施方案中,pde7b抑制剂的ic
50
约为0.1nm至约500nm。在一个实施方案中,pde7b抑制剂足以强效抑制pde7b的酶活性,其ic
50
约为0.1nm至约500nm。在一个实施方案中,pde7b抑制剂的ic
50
约为1nm至约100nm。用于测定pde7b抑制剂ic
50
的方法是本领域众所周知的,例如bardelle等人(同上)披露的测定法。用于测定pde7ab抑制的示例性测定法见实施例1。
137.在一些实施方案中,pde7抑制剂具有针对pde7b的同工酶选择性活性。pde7b选择性抑制剂降低pde7b活性超过降低pde7a活性的至少2倍,更优选至少10倍、至少20倍、至少50倍或至少100倍。在一些实施方案中,pde7b抑制剂是抑制pde7b活性的选择性超过任何其它pde(pde1-6、7a和8-11)酶活性的至少10倍(例如至少20倍或至少50倍或至少100倍)的抑制剂。
138.与其它pde相比的pde7的选择性
139.在一些实施方案中,抑制pde1b活性的pde7抑制剂的ic
50
是抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者的5倍以上(例如至少10倍、至少20倍或至少50倍或至少100倍)。换句话说,pde7抑制剂在抑制pde7a或pde7b活性(pde7抑制剂对pde7a或pde7b同工酶哪一个具有最大作用)时,比它在抑制pde1b活性时更强效(达5倍、10倍、20倍、50倍或100倍)。举例来说,对于本说明书的目的,该性质可甚至更简单地表述为pde7抑制剂在抑制pde7活性时比它抑制pde1b活性时更强效(达5倍、10倍、20倍、50倍或100倍)。
140.根据小鼠中pde1b基因缺失刺激多巴胺代谢并使动物对多巴胺能激动剂的作用变得敏感的报道(siuciak等,neuropharmacology53(1):113-23(2007)),对pde7和pde1b两者的双重抑制可在运动障碍治疗中提供额外益处。
141.在一些实施方案中,pde7抑制剂抑制pde10活性的ic
50
是抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者的5倍以上(例如至少10倍或至少20倍或至少50倍或至少100倍)。根据pde10选择性抑制剂引起纹状体中camp水平升高的报道(siuciak j.a.等,neuropharmacology 51(2):386-96(2006)),对pde7和pde10两者的双重抑制可在运动障碍治疗中提供额外益处。
142.在一些实施方案中,pde7抑制剂抑制pde3活性的ic
50
是抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者的10倍以上(例如至少20倍、至少50倍或至少100倍)。这是因
为有研究表明给予心力衰竭患者pde3选择性抑制剂增加他们的死亡率(packer m.等,n engl j med.325(21):1468-75(1991))。
143.在一些实施方案中,pde7抑制剂抑制pde4活性的ic
50
是抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者的10倍以上(例如至少20倍、至少50倍或至少100倍)。这是因为研究显示,小鼠pde4基因之一的缺失导致心肌病(lehnart s.e.等,cell123(1):25-35(2005))。
144.在一些实施方案中,pde7抑制剂在pde4抑制作用的体内试验(例如在内毒素处理后的tnfα水平的镇静或抑制作用)的半数最大有效剂量("ed50")是pde7a和pde7b的抑制作用的体内试验(例如在mptp-处理的动物中步长(stride length)的恢复)的ed50中的较小者的10倍以上(例如至少20倍、至少50倍或至少100倍)。依据此类实施方案,已确定某些具有pde4/pde7双重抑制活性的化合物在体内对pde7的选择性抑制作用大于对pde4的选择性抑制作用,如同在体外试验中所测定的化合物的pde4/pde7选择性进行比较的结果。在一些实施方案中,pde7抑制剂抑制pde3活性和pde4活性的ic
50
是抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者的10倍以上(例如至少20倍、至少50倍或至少100倍)。
145.在一些实施方案中,pde7抑制剂抑制pde8活性的ic
50
是抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者的10倍以上(例如至少20倍、至少50倍或至少100倍)。
146.在一些实施方案中,pde7抑制剂抑制pde4活性和pde8活性的ic
50
是抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者的10倍以上(例如至少20倍、至少50倍或至少100倍)。按照该实施方案,已知特异性/优先水解camp的pde家族包括pde4、pde7和pde8。
147.在一些实施方案中,pde7抑制剂抑制pde1、pde2、pde3、pde4、pde8、pde10和pde11活性的ic
50
是抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者的10倍以上。按照该实施方案,已知特异性/优先水解camp的pde家族包括pde4、pde7和pde8,且pde1、pde2、pde3、pde10,pde11家族针对camp和cgmp显示出相当大的活性。
148.在一些实施方案中,pde抑制剂是这样的选择性pde7抑制剂,即抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者是抑制pde1-6和pde8-11酶家族的任何其它pde酶的抑制剂ic
50
的1/10以下(例如1/20、1/50、或1/100)。
149.例如,可通过将药物抑制pde7(pde7a、pde7b或pde7a与pde7b)酶活性的能力与其抑制其它pde家族的pde酶的能力进行比较,来鉴定选择性pde7抑制剂。例如,可测定药物抑制pde7活性以及pde1、pde2、pde3、pde4、pde5、pde6、pde8、pde9、pde10和pde11的能力,来对药物进行评价。本文实施例2中提供了将药物抑制pde7酶活性的能力与抑制其它pde家族的pde酶的能力进行比较的示例性方法。可通过标准体外、体内或离体测定法,例如本文所述测定法,来测定抑制各个pde(1-6和8-11)同工酶的ic
50
与抑制pde7的ic
50
的比率(即较敏感的pde7a或pde7b)。
150.在一些实施方案中,pde7抑制剂对pde7具有选择性并且对抑制其它pdes(例如,pde1、pde2、pde3、pde4,和pde8、pde10,和pde11)基本没有活性,这是由于pde7抑制剂靶向一个或多个靶组织,例如脑和/或骨骼肌。如本文所描述的,相对于其它pde家族,pde7唯一地局限于哺乳动物受试者中。在脑内,pde7a广泛地分布于神经元和非神经元细胞群体中,包括基底神经节和黑质(miro等,synapse 40:201,2001)。pde7b表达于脑纹状体中(reyes-irisarri等,neuroscience 132:1173,2005)。
151.与已知涉及神经性运动障碍的其它非pde分子靶标相比的pde7选择性
152.在一些实施方案中,pde7抑制剂对pde7有选择性,且对已知或被认为是涉及神经性运动障碍病理的非pde分子靶标基本无活性。在一些实施方案中,pde7抑制剂是这样的pde7抑制剂,其抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者是对以下其它分子靶标具有抑制活性的药物ic
50
的1/2以下(例如1/5以下、1/10以下,例如1/20以下、1/50以下或1/100以下):(i)已知涉及选自以下神经性运动障碍病理的靶标:帕金森病、脑炎后帕金森综合征、多巴胺反应性肌张力障碍、夏-德雷格综合征、周期性肢体运动障碍(plmd)、睡眠中周期性肢体运动(plms)和腿多动综合征(rls),或(ii)治疗上有效的治疗疾病的其它药物所作用的分子靶标。
153.在其它实施方案中,pde7抑制剂对pde7有选择性,且对已知涉及帕金森病病理的非pde分子靶标基本无活性。在一些实施方案中,pde7抑制剂是这样pde7抑制剂,其抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者是对以下其它分子靶标具有抑制活性的药物ic
50
的1/2以下(例如1/5以下、1/10以下、1/20以下、1/50以下或1/100以下):(i)已知涉及帕金森病病理的靶标,例如儿茶酚-o-甲基转移酶(comt)、单胺氧化酶b(mao-b)、多巴胺转运蛋白(dat)、酪氨酸羟化酶、多巴胺受体、腺苷a2a受体、毒蕈碱性乙酰胆碱受体、n-甲基d-天冬氨酸(nmda)受体、γ氨基丁酸(gaba)受体和加巴喷丁受体,或(ii)治疗上有效的治疗帕金森病的其它药物所作用的分子靶标。用于将某一药物抑制pde7酶活性的能力与其抑制已知涉及帕金森病病理的其它分子靶标的能力进行比较的示例性方法见本文实施例4。
154.在其它实施方案中,pde7抑制剂对pde7有选择性,且对已知与多巴胺信号转导途径有关的非pde分子靶标基本无活性。在一些实施方案中,pde7抑制剂是这样的pde7抑制剂,其抑制pde7a活性的ic
50
和抑制pde7b活性的ic
50
中的较小者具有对已知与多巴胺信号转导途径有关的以下其它分子靶标具有活性的药物ic
50
的1/2以下(例如1/5以下、1/10以下,例如1/20以下、1/50以下或1/100以下):例如儿茶酚-o-甲基转移酶(comt)、单胺氧化酶b(mao-b)、多巴胺转运蛋白(dat)、酪氨酸羟化酶、多巴脱羧酶、多巴胺受体、腺苷酸环化酶、蛋白激酶a(pka)、多巴胺和环腺苷酸调节的分子量为32,000的磷蛋白(darpp32)及蛋白质磷酸酶-1。用于将某一药物抑制pde7酶活性的能力与其抑制已知与多巴胺信号转导途径有关的其它分子靶标的能力进行比较的示例性方法见本文实施例4。
155.pde7抑制剂的类型
156.pde7抑制剂可以是任何类型的物质,包括但不限于化合物、蛋白质或多肽、肽模拟物、核酸分子或核酶。在一些实施方案中,pde7抑制剂是小分子抑制剂,包括具有低分子量(即小于约450g/摩尔)的天然物质和合成物质,例如肽、肽模拟物和非肽抑制剂,例如化合物。
157.化合物:
158.用于本发明方法的pde7抑制剂包括可以通过常规途径(例如口服、肌内、皮下、经皮、经口腔、静脉内等)进入血流并最终通过血管系统穿过血脑屏障以抑制脑内pde7的分子。因此,对于这些给药方法,pde7抑制剂具有穿过血脑屏障的能力。下文所述的具有穿过血脑屏障能力的这些pde抑制剂(例如分子量小于约450g/摩尔且是足够亲脂的pde抑制剂),当通过最终转运到脑部血流中的途径给予时,可用于本发明的方法。
159.下面描述了用于本发明方法的示例性pde7抑制剂。
160.在一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于ep 1454897、wo 2003/053975和us 20050148604的化合物,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0161][0162]
上述化合物的取代基的定义如下:
[0163]
a表示n或cr4,
[0164]
b表示氢原子或卤素原子,
[0165]
r1表示任选取代的c
3-7
环烷基或叔丁基,
[0166]
r2表示氢、甲基或乙基,
[0167]
r3表示氢、硝基、氰基或卤素原子、nr5r6、c(=x)r7、so2nr5r6、or8、nr8conr5r6、nr8so2r9、nr8co2r9、杂芳基、任选取代的c
1-3
烷基、任选取代的c
1-6
烯基或者任选取代的饱和或不饱和的杂环烷基,
[0168]
r4表示氢,或需要时被一个或多个氟原子取代的c
1-3
烷氧基,
[0169]
r5和r6相同或不同,表示氢原子、任选取代的c
1-6
烷基、任选取代的杂环烷基或任选取代的酰基,或者与它们所连接的氮原子一起形成氮杂环丁烷基、吡咯烷基、哌啶基、吗啉代、硫代吗啉代、哌嗪基或高哌嗪基,这些基团的每一个被以下基团任选取代:任选取代的c
1-4
烷基、oh、c
1-3
烷氧基、co2h、nr5r6、氧代基、nr9cor7或c(=o)r7,
[0170]
r7表示任选取代的c
1-6
烷基、oh、or8或nr5r6,
[0171]
r8表示氢、任选取代的c
1-6
烷基或任选取代的杂环烷基,
[0172]
r9表示任选取代的c
1-6
烷基,
[0173]
x表示o、s或nh。
[0174]
关于上述化合物,“任选取代(的)”是指任选取代的直链、支链或环状烷基,例如甲基、乙基、丙基或环己基;羟基;氰基;烷氧基,例如甲氧基或乙氧基;任选取代的氨基,例如氨基、甲基氨基或二甲基氨基;任选取代的酰基,例如乙酰基或丙酰基;羧基;任选取代的芳基,例如苯基或萘基;任选取代的杂芳基,例如吡啶基、噻唑基、咪唑基或吡唑基(pyrazyl);任选取代的饱和或不饱和的杂环烷基,例如哌嗪基或吗啉基(morphonyl);任选取代的氨基甲酰基;任选取代的酰氨基;卤素原子,例如氯、氟或溴;硝基;任选取代的磺基;任选取代的磺酰胺基;氧代基;脲基;以及任选取代的直链、支链或环状烯基,例如乙烯基、丙烯基或环己烯基。
[0175]
杂芳基如r3的实例包括5-7元具有2-8个碳原子并含有1-4个由氧原子、氮原子或硫原子组成的杂原子的单环杂芳基,以及包含两个或更多个这类相同或不同的单环化合物
稠合在一起的多环杂芳基,单环杂芳基和多环杂芳基的实例为吡咯基、呋喃基、噻吩基、咪唑基、噻唑基、吡啶基、吡唑基(pyrazyl)、吲哚基、喹啉基、异喹啉基和四唑基。
[0176]
在一个实施方案中,用于本发明的pde7抑制剂具有下列结构式:
[0177][0178]
实施例1和实施例2描述了化合物1在抑制所选的pdes中的活性。实施例5和实施例6描述了化合物1在mptp帕金森病模型中的疗效。
[0179]
在其它实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0180][0181]
在另一个实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0182][0183]
实施例1和实施例2描述了化合物2在抑制所选的pdes中的活性。实施例7描述了化合物2在mptp帕金森病模型中的疗效。
[0184]
在其它实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0185][0186]
上述化合物的制备参见ep 1454897、wo 2003/053975和us 20050148604。
[0187]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利文献的化合物:us 2002/0198198、wo 2002/076953、wo 2002/074754、wo 2006/092691、bioorganic&medicinal chemistry letters 14(2004)4623-4626以及bioorganic&medicinal chemistryletters 14(2004)4627-4631,所述各参考文献通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂或其互变异构体、其外消旋体、其异构体及其药学上可接受的衍生物具有下列结构式:
[0188][0189]
上述化合物的取代基的定义如下:
[0190]
(a)x1、x2、x3和x4相同或不同,且选自:
[0191]
n,前提条件是x1、x2、x3和x4中不超过2个基团同时表示氮原子,或者,
[0192]
c-r1,其中r1选自:
[0193]
q1,或者
[0194]
低级烷基、低级烯基或低级炔基,这些基团是未取代的或者被一个或若干个基团q2取代;
[0195]
基团x
5-r5,其中,
[0196]
x5选自:
[0197]
单键,
[0198]
低级亚烷基、低级亚烯基或低级亚炔基(alkynylene);被1-2个选自o、s、s(=o)、so2或n的杂原子任选分隔开,这些基团的碳原子是未取代的或者被一个或若干个相同或不同的选自以下的基团取代:sr6、or6、nr6r7、=o、=s或=nr6,其中r6和r7相同或不同,且选自氢或低级烷基,
[0199]
r5选自芳基;杂芳基;被c(=o)或被1、2或3个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烷基;被c(=o)或者被1、2或3个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烯基;或二环基,这些基团是未取代的或者被一个或若干个选自q3、杂芳基的基团取代,或者是被q3任选取代的低级烷基;
[0200]
其中q1、q2和q3相同或不同,且选自:
[0201]
氢、卤素、cn、no2、so3h、p(=o)(oh)2、or2、oc(=o)r2、c(=o)or2、sr2、s(=o)r2、
nr3r4、q-r2、q-nr3r4、nr
2-q-nr3r4或nr
3-q-r2,其中q选自c(=nr)、c(=o)、c(=s)或so2,r选自氢或低级烷基,r2、r3和r4相同或不同,且选自:
[0202]
氢、被c(=o)任选分隔开的低级烷基、(ch2)
n-芳基、(ch2)
n-杂芳基、被c(=o)或者被1或2个选自o、s、s(=o)、so2或n的杂原子任选分隔开的(ch2)
n-环烷基,其中n为选自0、1、2、3或4的整数;
[0203]
这些基团是未取代的或者被一个或若干个选自以下的基团取代:低级烷基、卤素、cn、ch3、so3h、so2ch3、cf3、c(=o)nhso2ch3、or6、coor6、c(=o)r6、nr6r7、c(=o)nr6r7或so2nr6r7,其中r6和r7相同或不同,且选自氢或者被一个或两个选自or、coor或nrr8的基团任选取代的低级烷基,其中r和r8为氢或低级烷基,
[0204]
r6和r7和/或r3和r4,可与它们所连接的氮原子一起形成4-8元杂环,该环可含有一个或两个选自o、s、s(=o)、so2或n的杂原子,并可被以下基团取代,
[0205]
4-8元杂环,该环可含有一个或两个选自o、s或n的杂原子,并可被低级烷基取代,或者,
[0206]
被or'、nr'r”、c(=o)nr'r”或coor'任选取代的低级烷基,其中r'和r”相同或不同,且选自h、被or或coor任选取代的低级烷基,其中r为氢或低级烷基,且r'和r”可与它们所连接的氮原子一起形成4-8元杂环,该环可含有一个或两个选自o、s或n的杂原子;或者,
[0207]
(b)x为o、s或nr9,其中r9选自氢、cn、oh、nh2、低级烷基、低级烯基或低级炔基,这些基团是未取代的或者被以下基团取代:被1或2个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烷基;被1或2个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烯基;芳基;杂芳基;or
10
或nr
10r11
,其中r
10
和r
11
相同或不同,且选自氢或低级烷基;
[0208]
(c)y选自o、s或n-r
12
,其中r
12
选自氢、cn、oh、nh2、低级烷基、低级烯基或低级炔基,这些基团是未取代的或者被以下基团取代:被1或2个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烷基;被1或2个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烯基;芳基;杂芳基;or
10
或nr
10r11
,其中r
10
和r
11
相同或不同,且选自氢或低级烷基;
[0209]
(d)z选自ch-no2、o、s或nr
13
,其中r
13
选自氢;cn;oh;nh2;芳基;杂芳基;被一个或若干个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烷基;被一个或若干个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烯基;c(=o)r
14
;c(=o)nr
14r15
;or
14
或者未取代或被一个或若干个相同或不同且选自or
14
或nr
14r15
的基团取代的低级烷基;
[0210]r14
和r
15
独立选自氢或低级烷基,或者,r
14
和r
15
可与它们所连接的氮原子一起形成4-8元杂环,该杂环可含有一个或两个选自o、s或n的杂原子,并可被低级烷基取代;
[0211]
(e)z1选自h、ch3或nr
16r17
,其中r
16
和r
17
相同或不同,且选自氢;cn;芳基;杂芳基;被一个或若干个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烷基;被一个或若干个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烯基;c(=o)r
14
;c(=o)nr
14r15
;or
14
或者未取代或被一个或若干个选自or
14
或nr
14r15
的基团取代的低级烷基,
[0212]r14
和r
15
选自氢或低级烷基,且r
14
和r
15
和/或r
16
和r
17
可与它们所连接的氮原子一起形成4-8元杂环,该杂环可含有一个或两个选自o、s或n的杂原子,并可被低级烷基取代;
[0213]
(f)a为选自以下的环:
[0214][0215]
其中
[0216]
a1、a2、a3、a4、a5和a6相同或不同,选自o、s、c、c(=o)、so、so2或nr
18
,其中r
18
选自氢;芳基;杂芳基;被一个或若干个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烷基;被一个或若干个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烯基;未取代或被芳基、杂芳基取代的低级烷基;被一个或若干个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烷基;被一个或若干个选自o、s、s(=o)、so2或n的杂原子任选分隔开的环烯基;cn;nr
19r20
;c(=o)nr
19r20
;or
19
;c(=o)r
19
或c(=o)or
19
,其中r
19
和r
20
相同或不同,且选自氢或低级烷基;
[0217]
*表示由a环和含有x和/或y的主环共用的碳原子;
[0218]
a环中的各个碳原子是未取代或者被1或2个相同或不同选自以下的基团取代:被or
21
、nr
21r22
、coor
21
或conr
21r22
任选取代的低级烷基、低级卤代烷基、cn、f、=o、so2nr
19r20
、or
19
、sr
19
、c(=o)or
19
、c(=o)nr
19r20
或nr
19r20
,其中r
19
和r
20
相同或不同,且选自氢或被or
21
、nr
21r22
、coor
21
或conr
21r22
任选取代的低级烷基,其中r
21
和r
22
相同或不同,且选自氢或低级烷基,且r
19
和r
20
和/或r
21
和r
22
可与它们所连接的氮原子一起形成4-8元杂环;
[0219]
a环中不相邻的两个原子可通过2、3或4个碳原子链连接,该链由1个选自o、s或n的杂原子分隔开;前提条件是a1、a2、a3、a4、a5和a6中不超过2个基团同时表示杂原子。
[0220]
对于上述化合物,卤素包括氟、氯、溴和碘。优选的卤素为f和cl。低级烷基包括具有1-6个碳原子的直链和支链碳链。这类烷基的实例包括甲基、乙基、异丙基和叔丁基。低级烯基包括具有2-6个碳原子和至少一个双键的直链和支链烃基。这类烯基的实例为乙烯基、3-丁烯-1-基、2-乙烯基丁基和3-己烯-1-基。低级炔基包括具有2-6个碳原子和至少一个三键的直链和支链烃基。这类炔基的实例为乙炔基、3-丁炔-1-基、丙炔基、2-丁炔-1-基和3-戊炔-1-基。低级卤代烷基包括被一个或若干个卤素取代如上定义的的低级烷基。卤代烷基的实例为三氟甲基。芳基应理解为是指含有介于6和10个碳原子的芳香碳环。芳基的实例为苯基。杂芳基包括具有5-10环原子,其中1-4个独立选自o、s和n的芳环。代表性杂芳基在5或6元芳环中具有1、2、3或4个杂原子。这类基团的实例为四唑、吡啶基和噻吩基。代表性环烷基含有3-8个碳原子。这类基团的实例为环丙基、环丁基、环戊基、环己基、环庚基和环辛基。术语“被分隔开”是指在主链上,碳原子被杂原子或如本文定义的基团置换。例如,在“被c(=o)或者被1个选自o、s、s(=o)so2或n的杂原子任选分隔开的环烷基或环烯基”中,术语“被分隔开”是指c(=o)或杂原子可置换环中的碳原子。这类基团的实例为吗啉或哌嗪。环烯基包括3-10元含有至少一个双键的环烷基。杂环包括如上定义的杂芳基和如上定义的环烷基或环烯基,被1、2或3个选自o、s、s(=o)、so2或n的杂原子分隔开来。二环取代基是指相同或不同且选自芳基、杂环基环、环烷基或环烯基的两个环稠合在一起形成所述二环取代基。二环取代基的实例为吲哚基。
[0221]
在一个实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0222][0223]
实施例1和实施例2描述了化合物3在抑制所选的pdes中的活性。实施例7描述了化合物3在mptp帕金森病模型中的疗效。
[0224]
在其它实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0225][0226]
上述化合物的制备参见us 2002/0198198、wo 2002/076953、wo2002/074754、wo 2006/092691、bioorganic&medicinal chemistry letters 14(2004)4623-4626以及bioorganic&medicinal chemistry letters 14(2004)4627-4631。
[0227]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利文献的化合物:ep 1193261、wo 2002/28847、us 20030045557、美国专利第7,122,565号、bioorganic&medicinal chemistry letters 14(2004)4607-4613以及bioorganic&medicinal chemistryletters 14(2004)4615-4621,所述各参考文献通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂或其药学上可接受的衍生物具有下列结构式:
[0228][0229]
上述化合物的取代基的定义如下:
[0230]
y为s或o;
[0231]
r1为c
1-c
10
烷基、c
2-c
10
烯基、c
2-c
10
炔基、环烷基、环烯基、杂环、芳基或多环基团;各自被一个或若干个相同或不同的基团x
1-r4任选取代,其中x1为单键、低级亚烷基、c
2-c6亚烯基、亚环烷基、亚芳基或二价杂环,r4为:
[0232]
(1)h、=o、no2、cn、卤素、低级卤代烷基、低级烷基、羧酸生物电子等排体(carboxylic acidbioisostere);
[0233]
(2)coor5、c(=o)r5、c(=s)r5、so2r5、sor5、so3r5、sr5、or5;
[0234]
(3)c(=o)nr7r8、c(=s)nr7r8、c(=ch-no2)nr7r8、c(=n-cn)nr7r8、c(=n-so2nh2)nr7r8、c(=nr7)nhr8、c(=nr7)r8、c(=nr9)nhr8、c(=nr9)r8、so2nr7r8或nr7r8,其中r7和r8相同或不同,且选自oh、r5、r6、c(=o)nr5r6、c(=o)r5、so2r5、c(=nr9)nhr
10
、c(=nr9)r
10
、c(=ch-no2)nr9r
10
、c(=n-so2nh2)nr9r
10
、c(=n-cn)nr9r
10
或c(=s)nr9r
10
;
[0235]
r2为低级烷基、c
2-c
10
烯基、c
2-c
10
炔基、环烷基、环烯基、杂环、芳基;各自被一个或若干个相同或不同且选自以下的基团任选取代:
[0236]
(1)h、羧酸生物电子等排体、低级卤代烷基、卤素,
[0237]
(2)coor5、or5、so2r5,
[0238]
(3)so2nr
11r12
、c(=o)nr
11r12
、nr
11r12
,其中r
11
和r
12
相同或不同,且选自oh、r5、r6、c(=o)nr5r6、c(=o)r5、so2r5、c(=s)nr9r
10
、c(=ch-no2)nr9r
10
、c(=n-cn)nr9r
10
、c(=n-so2nh2)nr9r
10
、c(=nr9)nhr
10
或c(=nr9)r
10
;
[0239]
r3为x
2-r'3,其中x2为单键或选自以下的基团:c
1-c4亚烷基、c
2-c6亚烯基、c
2-c6亚炔基,各自被一个或若干个相同或不同且选自以下的基团任选取代:
[0240]
(1)h、c
1-c3烷基、c
3-c4环烷基、芳基、杂环、=o、cn,
[0241]
(2)or5、=nr5;或者
[0242]
(3)nr
13r14
,其中r
13
和r
14
相同或不同,且选自r5、r6、c(=o)nr5r6、c(=o)r5、so2r5、c(=s)nr9r
10
、c(=ch-no2)nr9r
10
、c(=nr9)nhr
10
或c(=nr9)r
10
;
[0243]
r'3为环烷基、环烯基、芳基、杂环或多环基团;各自被一个或若干个基团x
3-r
17
任选取代,其中x3为单键、低级亚烷基、c
2-c6亚烯基、c
2-c6亚炔基、亚环烷基、亚芳基、二价杂环或二价多环基团,且r
17
为:
[0244]
(1)h、=o、no2、cn、低级卤代烷基、卤素、羧酸生物电子等排体、环烷基,
[0245]
(2)coor5、c(=o)r5、c(=s)r5、so2r5、sor5、so3r5、sr5、or5;
[0246]
(3)c(=o)nr
15r16
、c(=s)nr
15r16
、c(=n-cn)nr
15r16
、c(=n-so2nh2)nr
15r16
、c(=ch-no2)nr
15r16
、so2nr
15r16
、c(=nr
15
)nhr
16
、c(=nr
15
)r
16
、c(=nr9)nhr
16
、c(=nr9)r
16
或nr
15r16
,其中r
15
和r
16
相同或不同,且选自oh、r5、r6、c(=o)nr5r6、c(=o)r5、so2r5、c(=s)
nr9r
10
、c(=ch-no2)nr9r
10
、c(=n-cn)nr9r
10
、c(=n-so2nh2)nr9r
10
、c(=nr9)nhr
10
或c(=nr9)r
10
,
[0247]
(4)被一个或若干个基团r5任选取代的杂环;
[0248]
其中r5和r6相同或不同,且选自h、低级烷基、c
2-c6烯基、c
2-c6炔基、x
4-环烷基、x
4-环烯基、x
4-芳基、x
4-杂环或x
4-多环基团,其中x4为单键、低级亚烷基或c
2-c6亚烯基;各自被一个或若干个相同或不同且选自以下的基团任选取代:卤素、=o、coor
20
、cn、or
20
、被or
20
任选取代的o-低级烷基、c(=o)-低级烷基、低级卤代烷基,
[0249][0250]
其中x5为单键或低级亚烷基,r
18
、r
19
和r
20
相同或不同,且选自h或低级烷基;
[0251]
x
6-杂环、x
6-芳基、x
6-环烷基、x
6-环烯基或x
6-多环基团,其中x6为单键或低级亚烷基,这些基团被一个或若干个相同或不同选自以下的基团任选取代:卤素、coor
21
、or
21
或(ch2)nnr
21r22
,其中n为0、1或2,r
21
和r
22
相同或不同,且选自h或低级烷基;
[0252]
r9选自h、cn、oh、低级烷基、o-低级烷基、芳基、杂环、so2nh2,或者
[0253][0254]
其中x5为单键或低级亚烷基,r
18
和r
19
相同或不同,且选自h或低级烷基;
[0255]r10
选自氢、低级烷基、环丙基或杂环。
[0256]
对于上述化合物,芳基是指在环结构中只含有碳原子的不饱和碳环,碳原子数介于5和10之间,包括苯基、萘基或四氢萘基。杂环是指不饱和或饱和的单环,在环结构中含有介于1和7个之间的碳原子,且环结构中有至少一个杂原子(例如氮、氧或硫),优选为1-4个相同或不同的选自氮、硫和氧原子的杂原子。合适的杂环包括吗啉基、哌嗪基、吡咯烷基、哌啶基、嘧啶基、2-呋喃基和3-呋喃基、2-噻吩基和3-噻吩基、2-吡啶基、2-吡喃基和3-吡喃基、羟基吡啶基、吡唑基、异唑基、四唑、咪唑、三唑等。多环基团包括至少两个相同或不同的选自芳基、杂环、环烷基、环烯基的环稠合在一起形成的所述多环基团,例如2-苯并噻吩基和3-苯并噻吩基、2-苯并呋喃基和3-苯并呋喃基、2-吲哚基、2-喹啉基和3-喹啉基、吖啶基、喹唑啉基、吲哚基苯并[1,3]二氧杂环戊烯基和9-硫代氧杂蒽烷基(thioxantanyl)。二环基是指相同或不同且选自芳基、杂环、环烷基或环烯基的两个环稠合在一起形成的所述二环基。卤素是指氟、氯、溴或碘。低级烷基是指含有1-6个碳原子的直链或支链烷基。低级烷基的实例包括甲基、乙基、丙基、丁基、异丙基、叔丁基、异丁基、正丁基、戊基、己基等。烯基是指含有一个或若干个双键、优选一个或两个双键的直链或支链的不饱和碳原子链。炔基是指含有一个或若干个三键、优选一个或两个三键的直链或支链的不饱和碳原子链。低级卤代烷基是指被一个或若干个卤素取代的低级烷基;优选的低级卤代烷基包括全卤代烷基,例如cf3。环烷基是指含有3-10个碳原子的饱和单碳环;包括环丙基、环丁基、环戊基、环己基和环庚基。环烯基是指含有3-10个碳原子的不饱和单碳环。合适的环烯基的实例为3-环己烯和3-环戊烯。羧酸生物电子等排体具有典型含义;常见的羧酸生物电子等排体为四唑-5-基、c(=o)n(h)oh、异唑-3-基、羟基噻二唑基、磺酰胺基、磺酰基甲酰氨基、膦酸、膦酰基氨基(phosphonamido)、次膦酸、磺酸、酰基磺酰胺基、巯基唑(mercaptoazole)、酰基氨
腈。
[0257]
在一个实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0258][0259]
实施例1和实施例2描述了化合物4在抑制若干pdes中的活性。实施例7描述了化合物4在mptp帕金森病模型中的疗效。
[0260]
在其它实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0261][0262]
上述化合物的制备参见ep 1193261、wo 02/28847、us 20030045557、美国专利第7,122,565号、bioorganic&medicinal chemistry letters 14(2004)4607-4613以及bioorganic&medicinal chemistryletters 14(2004)4615-4621。
[0263]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:wo 2004/111054、us 20060128728和us 20070270419,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂或其药学上可接受的盐或溶剂化物具有下列结构式:
[0264][0265]
上述化合物的取代基的定义如下:
[0266]
r1是取代或未取代的c
3-8
环烷基或叔丁基;
[0267]
r2为氢原子或c
1-3
烷基;
[0268]
r3为基团:nr5r6、c(=o)r7或s(o)
0-2
r8;
[0269]
r4为氢原子或为未取代的或被一个或多个氟原子取代的c
1-3
烷氧基;
[0270]
r5和r6彼此相同或不同,为氢原子、取代或未取代的c
1-6
烷基、取代或未取代的酰基、取代或未取代的杂环烷基及与结合r5和r6的氮原子一起形成的取代或未取代的杂环烷基环;
[0271]
r7为基团:or9或nr5r6;
[0272]
r8为氢原子、卤素原子、基团:nr5r6、取代或未取代的c
1-6
烷基或者取代或未取代的
芳基;
[0273]
r9为氢原子或者取代或未取代的c
1-6
烷基。
[0274]
对于上述化合物,术语“c
1-c3烷基”包括具有1-3个碳原子的直链或支链烷基。术语“c
3-c8环烷基”包括具有3-8个碳原子的环烷基,例如环丙基、环丁基、环戊基、环己基和环辛基。术语“杂环烷基”为3-7元含有1-4个相同或不同的例如氧、氮或硫原子的杂原子的杂环基,实例可包括吡咯烷基、哌啶基、哌嗪基、高哌嗪基、四氢呋喃基、四氢吡喃基、吗啉基和氮杂环丁烷基。术语“c
1-c3烷氧基”是指具有1-3个碳原子的烷氧基。术语“酰基”是指具有1-8个碳原子的酰基。术语“芳基”为具有6-12个碳原子的苯基、萘基、联苯基。术语“杂芳基”为5-7元含有2-8个碳原子和1-4个相同或不同的例如氧、氮、硫原子的杂原子的单环或其多环基团。实例包括吡咯基、呋喃基、噻吩基、咪唑基、噻唑基、吡嗪基、吲哚基、喹啉基、异喹啉基、四唑基、吡啶基、吡唑基、哒嗪基和嘧啶基。“取代或未取代的c
1-c6烷基”的合适取代基的实例包括羟基和卤素原子,“取代或未取代的酰基”的合适取代基的实例包括卤素原子和硝基。此外,“取代或未取代的芳基”的合适取代基的实例包括c
1-c3烷基、卤素原子、氨基、酰基、酰胺基、羟基、酰氨基、羧基和磺酰基。“取代或未取代的c
3-c8环烷基”的合适取代基的实例为c
1-c3烷基、羟基和氧代基,“取代或未取代的杂环烷基”的合适取代基的实例可包括羧基、酰基、烷氧基、氨基、烷基氨基、酰氨基、羟基、氧代基、亚乙基二氧基、甲基、乙基和羟基乙基。
[0275]
在其它实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0276]
[0277][0278]
上述化合物的制备参见wo 2004/111054、us 20060128728和us20070270419。
[0279]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:美国专利第6,903,109号、us20040082578、wo 2003/088963和us 20060154949,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂及其药学上可接受的盐、酯和前药形式具有下列结构式:
[0280][0281]
上述化合物的取代基的定义如下:
[0282]
(a)r1选自:
[0283]
(i)cor5,其中r5选自h、任选取代的c
1-8
直链或支链烷基、任选取代的芳基和任选取代的芳基烷基;其中烷基、芳基和芳基烷基上的取代基选自c
1-8
烷氧基、苯乙酰基氧基、羟基、卤素、对甲苯磺酰氧基、甲磺酰氧基、氨基、氰基、烷氧羰基或nr
20r21
,其中r
20
和r
21
独立选自氢、c
1-8
直链或支链烷基、c
3-7
环烷基、苄基或芳基;
[0284]
(ii)coor6,其中r6选自h、任选取代的c
1-8
直链或支链烷基、任选取代的芳基和任选取代的芳基烷基;其中烷基、芳基和芳基烷基上的取代基选自c
1-8
烷氧基、苯乙酰基氧基、羟
基、卤素、对甲苯磺酰氧基、甲磺酰氧基、氨基、氰基、烷氧羰基或nr
20r21
,其中r
20
和r
21
独立选自氢、c
1-8
直链或支链烷基、c
3-7
环烷基、苄基或芳基;
[0285]
(iii)氰基;
[0286]
(iv)与r4形成的内酯或内酰胺;
[0287]
(v)conr7r8,其中r7和r8独立选自h、c
1-8
直链或支链烷基、c
3-7
环烷基、三氟甲基、羟基、烷氧基、酰基、烷基羰基、羧基、芳基烷基、芳基、杂芳基和杂环基;其中烷基、环烷基、烷氧基、酰基、烷基羰基、羧基、芳基烷基、芳基、杂芳基和杂环基可被以下基团取代:羧基、烷基、芳基、取代芳基、杂环基、取代杂环基、杂芳基、取代杂芳基、异羟肟酸、磺酰胺基、磺酰基、羟基、巯基、烷氧基或芳基烷基;
[0288]
或者r7和r8与它们所连接的氮一起形成杂环基或杂芳基;
[0289]
(vi)包括任选取代的杂芳基的羧酸酯或羧酸生物电子等排体;
[0290]
(b)r2选自任选取代的烷基、任选取代的芳基、任选取代的杂芳基、任选取代的c
3-7
环烷基、任选取代的杂环基,其中杂环基为1,3-二氧杂环戊烷或呋喃,或者r2为
[0291]
(c)r3为1-4个独立选自以下的基团:
[0292]
(i)氢、卤素、c
1-8
直链或支链烷基、芳基烷基、c
3-7
环烷基、c
1-8
烷氧基、氰基、c
1-4
烷氧羰基、三氟甲基、c
1-8
烷基磺酰基、卤素、硝基、羟基、三氟甲氧基、c
1-8
羧酸基(carboxylate)、芳基、杂芳基和杂环基;
[0293]
(ii)nr
10r11
,其中r
10
和r
11
独立选自h、c
1-8
直链或支链烷基、芳基烷基、c
3-7
环烷基、羧基烷基、芳基、杂芳基或杂环基,或者r
10
和r
11
与它们所连接的氮一起形成杂环基或杂芳基;
[0294]
(iii)nr
12
cor
13
,其中r
12
选自氢或烷基,r
13
选自氢、烷基、取代烷基、c
1-3
烷氧基、羧基烷基、r
30r31
n(ch2)
p
、r
30r31
nco(ch2)
p
、芳基、芳基烷基、杂芳基或杂环基,或者r
12
和r
13
与羰基一起形成含有杂环基的羰基,其中r
30
和r
31
独立选自h、oh、烷基和烷氧基,p为1-6的整数,其中烷基可被以下基团取代:羧基、烷基、芳基、取代芳基、杂环基、取代杂环基、杂芳基、取代杂芳基、异羟肟酸、磺酰胺、磺酰基、羟基、巯基、烷氧基或芳基烷基;
[0295]
(d)r4选自(i)氢,(ii)c
1-3
直链或支链烷基,(iii)苄基,(iv)nr
13r14
,其中r
13
和r
14
独立选自氢和c
1-6
烷基;其中c
1-3
烷基和苄基被一个或多个选自以下的基团任选取代:c
3-7
环烷基、c
1-8
烷氧基、氰基、c
1-4
烷氧羰基、三氟甲基、c
1-8
烷基磺酰基、卤素、硝基、羟基、三氟甲氧基、c
1-8
羧酸、氨基、nr
13r14
、芳基和杂芳基;
[0296]
(e)x选自s和o。
[0297]
在一个替代实施方案中,r1、r3和r4如上定义,r2为nr
15r16
,其中r
15
和r
16
独立选自氢、c
1-8
直链或支链烷基、芳基烷基、c
3-7
环烷基、芳基、杂芳基和杂环基,或者r
15
和r
16
与它们所连接的氮一起形成杂环基或杂芳基。
[0298]
对于上述化合物,“烷基”是指直链、环状和支链烷基。烷基可被一个或多个以下基团任选取代:例如卤素、oh、cn、巯基、硝基、氨基、c
1-c
8-烷基、c
1-c
8-烷氧基、c
1-c
8-烷硫基、c
1-c
8-烷基-氨基、二(c
1-c
8-烷基)氨基、(一、二、三和全)卤代-烷基、甲酰基、羧基、烷氧基羰基、c
1-c
8-烷基-co-o-、c
1-c
8-烷基-co-nh-、氨甲酰、异羟肟酸、磺酰胺、磺酰基、巯基、芳
基、芳基(c
1-c8)烷基、杂环基和杂芳基。术语“生物电子等排体”定义为“具有产生大体类似的生物学性质的理化性质的基团或分子”(burger's medicinal chemistry and drug discovery,m.e.wolff编著,第5版,第1卷,1995,第785页)。本文所用术语“酰基”,不论是单独使用还是作为取代基的部分,是指通过脱去羟基由有机酸衍生的具有2-6个碳原子有机基团(支链或直链)。“芳基”或“ar”不论是单独使用还是作为取代基的部分是指碳环芳基,包括但不限于苯基、1-萘基或2-萘基等。碳环芳基上的1-5个氢原子可被以下基团独立置换:卤素、oh、cn、巯基、硝基、氨基、c
1-c
8-烷基、c
1-c
8-烷氧基、c
1-c
8-烷硫基、c
1-c
8-烷基-氨基、二(c
1-c
8-烷基)氨基、(一、二、三和全)卤代-烷基、甲酰基、羧基、烷氧基羰基、c
1-c
8-烷基-co-o-、c
1-c
8-烷基-co-nh-或氨甲酰。示例性芳基包括例如苯基、萘基、联苯、氟苯基、二氟苯基、苄基、苯甲酰氧基苯基、乙氧羰基苯基、乙酰基苯基、乙氧基苯基、苯氧基苯基、羟基苯基、羧基苯基、三氟甲基苯基、甲氧基乙基苯基、乙酰氨基苯基、甲苯基、二甲苯基、二甲基氨基甲酰基苯基等。术语“杂芳基”是指具有5-10个环原子的环状完全不饱和基团,其中一个环原子选自s、o和n;0-2个环原子为独立选自s、o和n的额外杂原子;其余环原子为碳。该基团可通过任何环原子与分子其余部分连接起来。术语“杂环基(heterocycle)”、“杂环的”和“杂环(heterocycle)”是指任选取代的完全饱和或部分饱和的环状基团,例如在至少一个含碳原子环中具有至少一个杂原子的4-7元单环、7-11元双环或10-15元三环系统。含杂原子杂环基中的各个环可具有1、2或3个选自氮原子、氧原子和硫原子的杂原子,其中氮杂原子和硫杂原子还可任选被氧化。氮原子可任选被季铵化。杂环基可连接在任何杂原子或碳原子上。
[0299]
在其它实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0300][0301]
上述化合物的制备参见美国专利第6,903,109号、us 20040082578、wo 2003/088963和us 20060154949。
[0302]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:美国专利第6,958,328号、wo 2002/085894和us 20030212089,所述各专利和专利申请通过引用以其整体明确结合到本文中。这些pde7抑制剂具有与上述专利(例如美国专利第6,903,109号)相同的结构式,只是r1不是羧酸酯或羧酸生物电子等排体。这些化合物的制备参见美国专利第6,958,328号、us 20030212089和wo 2002/085894。
[0303]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献的化合物:wo 2006/004040和ep 1775298,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂或其药学上可接受的盐或溶剂化物具有下列结构式:
[0304][0305]
上述化合物的取代基的定义如下:
[0306]
r1为取代或未取代的c
3-8
烷基、取代或未取代的环烷基或者取代或未取代的杂环烷基(例如环己基、环庚基或四氢吡喃基);
[0307]
r2为氢原子或者取代或未取代的c
1-3
烷基(例如甲基);
[0308]
r3为氢原子、取代或未取代的c
1-3
烷基或卤素原子;
[0309]
r4为取代或未取代的芳基、取代或未取代的杂芳基或基团conr5r6或co2r7,
[0310]
其中r5和r6彼此相同或不同,为氢原子、可被卤素原子取代的c
1-6
烷基、取代或未取代的芳基、取代或未取代的杂芳基、取代或未取代的杂环烷基、取代或未取代的环烷基、基团nr7cor8、cor8、nr9r
10
、取代或未取代的环烷基、取代或未取代的杂环烷基、取代或未取代的芳基、取代或未取代的杂芳基,或者取代或未取代的杂环烷基,其中与结合r5和r6的氮原子一起形成所述环;
[0311]
其中r7为氢原子或者取代或未取代的c
1-3
烷基;
[0312]
其中r8为取代或未取代的杂环烷基,或基团oh、or7或nr9r
10
;
[0313]
其中r9和r
10
彼此相同或不同,为氢原子、取代或未取代的c
1-3
烷基、取代或未取代的杂环烷基、取代或未取代的酰基、基团so2r7,或者取代或未取代的杂环烷基,其中与结合r5和r6的氮原子一起形成所述环。
[0314]
对于上述化合物,术语“环烷基”是指具有3-8个碳原子的环烷基。术语“杂环烷基”可为3-7元含有1-4个相同或不同的例如氧、氮或硫原子的杂原子的单环或多环杂环基,实例可包括哌啶基、吡咯烷基、哌嗪基、四氢呋喃基、四氢吡喃基、吗啉基、氮杂环丁烷基、咪唑烷基、唑烷基、六氢吡咯烷基、octahydroindolidinyl、octahydroquinolidinyl、八氢吲哚基,及其氧代-衍生物。术语“芳基”可为芳烃基,由单苯环或者结合或稠合苯环组成,例如苯基、萘基、联苯等;芳基还可为二环或三环基团,由苯环与环烷基或杂环稠合组成,例如1,2,3,4-四氢萘、2,3-二氢茚、二氢吲哚、苯并呋喃等。术语“杂芳基”可为5-7元单环杂芳基或多环杂芳基,具有2-8个碳原子与1-4个例如氧、氮、硫原子的杂原子,其中多环杂芳基具有由相同或不同的单环杂芳基或苯环彼此稠合的环系;或者多环基团由杂芳基与环烷基或杂环烷基环稠合组成。本发明的合适取代基的实例可包括可被一个或多个以下基团取代的直链、支链或环状c
1-c8烷基:甲基、乙基、丙基、异丙基、正丁基、叔丁基、环己基、环庚基、甲氧基甲基、羟基甲基、三氟甲基、c
1-c3烷氧基、卤素原子和羟基;羟基;氰基;取代或未取代的烷氧基,例如甲氧基、乙氧基;可被c
1-c6烷基或酰基取代的氨基,例如氨基、甲基氨基、乙基氨
基、二甲基氨基、酰基氨基等;羧基;取代或未取代的酯基;磷酸基;磺基;取代或未取代的芳基;取代或未取代的杂芳基;可被取代的饱和或不饱和杂环烷基;取代或未取代的氨基甲酰基;取代或未取代的酰胺基;取代或未取代的硫代酰胺基;卤素原子;硝基;取代或未取代的磺基;取代或未取代的磺酰基酰胺基;氧代基;取代或未取代的脲基;直链、支链或环烯基,例如乙烯基、丙烯基、环己烯基等。
[0315]
在其它实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0316]
[0317][0318]
上述化合物的制备参见ep 1775298和wo 2006/004040。
[0319]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下
专利的化合物:wo 2004/111053和us 20060128707,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂或其药学上可接受的盐或溶剂化物具有下列结构式:
[0320][0321]
上述化合物的取代基的定义如下:
[0322]
a为n或cr4;
[0323]
b为n或ch;
[0324]
r1为取代或未取代的c
3-8
环烷基或叔丁基;
[0325]
r2为氢原子或c
1-6
烷基;
[0326]
r3为氢原子;硝基;氰基;卤素原子;杂芳基;取代或未取代的c
1-6
烷基;取代或未取代的c
2-6
烯基;被取代或未取代的饱和或不饱和杂环烷基;基团:nr5r6、c(o)r7、so2r7、or8、nr8cor7、nr8so2r7;
[0327]
r4为氢原子或者未取代或被一个或多个氟原子取代的c
1-3
烷氧基;
[0328]
r5和r6彼此相同或不同,为氢原子;取代或未取代的c
1-6
烷基;取代或未取代的酰基;或者取代或未取代的杂环烷基;
[0329]
r7为氢原子;取代或未取代的c
1-6
烷基;取代或未取代的杂环烷基;oh;or8或nr5r6;
[0330]
r8为氢原子、取代或未取代的c
1-6
烷基或者取代或未取代的杂环烷基。
[0331]
对于上述化合物,术语“c
1-c6烷基”是指具有1-6个碳原子的直链或支链烷基,术语“c
2-c6烯基”是指具有2-6个碳原子的直链或支链烯基。术语“环烷基”是指具有3-8个碳原子的环烷基,例如环丙基、环丁基、环戊基、环己基、环庚基和环辛基。术语“杂环烷基”为3-7元含有1-4个相同或不同的例如氧、氮或硫原子的杂原子的杂环基,实例可包括哌啶基、吡咯烷基、哌嗪基、四氢呋喃基、四氢吡喃基、吗啉基、氮杂环丁烷基和高哌嗪基。术语“杂芳基”为5-7元含有2-8个碳原子和1-4个相同或不同的例如氧、氮或硫原子的杂原子的单环或其多环基团。实例包括吡咯、呋喃基、噻吩基、咪唑基、噻唑基、吡嗪基、吲哚基、喹啉基、异喹啉基、四唑基、吡啶基、吡唑基、哒嗪基和嘧啶基。“卤素原子”包括氟、氯、溴和碘。“取代或未取代的c
1-c6烷基”、“取代或未取代的c
3-c8环烷基”、“取代或未取代的烯基”、“取代或未取代的杂环烷基”和“取代或未取代的酰基”的合适取代基的实例包括直链或支链、或者取代或未取代的烷基,例如甲基、乙基、丙基、异丙基、正丁基、叔丁基;取代或未取代的环烷基,例如环丙基、环丁基、环戊基、环己基和环庚基;羟基;氰基;烷氧基,例如甲氧基和乙氧基;取代或未取代的氨基,例如氨基、甲基氨基、乙基氨基和二甲基氨基;取代或未取代的酰基,例如乙酰基和丙酰基;取代或未取代的芳基;取代或未取代的杂芳基;取代或未取代的饱和或不饱和杂环烷基;取代或未取代的氨基甲酰基;取代或未取代的酰胺基;卤素原子;硝基;取代或未取代的磺基;氧代基;脲基;取代或未取代的直链或支链或环烯基,例如乙烯基、丙烯
基和环己烯基。
[0332]
在其它实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0333][0334]
上述化合物的制备参见us 20060128707和wo 2004/111053。
[0335]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利文献的化合物:美国专利第6,617,357号、us20020156064和molecular pharmacology,66:1679-1689,2004,所述各参考文献通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂或其药学上可接受的盐或溶剂化物具有下列结构式:
[0336][0337]
上述化合物的取代基的定义如下:
[0338]
r1为nrarb,其中ra和rb独立地为h或c
1-6
烷基,或者表示由碳或者碳与一个或多个选自o、n或s的额外杂原子组成的5-7元环;
[0339]
r2为h、c
1-8
烷基、c
1-3
烷基-ar、c
1-3
烷基-c
3-6
环烷基、c
2-8
烯基、c
2-4
烯基-ar或c
2-4
烯基-c
3-6
环烷基,其中ar为取代或未取代的苯基;
[0340]
r3为no2、卤素、cn、c(o)or7、cor1或nrarb,其中ra和rb独立地为h或c
1-6
烷基;
[0341]
r4为h、oc
1-6
烷基、卤素、c(o)nrarb、c(o)or7、c
1-8
烷基、ochf2、ch2or8、oc
1-3
烷基-ar或ch2nhc(o)ch3;
[0342]
r5为h、卤素或烷基;
[0343]
r6为c
1-8
烷基、oc
1-4
烷基或卤素;
[0344]
r7为氢或为酯或酰胺-形成基团;
[0345]
r8为氢或c
1-6
烷基。
[0346]
在一个实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0347][0348]
上述化合物的制备参见美国专利第6,617,357号、us 20020156064和molecularpharmacology,66:1679-1689,2004。
[0349]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献的化合物:美国专利第6,852,720号、ep 1348433和wo 2003/082277,所述各专利和专
利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂其外消旋体、其异构体、其n-氧化物或其药学上可接受的酸式盐或碱式盐具有下列结构式:
[0350][0351]
上述化合物的取代基的定义如下:
[0352]
r1为选自环烷基、杂环烷基、芳基和杂芳基的基团,这些基团被一个或多个相同或不同彼此独立选自以下的基团任选取代:卤素、三氟甲基、硝基、氰基、氧代、nr4r5、co2r4、conr4r5、or4、s(o)nr4、s(o)nr4r5、四唑基和被1-3个相同或不同彼此独立选自以下的基团任选取代的(c
1-c6)烷基:or4、nr4r5和co2r4;其中n为0-2的整数,0和2也包括在内,r4和r5相同或不同且彼此独立地为氢原子或者式x
1-ra基团,其中x1为单键或(c
1-c6)亚烷基,ra为选自(c
1-c6)烷基、环烷基、杂环烷基、芳基和杂芳基的基团,
[0353]
r2为选自(c
1-c6)烷基、(c
2-c6)烯基、(c
2-c6)炔基、芳基和环烷基的基团,
[0354]
r3为选自环烷基、杂环烷基、芳基和杂芳基的基团,这些基团被一个或多个相同或不同彼此独立选自以下的基团任选取代:卤素、硝基、氰基、三氟甲基、氧代、(c
1-c6)烷基、or6、nr6r7、cor6、co2r6、conhoh、conr6r7、s(o)mr6、s(o)mnr6r7、nr6cor7、nr6so2r7、n(so2r7)2、nr6conr7r8、c(=n-cn)nr6r7、nr8c(=n-cn)nr6r7和被(c
1-c4)烷基任选取代的四唑基,其中m为0-2的整数,0和2也包括在内,r6和r7相同或不同且彼此独立地为氢原子或者式x2rb基团,其中x2为单键或(c
1-c6)亚烷基,rb为选自(c
1-c6)烷基、环烷基、杂环烷基、芳基和杂芳基的基团,这些基团被1-3个相同或不同彼此独立选自以下的基团任选取代:羟基、(c
1-c6)烷氧基、(c
1-c6)烷基、氨基、一(c
1-c6)烷基氨基、二(c
1-c6)烷基氨基(各烷基氨基相同或不同,彼此独立)、羧基、(c
1-c6)烷氧基羰基和苄基,r8表示氢原子或(c
1-c6)烷基。
[0355]
上述化合物的制备参见美国专利第6,852,720号、ep 1348433和wo 2003/082277。
[0356]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:美国专利第6,753,340号、us20030191167、ep 1348701和wo 2003/082839,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂其外消旋体、其异构体、其n-氧化物或其药学上可接受的酸式盐或碱式盐具有下列结构式:
[0357][0358]
上述化合物的取代基的定义如下:
[0359]r1a
为基团选自氢、(c
1-c6)烷基和芳基(c
1-c6)烷基,
[0360]r1b
为选自环烷基、杂环烷基、芳基和杂芳基的基团,这些基团被一个或多个相同或不同彼此独立选自以下的基团任选取代:卤素、三氟甲基、硝基、氰基、氧代、nr4r5、co2r4、conr4r5、or4、s(o)nnr4、s(o)nnr4r5、四唑基和被1-3个相同或不同彼此独立选自以下的基团任选取代的(c
1-c6)烷基:or4、nr4r5和co2r4,其中n为0-2的整数,0和2也包括在内,r4和r5相同或不同且彼此独立地为氢原子或者式x
1-ra基团,其中x1为单键或(c
1-c6)亚烷基,ra为选自以下的基团:(c
1-c6)烷基、环烷基、杂环烷基、芳基和杂芳基,
[0361]
r2为选自(c
1-c6)烷基、(c
2-c6)烯基、(c
2-c6)炔基、芳基和环烷基的基团,
[0362]
r3为选自环烷基、杂环烷基、芳基和杂芳基的基团,这些基团被一个或多个相同或不同彼此独立选自以下的基团任选取代:卤素、硝基、氰基、三氟甲基、氧代、(c
1-c6)烷基、or6、nr6r7、cor6、co2r6、conhoh、conr6r7、s(o)mr6、s(o)mnr6r7、nr6cor7、nr6so2r7、n(so2r7)2、nr6conr7r8、c(=n-cn)nr6r7、nr8c(=n-cn)nr6r7和被(c
1-c4)烷基任选取代的四唑基,其中m为0-2的整数,0和2也包括在内,r6和r7相同或不同且彼此独立地为氢原子或式x
2-rb基团,其中x2为单键或(c
1-c6)亚烷基,rb为选自(c
1-c6)烷基、环烷基、杂环烷基、芳基和杂芳基的基团,这些基团被1-3个相同或不同彼此独立选自以下的基团任选取代:羟基、(c
1-c6)烷氧基、(c
1-c6)烷基、氨基、一(c
1-c6)烷基氨基、二(c
1-c6)烷基氨基(各烷基氨基相同或不同、彼此独立)、羧基、(c
1-c6)烷氧基羰基和苄基,r8为氢原子或(c
1-c6)烷基。
[0363]
这些化合物的制备参见美国专利第6,753,340号、us 20030191167、ep 1348701和wo 2003/082839。
[0364]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:美国专利第6,849,638号、us 20030119829和wo 2002/088138,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂及其药学上可接受的盐具有下列结构式:
[0365][0366]
上述化合物的取代基的定义如下:
[0367]
r1和r2独立选自氢、1-8个碳原子的烷基、2-8个碳原子的烯基、2-8个碳原子的炔基、3-7个碳原子的环烷基、2-6个碳原子和1-2个选自nh、s和o的杂原子的完全饱和杂环、6-12个碳原子的芳基,该芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基;6-12个碳原子的芳基或4-11个碳原子和1、2个选自n、s和o的杂原子的杂芳基、4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,该杂芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基;6-12个碳原子的芳基或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基及r
4-r5,或者r1和r2与它们所连接的氮
原子结合在一起形成5-7元可含有1-2个选自nh、nr8、s和o的额外杂原子的饱和环,或者与它们所连接的氮原子结合在一起形成5-7元可含有1-2个选自n、s和o的额外杂原子的不饱和环;
[0368]
其中所述饱和或不饱和环可被选自以下的1-2个取代基取代:oh、1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、3-7个碳原子的环烷基、2-6个碳原子和1-2个选自nh、s和o的杂原子的完全饱和杂环、卤素、1-2个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子的烷氧基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基及r
9-r
10
;或者
[0369]
r1和r2与它们所连接的氮原子结合在一起形成8-10元饱和二环;
[0370]
r3选自nh、s、s(=o)2和o;
[0371]
r4选自1-8个碳原子的烷基、2-8个碳原子的烯基、2-8个碳原子的炔基、c(=c)、s(=o)2和c(=o)o;
[0372]
r5选自氢、oh、1-8个碳原子的烷基、2-8个碳原子的烯基、2-8个碳原子的炔基、1-8个碳原子的烷氧基、6-12个碳原子的芳基,该芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基;6-12个碳原子的芳基及4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基;4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,该杂芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基、6-12个碳原子的芳基及4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基、3-7个碳原子的环烷基、2-6个碳原子和1-2个选自nh、s和o的杂原子的完全饱和杂环及nr6r7,
[0373]
r6和r7独立选自氢、1-8个碳原子的烷基、2-8个碳原子的烯基和2-8个碳原子的炔基,或者r6和r7与它们所连接的氮原子结合在一起形成5-7元可含有1-2个选自n、s和o的额外杂原子的不饱和环,或者形成5-7元可含有1-2个选自nh、s和o的额外杂原子的饱和环;
[0374]
r8选自1-8个碳原子的烷基、2-8个碳原子的烯基、2-8个碳原子的炔基、r
11-r
12
、3-7个碳原子的环烷基、2-6个碳原子和1-2个选自nh、s和o的杂原子的完全饱和杂环、6-12个碳原子的芳基,该芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基、6-12个碳原子的芳基或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基、4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,该杂芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基、6-12个碳原子的芳基或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基;
[0375]
r9选自1-8个碳原子的烷基、2-8个碳原子的烯基和2-8个碳原子的炔基;
[0376]r10
选自oh、6-12个碳原子的芳基,该芳基可被以下基团取代:1-6个碳原子的烷基、
2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基、6-12个碳原子的芳基或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,以及4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,该杂芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基、6-12个碳原子的芳基或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基;
[0377]r11
选自1-8个碳原子的烷基、2-8个碳原子的烯基和2-8个碳原子的炔基;
[0378]r12
选自3-7个碳原子的环烷基、2-6个碳原子和1-2个选自nh、s和o的杂原子的完全饱和杂环、6-12个碳原子的芳基,该芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基、6-12个碳原子的芳基或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,以及4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,该杂芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基、6-12个碳原子的芳基或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基。
[0379]
这些化合物的制备参见美国专利第6,849,638号、us 20030119829和wo 2002/088138。
[0380]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:us 2005222138和wo 2003/064389,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0381][0382]
上述化合物的取代基的定义如下:
[0383]
r1和r2各自独立地为:(1)氢原子,或(2)c
1-8
烷基,或者
[0384]
r1和r2可与它们所连接的碳原子一起形成环1(cyc1),
[0385]
其中r1和r2不同时表示氢原子;
[0386]
z为(1)cr3r4,(2)o,(3)s,或(4)化学键;
[0387]
r3和r4各自独立地为:(1)氢原子,(2)c
1-8
烷基,(3)c
1-8
烷氧基,或(4)羟基,或者
[0388]
r3和r4可与它们所连接的碳原子一起形成环1或c(o);
[0389]
r5和r6各自独立地为:(1)氢原子,或(2)c
1-8
烷基,或者
[0390]
r5和r6可与它们所连接的碳原子一起形成环1;
[0391]
由r1和r2、r3和r4、r5和r6表示的环1各自独立地为(1)c3-10环烷基,或(2)3-10元含有1-2个选自氧、氮和硫的杂原子的单环杂环,环1可被r
10
取代;
[0392]r10
为(1)c
1-8
烷基,(2)c
1-8
烷氧基,(3)羟基,(4)coor
11
,(5)氧代,(6)so2r
12
,或(7)cor
13
;
[0393]r11
为氢原子或c
1-8
烷基;
[0394]r12
和r
13
为(1)c
1-8
烷基,或(2)可被c
1-8
烷基取代的苯基;
[0395]
r7和r8各自独立地为:(1)氢原子,(2)c
1-8
烷基,(3)c
1-8
烷氧基,(4)羟基,(5)氰基,(6)卤素原子,(7)coor
14
,(8)conr
15r16
,(9)环2,(10)c
2-8
烯基,(11)c
2-8
炔基,(12)nr
51r52
,(13)硝基,(14)甲酰基,(15)c
2-8
酰基,(16)被以下基团取代的c
1-8
烷基:羟基、c
1-8
烷氧基、环2、nr
51r52
或nr
53-环2,(17)nr
54
cor
55
,(18)nr
56
so2r
57
,(19)so2nr
58r59
,(20)被coor
14
取代的c
2-8
烯基,(21)ch=n-oh,(22)c
1-8
亚烷基-nr
60-(c
1-8
亚烷基)-r
61
,(23)c
1-8
烷硫基,(24)被1-3个卤素原子取代的c
1-8
烷基,(25)被1-3个卤素原子取代的c
1-8
烷氧基,(26)被环2取代的c
1-8
烷氧基,(27)o-环2,(28)oso2r
65
,或(29)ch=n-or
137
;
[0396]r14
为氢原子或c
1-8
烷基;
[0397]r15
和r
16
各自独立地为氢原子或c
1-8
烷基;
[0398]r51
和r
52
、r
58
和r
59
各自独立地为氢原子或c
1-8
烷基;
[0399]r53
、r
54
、r
56
和r
60
各自独立地为氢原子或c
1-8
烷基;
[0400]r55
为氢原子、c
1-8
烷基或c
1-8
烷氧基;r
57
为c
1-8
烷基;
[0401]r61
为nr
62r63
或羟基;
[0402]r62
和r
63
各自独立地为氢原子或c
1-8
烷基;
[0403]r65
为c
1-8
烷基;
[0404]r137
为c
1-8
烷基;
[0405]
(下文中简称环)为环2,其中与羰基连接的基团为碳;
[0406]
由环表示的r7、r8和环2各自独立地为:(1)c
3-15
一环、二环或三环(稠合或螺)碳环,或(2)含有1-4个选自氧、氮和硫的杂原子的3-15元一环、二环或三环(稠合或螺)杂环;
[0407]
环2可被1-5个r
17
或r
17'
取代;
[0408]r17
为(1)c
1-8
烷基,(2)c
2-8
烯基,(3)c
2-8
炔基,(4)c
1-8
烷氧基,(5)c
1-8
烷硫基,(6)羟基,(7)卤素原子,(8)硝基,(9)氧代,(10)羧基,(11)甲酰基,(12)氰基,(13)nr
18r19
,(14)可被1-5个r
20
取代的苯基、苯氧基或苯硫基,(15)可被1-5个r
21
取代的c
1-8
烷基、c
2-8
烯基、c
1-8
烷氧基或c
1-8
烷硫基,(16)ocor
22
,(17)conr
23r24
,(18)so2nr
25r26
,(19)coor
27
,(20)cocoor
28
,(21)cor
29
,(22)cocor
30
,(23)nr
31
cor
32
,(24)so2r
33
,(25)nr
34
so2r
35
或(26)sor
64
;
[0409]r18
和r
19
、r
31
和r
34
各自独立地为氢原子或c
1-8
烷基;
[0410]r20
和r
21
为c
1-8
烷基、c
1-8
烷氧基、羟基、卤素原子、硝基或coor
36
;
[0411]r22
和r
64
各自独立地为c
1-8
烷基;
[0412]r23
、r
24
、r
25
和r
26
各自独立地为氢原子、c
1-8
烷基或苯基;
[0413]r27
、r
28
、r
29
、r
30
、r
32
、r
33
和r
35
为(1)c
1-8
烷基,(2)c
2-8
烯基,(3)被1-5个r
37
取代的c
1-8
烷基,(4)二苯基甲基,(5)三苯基甲基,(6)环3,(7)被环3取代的c
1-8
烷基或c
2-8
烯基,(8)被o-环3、s-环3或so
2-环3取代的c
1-8
烷基;
[0414]r36
为氢原子或c
1-8
烷基;
[0415]r37
为c
1-8
烷氧基、c
1-8
烷硫基、苄氧基、卤素原子、硝基或coor
38
;
[0416]r38
为氢原子、c
1-8
烷基或c
2-8
烯基;
[0417]
环3为(1)c
3-15
一环、二环或三环(稠合或螺)碳环,或(2)3-15元含有1-4个选自氧、氮和硫的杂原子的一环、二环或三环(稠合或螺)杂环;
[0418]
环3可被1-5个r
39
取代;
[0419]r39
为(1)c
1-8
烷基,(2)c
2-8
烯基,(3)c
2-8
炔基,(4)c
1-8
烷氧基,(5)c
1-8
烷硫基,(6)羟基,(7)卤素原子,(8)硝基,(9)氧代,(10)氰基,(11)苄基,(12)苄氧基,(13)被1-5个r
40
取代的c
1-8
烷基、c
1-8
烷氧基或c
1-8
烷硫基,(14)可被1-5个r
41
取代的苯基、苯氧基、苯硫基、苯基磺酰基或苯甲酰基,(15)ocor
42
,(16)so2r
43
,(17)nr
44
cor
45
,(18)so2nr
46r47
,(19)coor
48
或(20)nr
49r50
;
[0420]r40
为卤素原子;
[0421]r41
为c
1-8
烷基、c
1-8
烷氧基、卤素原子或硝基;
[0422]r42
、r
43
和r
45
为c
1-8
烷基;
[0423]r44
和r
48
为氢原子或c
1-8
烷基;
[0424]r46
和r
47
、r
49
和r
50
各自独立地为氢原子或c
1-8
烷基;
[0425]r17'
为(1)sh,(2)nr
66
cho,(3)环5,(4)被环5取代的c
1-8
烷基、c
2-8
烯基或c
2-8
炔基,(5)co-(nh-氨基酸残基-co)
n-oh,(6)nr
67
conr
68r69
,(7)conr
70
nr
71r72
,(8)conr
73
or
74
,(9)conr
75
cor
76
,(10)c(s)nr
77r78
,(11)conr
79
c(s)coor
80
,(12)nr
81
cocoor
82
,(13)nr
83
coor
84
,(14)conr
85
c(s)r
86
,(15)ocor
87
,(16)sor
88
,(17)conr
89r90
,(18)so2nr
91r92
,(19)coor
93
,(20)cocoor
94
,(21)cor
95
,(22)cocor
96
,(23)nr
97
cor
98
,(24)so2r
99
,(25)nr
100
so2r
101
或(26)nr
102r103
;
[0426]
n为1或2的整数;
[0427]r66
、r
73
、r
75
、r
77
、r
79
、r
81
、r
83
、r
85
、r
97
、r
100
和r
102
为氢原子或c
1-8
烷基;
[0428]r67
和r
68
、r
70
和r
71
各自独立地为氢原子或c
1-8
烷基;
[0429]r89
和r
91
为(1)氢原子,(2)c
1-8
烷基,(3)苯基,或(4)被氰基或c
1-8
烷氧基取代的c
1-8
烷基;
[0430]r103
为环6;
[0431]r69
、r
72
、r
74
、r
76
、r
78
、r
80
、r
82
、r
84
、r
86
、r
87
、r
88
、r
90
和r
92
为(1)氢原子,(2)c
1-8
烷基,(3)c
2-8
烯基,(4)c
2-8
炔基,(5)被1-5个r
104
取代的c
1-8
烷基,(6)二苯基甲基,(7)三苯基甲基,(8)环6,(9)被环6取代的c
1-8
烷基或c
2-8
烯基,或(10)被o-环6、s-环6或so
2-环6取代的c
1-8
烷基;
[0432]r104
为(1)c
1-8
烷氧基,(2)c
1-8
烷硫基,(3)苄氧基,(4)卤素原子,(5)硝基,(6)coor
105
,(7)氰基,(8)nr
106r107
,(9)nr
108
cor
109
,(10)羟基,(11)sh,(12)so3h,(13)s(o)oh,(14)oso3h,(15)c
2-8
烯氧基,(16)c
2-8
炔氧基,(17)cor
110
,(18)so2r
111
,或(19)被羟基取代的c1-8
烷氧基或c
1-8
烷硫基;
[0433]r105
为氢原子、c
1-8
烷基或c
2-8
烯基;
[0434]r106
和r
107
各自独立地为氢原子或c
1-8
烷基;
[0435]r108
为氢原子或c
1-8
烷基;
[0436]r109
和r
111
为c
1-8
烷基;
[0437]r110
为c
1-8
烷基或卤素原子;
[0438]r93
、r
94
、r
95
、r
96
、r
98
、r
99
和r
101
为(1)c
2-8
炔基,(2)被r
128
取代的c
1-8
烷基,r
128
可被1-4个r
29
取代,(3)环8,(4)被环8取代的c
1-8
烷基或c
2-8
烯基,或(5)被o-环8、s-环8或so
2-环8取代的c
1-8
烷基;r
128
为(1)氰基,(2)nr
106r107
,(3)nr
108
cor
109
,(4)羟基,(5)sh,(6)so3h,(7)s(o)oh,(8)oso3h,(9)c
2-8
烯氧基,(10)c
2-8
炔氧基,(11)cor
110
,(12)so2r
111
,或(13)被羟基取代的c
1-8
烷氧基或c
1-8
烷硫基;
[0439]r129
具有与r
104
相同的含义;
[0440]
环5和环6可被1-5个r
112
取代;
[0441]r112
为(1)c
1-8
烷基,(2)c
2-8
烯基,(3)c
2-8
炔基,(4)c
1-8
烷氧基,(5)c
1-8
烷硫基,(6)羟基,(7)卤素原子,(8)硝基,(9)氧代,(10)氰基,(11)苄基,(12)苄氧基,(13)被1-5个r
113
取代的c
1-8
烷基、c
1-8
烷氧基或c
1-8
烷硫基,(14)可被1-5个r
114
取代的苯基、苯氧基、苯硫基或苯甲酰基,(15)cor
115
,(16)so2r
116
,(17)nr
117
cor
118
,(18)so2nr
119r120
,(19)coor
121
,(20)nr
122r123
,(21)cor
124
,(22)conr
125r126
,(23)sh,(24)被羟基或nr
127-苯甲酰基取代的c
1-8
烷基,或(25)环7;
[0442]r113
为卤素原子;
[0443]r114
为c
1-8
烷基、c
1-8
烷氧基、卤素原子或硝基;
[0444]r115
、r
116
和r
118
为c
1-8
烷基;
[0445]r117
、r
121
、r
124
和r
127
为氢原子或c
1-8
烷基;
[0446]r119
和r
120
、r
122
和r
123
、r
125
和r
126
各自独立地为氢原子或c
1-8
烷基;
[0447]
环7可被1-5个选自以下的基团取代:(1)c
1-8
烷基,(2)c
1-8
烷氧基,(3)卤素原子,或(4)硝基;
[0448]
环8可被r
130
取代,且可被1-4个r
131
进一步取代;
[0449]r130
为(1)cor
124
,(2)conr
125r126
,(3)sh,(4)被羟基或nr
127-苯甲酰基取代的c
1-8
烷基,或(5)环7;
[0450]r131
具有与r
112
相同的含义;
[0451]
环5、环6、环7和环8为(1)c
3-15
一环、二环或三环(稠合或螺)碳环,或(2)3-15元含有1-4个选自1-4氧、氮或硫的杂原子的一环、二环或三环(稠合或螺)杂环;
[0452]
其中当r
17'
为环5时,则环5不是可被1-5个选自以下基团取代的苯基:c
1-8
烷基、c
1-8
烷氧基、羟基、卤素原子、硝基、cooh或coo(c
1-8
烷基);
[0453]
其中环7不是苯基;
[0454]
环4为(1)c
5-7
单环碳环,或(2)5-7元含有1-2个选自氧、氮和硫的杂原子的单环杂环;(下文简称虚线a;)和(下文简称虚线b;)为(1)化学键或(2)双键;
[0455]
r9(1)不存在或(2)为氢原子;
[0456]
其中
[0457]
(1)当虚线a为化学键时,虚线b为双键,r9不存在,
[0458]
(2)当虚线a为双键,则虚线b为化学键,r9为氢原子,r6不存在,
[0459]
(3)不包括2-(3,3-二甲基-3,4-二氢-(2h)-异喹啉-1-亚基)-1-苯基乙-1-酮或其药学上可接受的盐。
[0460]
这些化合物的制备参见us 2005222138和wo 2003/064389。
[0461]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于wo 2003/057149的化合物,该专利通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂及其药学上可接受的盐具有下列结构式:
[0462][0463]
上述化合物的取代基的定义如下:
[0464]
(1)x选自卤素和nr1r2,
[0465]
(2)y选自nr3、s和o,前提条件是当x为cl时,y不为s,
[0466]
(3)r1和r2独立选自氢、1-8个碳原子的烷基、2-8个碳原子的烯基、2-8个碳原子的炔基、3-7个碳原子的环烷基、5-9个碳原子的多环烷基、2-6个碳原子和1-2个选自nh、s和o的杂原子的杂环烷基、6-12个碳原子的芳基,该芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基、6-12个碳原子的芳基或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基、4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,该杂芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基、6-12个碳原子的芳基或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,及r4r5,或者r1和r2与它们所连接的氮原子结合在一起形成5-7元单环饱和环,任选含有1-2两个选自nh、nr6、s和o的额外杂原子,或者r1和r2与它们所连接的氮原子结合在一起形成6-10元稠合多环饱和环,任选含有1-2个选自nh、nr6、s和o的额外杂原子,或者r1和r2与它们所连接的氮原子结合在一起形成5-7元不饱和环,任选含有1-2个选自n、s和o的额外杂原子,其中所述单环饱和环、多环饱和环或不饱和环可被1-2个选自以下取代基取代:oh、1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、3-7个碳原子的环烷基、2-6个碳原子和1-2个选自nh、s和o的杂原子的杂环烷基、卤素、1-2个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子的烷氧基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基,及r7r8,
[0467]
(4)r3选自氢、1-8个碳原子的烷基、2-8个碳原子的烯基、2-8个碳原子的炔基、3-7个碳原子的环烷基及4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,该杂芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原
子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基、6-12个碳原子的芳基或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,
[0468]
(5)r4选自1-8个碳原子的烷基、2-8个碳原子的烯基、2-8个碳原子的炔基、c(=o)、s(=o)2和c(=o)o,
[0469]
(6)r5选自氢、oh、1-8个碳原子的烷基、2-8个碳原子的烯基、2-8个碳原子的炔基、1-8个碳原子的烷氧基、1-8个碳原子的烷硫基(thioxy)、6-12个碳原子的芳基,该芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基、6-12个碳原子的芳基或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基、4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,该杂芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基、6-12个碳原子的芳基、或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基、3-7个碳原子的环烷基、2-6个碳原子和1-2个选自nh、s和o的杂原子的杂环烷基,及nr9r
10
,
[0470]
(7)r6和r7独立选自1-8个碳原子的烷基、2-8个碳原子的烯基和2-8个碳原子的炔基,
[0471]
(8)r8选自oh、6-12个碳原子的芳基,该芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷氧基、6-12个碳原子的芳基或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基,该杂芳基可被以下基团取代:1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、1-6个碳原子和最高达到全卤代水平的一定数量的卤素原子的卤代烷基、6-12个碳原子的芳基或4-11个碳原子和1-2个选自n、s和o的杂原子的杂芳基;
[0472]
(9)r9和r
10
独立选自氢、1-8个碳原子的烷基、2-8个碳原子的烯基和2-8个碳原子的炔基,或者r9和r
10
与它们所连接的氮原子结合在一起形成5-7元可含有1-2个选自n、s和o的额外杂原子的不饱和环,或者形成5-7元可含有1-2个选自nh、nr
11
、s和o的额外杂原子的饱和环;
[0473]
(10)r1选自1-8个碳原子的烷基、2-8个碳原子的烯基和2-8个碳原子的炔基。
[0474]
这些化合物的制备参见wo 2003/057149。
[0475]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:us 20030092721、美国专利第7,022,849号、wo 2002/102315和us 2006116516,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0476][0477]
上述化合物的取代基的定义如下:r1为h或烷基;
[0478]
r2为(a)杂芳基或杂环基,其中任一个可被基团t1、t2、t3中的1-3个任选取代;或者(b)与杂芳环或杂环稠合的芳基,该复合环系可被基团t1、t2、t3中的1-3个任选取代;
[0479]
l为(a)or4、c(o)r4、c(o)or4、sr4、nr3r4、c(o)nr3r4、nr3so2r
4b
、卤素、硝基或卤代烷基;或者(b)烷基、芳基、杂芳基、杂环基或环烷基,其中的任一个可被基团t1a、t2a和/或t3a中的1-3个任选取代;
[0480]
y1、y2和y3独立地为(a)氢、卤素或-or
4a
;或者(b)烷基、烯基或炔基,其中的任一个可被基团t1b、t2b和/或t3b中的1-3个任选取代;
[0481]
r3和r4独立地为h、烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,其中的任一个被基团t1a、t2a和/或t3a中的1-3个任选取代;或者
[0482]
r3和r4可与它们所连接的氮原子一起结合形成4-8元被基团t1a、t2a和/或t3a中的1-3个任选取代的杂环;
[0483]r4a
为氢、烷基、烯基、芳基、杂芳基、(芳基)烷基、(杂芳基)烷基、杂环基、(杂环基)烷基、环烷基或(环烷基)烷基,其中的任一个可被基团t1b、t2b和/或t3b中的1-3个任选取代;
[0484]r4b
为烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,其中的任一个被基团t1a、t2a和/或t3a中的1-3个任选取代;
[0485]
z为n或ch;
[0486]
t1-1b、t2-2b和t3-3b各自独立为;
[0487]
(1)氢或t6,其中t6为:(i)烷基、(羟基)烷基、(烷氧基)烷基、烯基、炔基、环烷基、(环烷基)烷基、环烯基、(环烯基)烷基、芳基、(芳基)烷基、杂环基、(杂环基)烷基、杂芳基或(杂芳基)烷基;(ii)本身又被一个或多个相同或不同的基团(i)取代的基团(i);或(iii)被一个或多个下列t1-1b、t2-2b和t3-3b中定义的基团(2)至基团(13)取代的基团(i)或基团(ii);
[0488]
(2)-oh或-ot6;
[0489]
(3)-sh或-st6;
[0490]
(4)-c(o)
t
h、-c(o)
t
t6或-o-c(o)t6,其中t为1或2;
[0491]
(5)-so3h、-s(o)
t
t6或s(o)
t
n(t9)t6;
[0492]
(6)卤素;
[0493]
(7)氰基;
[0494]
(8)硝基;
[0495]
(9)-t4-nt7t8;
[0496]
(10)-t4-n(t9)-t5-nt7t8;
[0497]
(11)-t4-n(t10)-t5-t6;
[0498]
(12)-t4-n(t10)-t5-h;和
[0499]
(13)氧代;
[0500]
t4和t5各自独立地为单键、t11s(o)
t
t12-、t11c(o)t12-、t11c(s)t12、t11ot12、t11st12、t11oc(o)t12、t11c(o)ot12、t11c(=nt9a)t12或t11c(o)c(o)t12;
[0501]
t7、t8、t9、t9a和t10为:
[0502]
(1)各自独立地为氢或t6定义中提供的基团,或
[0503]
(2)t7和t8可连在一起成为亚烷基或亚烯基,与它们所连接的原子一起构成3-8元饱和或不饱和环,该环是未取代的或者被一个或多个t1-1b、t2-2b和t3-3b的描述中所列的基团取代,或者
[0504]
(3)t7或t8连同t9可为亚烷基或亚烯基,与它们所连接的氮原子一起构成3-8元饱和或不饱和环,该环是未取代的或者被一个或多个t1-1b、t2-2b和t3-3b的描述中所列的基团取代,或者
[0505]
(4)t7和t8或t9和t10可与它们所连接的氮原子一起结合形成基团n=ct13t14,其中t13和t14独立地为h或t6定义中所提供的基团;t11和t12独立地为单键、亚烷基、亚烯基或亚炔基。
[0506]
这些化合物的制备参见us 20030092721、美国专利第7,022,849号、wo 2002/102315和us 2006116516。
[0507]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:美国专利第6,838,559号、u.s.20030100571和wo 2002/102314,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0508][0509]
上述化合物的取代基的定义如下:
[0510]
r1为h或烷基;
[0511]
r2为(a)杂芳基或杂环基,其中任一个可被基团t1、t2、t3中的1-3个任选取代;(b)被基团t1、t2、t3中的1-3个取代的芳基,前提条件是t1、t2、t3中至少一个不是h;或(c)与杂芳环或杂环稠合的芳基,其中该复合环系可被基团t1、t2、t3中的1-3个任选取代;
[0512]
y为烷基、烯基、炔基、环烷基、芳基、杂环基、杂芳基、(芳基)烷基或(杂芳基)烷基,其中任一个可被基团t1a、t2a、t3a中的1-3个任选取代;
[0513]
j为(a)氢、卤素或or4,或者(b)烷基、烯基、炔基、芳基、杂芳基、杂环基或环烷基,
其中任一个可被基团t1b、t2b、t3b中的1-3个任选取代;
[0514]
z为(a)or4、sr4、nr3r4、nr3so2r
4a
、卤素、硝基、卤代烷基;或者(b)烷基、芳基、杂芳基、杂环基或环烷基,其中任一个可被基团t1c、t2c、t3c中的1-3个任选取代;
[0515]
r3为h、烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,在化合价允许的情况下,其中任一基团可被基团t1c、t2c、t3c的1-3个独立地任选取代;
[0516]
r4为烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,在化合价允许的情况下,其中任一基团可被基团t1d、t2d或t3d的1-3个独立地任选取代;或者
[0517]
r3和r4可与它们所连接的氮原子一起结合形成4-8元被基团t1c、t2c或t3c中的1-3个任选取代的杂环;
[0518]r4a
为氢、烷基、烯基、芳基、杂芳基、(芳基)烷基、(杂芳基)烷基、杂环基、(杂环基)烷基、环烷基或(环烷基)烷基,其中任一个可被基团t1d、t2d或t3d中的1-3个任选取代;
[0519]
t1、t1a、t1b、t1c、t1d、t2、t2a、t2b、t2c、t2d、t3、t3a、t3b、t3c和t3d(下文亦简称t1-1d、t2-2d和t3-3d)独立地为:
[0520]
(1)氢或t6,其中t6为:
[0521]
(a)烷基、(羟基)烷基、(烷氧基)烷基、烯基、炔基、环烷基、(环烷基)烷基、环烯基、(环烯基)烷基、芳基、(芳基)烷基、杂环基、(杂环基)烷基、杂芳基或(杂芳基)烷基;
[0522]
(b)自身被一个或多个相同或不同的基团(a)取代的基团(a);或者
[0523]
(c)被一个或多个(优选1-3个)t1-1d、t2-2d和t3-3d中定义的下列基团(2)-(13)独立取代的基团(a)或基团(b),
[0524]
(2)oh或ot6,
[0525]
(3)sh或st6,
[0526]
(4)c(o)
t
h,c(o)
t
t6或oc(o)t6,其中t为1或2;
[0527]
(5)so3h、s(o)
t
t6或s(o)
t
n(t9)t6,
[0528]
(6)卤素,
[0529]
(7)氰基,
[0530]
(8)硝基,
[0531]
(9)t4nt7t8,
[0532]
(10)t4n(t9)-t5nt7t8,
[0533]
(11)t4n(t10)-t5-t6,
[0534]
(12)t4n(t10)-t5h,
[0535]
(13)氧代,
[0536]
t4和t5各自独立地为单键、t11-s(o)
t-t12、t11-c(o)-t12、t11-c(s)-t12、t11-o-t12、-t11s-t12、-t11oc(o)-t12、-t11-c(o)o-t12、-t11c(=nt9a)-t12或t11-c(o)-c(o)-t12;
[0537]
t7、t8、t9、t9a和t10为:
[0538]
(1)各自独立地为氢或t6定义中提供的基团,或者
[0539]
(2)t7和t8可连在一起成为亚烷基或亚烯基,与它们所连接的原子一起构成3-8元
饱和或不饱和环,该环是未取代的或被一个或多个t1-1d、t2-2d和t3-3d的描述中所列的基团取代,或者
[0540]
(3)t7或t8连同t9可为亚烷基或亚烯基,与它们所连接的氮原子一起构成3-8元饱和或不饱和环,该环是未取代的或被一个或多个t1-1d、t2-2d和t3-3d的描述中所列的基团取代,或者
[0541]
(4)t7和t8或t9和t10可与它们所连接的氮原子一起结合形成基团n=ct13t14,其中
[0542]
t13和t14各自独立地为h或t6定义中所提供的基团;和
[0543]
t11和t12各自独立地为单键、亚烷基、亚烯基或亚炔基。
[0544]
这些化合物的制备参见美国专利第6,838,559号、u.s.20030100571和wo 2002/102314。
[0545]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:美国专利第7,087,614号,u.s.20030162802和wo 2002/102313,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0546][0547]
以上化合物的取代基描述如下。
[0548]
在相关实施方案中,用于本发明方法的pde7抑制剂具有下式:
[0549][0550]
上述化合物的取代基的定义如下:
[0551]r1a
为氢或烷基;r
2a
为w为s;x1为烷氧基;x2为烷基;
[0552]
z*为卤素、卤代烷基、唑基、nr
3ar4a
、c(o)-n(h)-亚烷基-cooh、或者未取代或被杂芳基、co
t
h或co
t
t6取代的苯基;
[0553]r3a
为氢或烷基;
[0554]r4a
为烷基、烷氧基、未取代或取代的(杂芳基)烷基、未取代或取代的杂环基、未取代或取代的(杂环基)烷基,或(芳基)烷基,其中芳基被基团t1和/或t2的一个或两个取代和/或被基团t3进一步取代;或者r
3a
和r
4a
与它们所连接的氮原子一起结合形成未取代或取代的杂环;
[0555]r5a
为未取代或取代的(杂芳基)烷基,或(芳基)烷基,其中芳基被基团t1和/或t2的一个或两个取代和/或被基团t3进一步取代;或者r
5a
和r
6a
与它们所连接的氮原子一起结合形成未取代或取代的杂环;r
6a
为氢或烷基;j*为氢或烷基;t1和t2独立地为烷氧基、烷氧基羰基、杂芳基、so3h或so2r
8a
,其中r
8a
为烷基、氨基、烷基氨基或二烷基氨基;或者t1和t2与它们所连接的芳环一起结合形成二环;t3为h、烷基、卤素、卤代烷基或氰基;t为1或2;t6为烷基、卤代烷基、环烷基、烷氧基或杂芳基。
[0556]
这些化合物的制备参见美国专利第7,087,614号、u.s.20030162802和wo 2002/102313。
[0557]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:us 20030104974、wo 2002/088080和wo 2002/088079,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0558][0559]
上述化合物的取代基的定义如下:
[0560]
r1为h或烷基;r2为任选取代的杂芳基或4-取代芳基;r3为氢或烷基;r4为烷基、任选取代的(芳基)烷基、任选取代的(杂芳基)烷基、任选取代的杂环基或任选取代的(杂环基)烷基;或者r3和r4可与它们所连接的氮原子一起结合形成任选取代的杂环;r5为烷基、任选取代的(芳基)烷基或任选取代的(杂芳基)烷基;r6为氢或烷基。
[0561]
在一个相关实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0562][0563]
其中r
1a
为h或烷基;r
2a
为任选取代的杂芳基;z为卤素、烷基、取代烷基、卤代烷基或nr
3ar4a
;r
3a
为氢或烷基;r
4a
为烷基、任选取代的(杂芳基)烷基、任选取代的杂环基、任选取代的(杂环基)烷基或(芳基)烷基,其中芳基被基团t1和t2中的一个或两个取代并被基团t3任选进一步取代;或者r
3a
和r
4a
可与它们所连接的氮原子一起结合形成任选取代的杂环;r
5a
为(芳基)烷基,其中芳基被基团t1和t2中的一个或两个取代并被基团t3任选进一步取代;r
6a
为氢或烷基;r
7a
为氢或烷基;t1和t2独立地为烷氧基、烷氧基羰基、杂芳基或so2r
8a
,其中r8a
为烷基、氨基、烷基氨基或二烷基氨基;或者t1和t2与它们所连接的原子一起结合形成环(例如苯并二氧杂环戊烯);t3为h、烷基、卤素、卤代烷基或氰基。
[0564]
在另一个相关实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0565][0566]
其中r
1b
为h或烷基;r
2b
为任选取代的杂芳基;r
3b
为h或烷基;r
4b
为任选取代的(芳基)烷基;r
5b
为h、烷基或c(o)(ch2)voyr
6b
,其中y为化学键或c(o),r
6b
为氢或烷基,v为0-2的整数;j1和j2独立地为任选取代的c
1-13
亚烷基,前提条件是j1和j2不同时大于c2亚烷基;x4和x5为与j1和j2的一个或两个中的任何可用碳原子结合的任选取代基,独立选自氢、or7、nr8r9、烷基、取代烷基、烯基、取代烯基、炔基、取代炔基、环烷基、取代环烷基、芳基、取代芳基、杂环烷基或杂芳基;r7为氢、烷基、取代烷基、烯基、炔基、环烷基、取代环烷基、c(o)烷基、c(o)取代烷基、c(o)环烷基、c(o)取代环烷基、c(o)芳基、c(o)取代芳基、c(o)o-烷基、c(o)o-取代烷基、c(o)杂环烷基、c(o)杂芳基、芳基、取代芳基、杂环烷基和杂芳基;r8和r9独立选自氢、烷基、取代烷基、环烷基、取代环烷基、烯基、炔基、c(o)烷基、c(o)取代烷基、c(o)环烷基、c(o)取代环烷基、c(o)芳基、c(o)取代芳基、c(o)o烷基、c(o)o取代烷基、c(o)杂环烷基、c(o)杂芳基、s(o)2烷基、s(o)2取代烷基、s(o)2环烷基、s(o)2取代环烷基、s(o)2芳基、s(o)2取代芳基、s(o)2杂环烷基、s(o)2杂芳基、芳基、取代芳基、杂环烷基和杂芳基,或者r8和r9与它们所连接的氮原子一起构成任选取代的杂环烷基或杂芳环。
[0567]
在又一个相关的实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0568][0569]
其中r
1c
为h或烷基;r
2c
为任选取代的杂芳基;r
3c
为h或烷基;r
4c
为任选取代的(芳基)烷基;x4和x5为与j1和j2的一个或两个中的任何可用碳原子结合的任选取代基,独立选自氢、or7、nr8r9、烷基、取代烷基、烯基、取代烯基、炔基、取代炔基、环烷基、取代环烷基、芳基、取代芳基、杂环烷基或杂芳基。
[0570]
这些化合物的制备参见us 20030104974、wo 2002/088080和wo 2002/088079。
[0571]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:us 20030092908和wo2002/087513,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0572][0573]
上述化合物的取代基的定义如下:
[0574]
r1为氢或烷基;
[0575]
r2为:(a)杂芳基或杂环基,其中任一个可被基团t1、t2、t3中的1-3个任选取代;(b)被基团t1、t2、t3中的1-3个取代的芳基,前提条件是t1、t2、t3中至少一个不是h;或者(c)与杂芳环或杂环稠合的芳基,该复合环系可被基团t1、t2、t3中的1-3个任选取代;
[0576]
z为nr3r4、nr3so2r
4a
、or4、sr4、卤代烷基或卤素;
[0577]
r3和r4独立地为h、烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,化合价允许的情况下,其中任一基团可被基团t1a、t2a或t3a中1-3个独立地任选取代;或者
[0578]
r3和r4可与它们所连接的氮原子一起形成杂环或杂芳环,化合价允许的情况下,其可被基团t1a、t2a或t3a中1-3个独立地任选取代;
[0579]r4a
为烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,化合价允许的情况下,其中任一基团可被基团t1a、t2a或t3a中1-3个独立地任选取代;
[0580]r3b
和r
4b
独立地为h、烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基;
[0581]
r5为:
[0582]
(1)氢或氰基;
[0583]
(2)烷基、烯基、炔基、环烷基、(环烷基)烷基、芳基、(芳基)烷基、杂环基、(杂环基)烷基、杂芳基或(杂芳基)烷基,化合价允许的情况下,其中任一基团可被基团t1b、t2b或t3b中1-3个独立地任选取代;或
[0584]
(3)c(o)r6、c(o)or6、c(o)-c(o)or或so2r
6a
;
[0585]
r6为h、烷基、烯基、nr
3br4b
、杂环基、(杂环基)烷基、(羟基)烷基、(烷氧基)烷基、(芳氧基)烷基、(nr
3br4b
)烷基、杂芳基、芳基或(芳基)烷基,化合价允许的情况下,其中任一基团可被基团t1b、t2b或t3b中1-3个独立地任选取代;
[0586]r6a
为烷基、烯基、nr
3br4b
、杂环基、(杂环基)烷基、(羟基)烷基、(烷氧基)烷基、(芳氧基)烷基、(nr
3br4b
)烷基、杂芳基、芳基或(芳基)烷基,化合价允许的情况下,其中任一基团可被基团t1b、t2b或t3b中1-3个独立地任选取代;
[0587]
j1和j2独立地为任选取代的c
1-3
亚烷基,前提条件是j1和j2不同时大于c2亚烷基;
[0588]
t1-1b、t2-2b和t3-3b各自独立为:
[0589]
(1)氢或t6,其中t6为(i)烷基、(羟基)烷基、(烷氧基)烷基、烯基、炔基、环烷基、(环烷基)烷基、环烯基、(环烯基)烷基、芳基、(芳基)烷基、杂环基、(杂环基)烷基、杂芳基或
(杂芳基)烷基;(ii)本身又被一个或多个相同或不同的基团(i)取代的基团(i);或(iii)被一个或多个(优选1-3)t1-1b、t2-2b和t3-3b中定义的下列基团(2)-(13)独立取代的基团(i)或基团(ii),
[0590]
(2)oh或ot6,
[0591]
(3)sh或st6,
[0592]
(4)c(o)
t
h、c(o)
t
t6或oc(o)t6,其中t为1或2,
[0593]
(5)so3h、s(o)
t
t6或s(o)
t
n(t9)t6,
[0594]
(6)卤素,
[0595]
(7)氰基,
[0596]
(8)硝基,
[0597]
(9)t4-nt7t8,
[0598]
(10)t4-n(t9)-t5-nt7t8,
[0599]
(11)t4-n(t10)-t5-t6,
[0600]
(12)t4-n(t10)-t5h,
[0601]
(13)氧代,
[0602]
t4和t5各自独立地为(1)单键,(2)t11-s(o)
t-t12,(3)t11-c(o)-t12,(4)t11-c(s)-t12,(5)-t11-o-t12,(6)t11-s-t12,(7)t11-o-c(o)-t12,(8)t11-c(o)-o-t12,(9)t11-c(=nt9a)-t12,或(10)t11-c(o)-c(o)-t12,
[0603]
t7、t8、t9、t9a和t10,
[0604]
(1)各自独立地为氢或t6定义中提供的基团,或者
[0605]
(2)t7和t8可连在一起成为亚烷基或亚烯基,与它们所连接的原子一起构成3-8元饱和或不饱和环,该环是未取代的或被t1-1b、t2-2b和t3-3b的描述中所列的一个或多个基团取代,或者
[0606]
(3)t7或t8连同t9可为亚烷基或亚烯基,与它们所连接的氮原子一起构成3-8元饱和或不饱和环,该环是未取代的或被t1-1b、t2-2b和t3-3b的描述中所列的一个或多个基团取代,或者
[0607]
(4)t7和t8或t9和t10可与它们所连接的氮原子一起结合形成基团n=ct13t14,其中t13和t14各自独立地为h或t6定义中所提供的基团;
[0608]
t11和t12各自独立地为单键、亚烷基、亚烯基或亚炔基。
[0609]
这些化合物的制备参见us 20030092908和wo 2002/087513。
[0610]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:us 20040127707和wo2002/085906,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂及这些化合物的盐具有下列结构式:
[0611][0612]
上述化合物的取代基的定义如下:
[0613]
r1为1-2c-烷氧基或完全或主要被氟取代的1-2c-烷氧基,
[0614]
r2为氟、溴或氯,
[0615]
r3和r4同时为氢或一起形成额外的化学键,
[0616]
r5为r6、c
mh2m-r7、c
nh2n-c(o)r8、ch(r9)2、c
ph2p-y-芳基1(ary1)、r
12
或r
26
,其中:
[0617]
r6为1-8c-烷基、3-10c-环烷基、3-7c-环烷基甲基、3-7c-烯基、3-7c-炔基(alkinyl)、苯基-3-4c-烯基、7-10c-多环烷基、萘基、吡啶基、吡嗪基、哒嗪基、嘧啶基、喹唑啉基、喹喔啉基、噌啉基、异喹啉基、喹啉基、茚满基、吲唑基、苯并唑基、苯并噻唑基、唑基、噻唑基、n-甲基哌啶基、四氢吡喃基、6-甲基-3-三氟甲基-吡啶基-2-基、1,3,4-三甲基-1h-吡唑并[3,4-b]吡啶基-6-基、3-噻吩-2-基[1,2,4]噻二唑-5-基、1,1-二氧化(dioxide)-四氢噻吩-3-基、1-氧代-1,3-二氢-异苯并呋喃-5-基、4-(4-基-丁-1-氧基)苯甲酸或者未取代的或被r
61
和/或r
62
取代的苯基,其中:
[0618]r61
为羟基、1-4c-烷基、1-4c-烷氧基、硝基、氰基、卤素、羧基、羟基羰基-1-4c-烷基、1-4c-烷氧基羰基、羟基-1-4c-烷基、氨基、一-1-4c-烷基氨基或二-1-4c-烷基氨基、1-4c-烷基羰基氨基、氨基羰基、一-1-4c-烷基氨基羰基或二-1-4c-烷基氨基羰基、氨基磺酰基、一-1-4c-烷基氨基磺酰基或二-1-4c-烷基氨基磺酰基、4-甲基苯基磺酰胺基、咪唑基、四唑-5-基、2-(1-4c-烷基)四唑-5-基或2-苄基四唑-5-基,
[0619]r62
为1-4c-烷基、1-4c-烷氧基、硝基或卤素,
[0620]
r7为羟基、卤素、氰基、硝基、硝酰氧基(o-no2)、羧基、羧基苯基氧基、苯氧基、1-4c-烷氧基、3-7c-环烷氧基(cydoalkoxy)、3-7c-环烷基甲氧基、1-4c-烷基羰基、1-4c-烷基羰基氧基、1-4c-烷基羰基氨基、1-4c-烷氧基羰基、氨基羰基、一-1-4c-烷基氨基羰基或二-1-4c-烷基氨基羰基、氨基、一-1-4c-烷基氨基或二-1-4c-烷基氨基,或者未取代或被r
71
和/或r
72
取代的哌啶基、哌嗪基、吡咯烷基或吗啉基,其中:
[0621]r71
为羟基、1-4c-烷基、羟基-1-4c-烷基或1-4c-烷氧基羰基,
[0622]r72
为1-4c-烷基、羧基、氨基羰基或1-4c-烷氧基羰基,
[0623]
r8为未取代或被r
81
和/或r
82
取代的苯基、萘基、菲基或蒽基,其中:
[0624]r81
为羟基、卤素、氰基、1-4c-烷基、1-4c-烷氧基、羧基、氨基羰基、一-1-4c-烷基氨基羰基或二-1-4c-烷基氨基羰基、1-4c-烷基羰基氧基、1-4c-烷氧基羰基、氨基、一-1-4c-烷基氨基或二-1-4c-烷基氨基、1-4c-烷基羰基氨基或者完全或主要被氟取代的1-4c-烷氧基,
[0625]r82
为羟基、卤素、1-4c-烷基、1-4c-烷氧基或者完全或主要被氟取代的1-4c-烷氧基,
[0626]
r9为c
qh2q-苯基,
[0627]
y为化学键或o(氧),
[0628]
芳基1为未取代的苯基、萘基、吡啶基、吡嗪基、哒嗪基、嘧啶基、喹唑啉基、喹喔啉基、噌啉基、异喹啉基、喹啉基、香豆素基(coumarinyl)、苯并咪唑基、苯并唑基、苯并噻唑基、苯并三唑基、n-苯并琥珀酰亚氨基(benzosuccinimidyl)、咪唑基、吡唑基、唑基、噻唑基、呋喃基、噻吩基、吡咯基、2-(1-4c-烷基)-噻唑-4-基或被r
10
和/或r
11
取代的苯基,其中
[0629]r10
为羟基、卤素、硝基、氰基、1-4c-烷基、三氟甲基、1-4c-烷氧基、羧基、羟基羰基-1-4c-烷基、1-4c-烷基羰基氧基、1-4c-烷氧基羰基、氨基、一-1-4c-烷基氨基或二-1-4c-烷基氨基、1-4c-烷基羰基氨基、氨基羰基、一-1-4c-烷基氨基-羰基或二-1-4c-烷基氨基-羰基、咪唑基或四唑基,r
11
为羟基、卤素、硝基、1-4c-烷基或1-4c-烷氧基,
[0630]
m为1-8的整数,n为1-4的整数,p为1-6的整数,q为0-2的整数,
[0631]r12
为式(a)基团
[0632]
其中r
13
为s(o)
2-r
14
、s(o)
2-(ch2)
r-r
15
、(ch2)
s-s(o)2r
16
、c(o)r
17
、c(o)-(ch2)
r-r
18
、(ch2)
s-c(o)-r
19
、杂芳基1(hetaryl1)、芳基2或芳基
3-1-4c-烷基,r
14
为1-4c-烷基、5-二甲基氨基萘-1-基、n(r
20
)r
21
、苯基或被r
22
和/或r
23
取代的苯基,r
15
为n(r
20
)r
21
,r
16
为n(r
20
)r
21
,
[0633]r17
为1-4c-烷基、羟基羰基-1-4c-烷基、苯基、吡啶基、4-乙基-哌嗪-2,3-二酮-1-基、2-氧代-咪唑烷-1-基或n(r
20
)r
21
,r
18
为n(r
20
)r
21
、r
19
为n(r
20
)r
21
、苯基、被r
22
和/或r
23
和/或r
24
取代的苯基,r
20
和r
21
彼此独立地为氢、1-7c-烷基、3-7c-环烷基、3-7c-环烷基甲基或苯基,或者r
20
和r
21
一起并同时包括它们所连接的氮原子共同形成4-吗啉基-环、1-吡咯烷基-环、1-哌啶基-环、1-六氢氮杂基(hexahydroazepino)-环或式(b)的1-哌嗪基-环,
[0634]
其中r
25
为吡啶-4-基、吡啶-4-基甲基、1-4c-烷基-二甲基氨基、二甲基氨基羰基甲基、n-甲基-哌啶-4-基、4-吗啉代-乙基或四氢呋喃-2-基甲基-,r
22
为卤素、硝基、氰基、羧基、1-4c-烷基、三氟甲基、1-4c-烷氧基、1-4c-烷氧基羰基、氨基、单-1-4c-烷基氨基(alkyiamino)或二-1-4c-烷基氨基、氨基羰基1-4c-烷基羰基氨基、单-1-4c-烷基氨基羰-基或二-1-4c-烷基氨基羰基,r
23
为卤素、氨基、硝基、1-4c-烷基或1-4c-烷氧基,r
24
为卤素,
[0635]
杂芳基1为嘧啶-2-基、噻吩并-[2,3-d]嘧啶-4-基、1-甲基-1h-吡唑并-[3,4-d]嘧啶-4-基、噻唑基、咪唑基或呋喃基,芳基2为吡啶基、苯基或被r
22
和/或r
23
取代的苯基,芳基3为吡啶基、苯基、被r
22
和/或r
23
取代的苯基、2-氧代-2h-苯并吡喃-7-基或4-(1,2,3-噻二唑-4-基)苯基,
[0636]
r为1-4的整数,s为1-4的整数,
[0637]r26
为式(c)的基团,
[0638]
其中r
27
为c(o)r
28
、(ch2)
t-c(o)r
29
、(ch2)
ur30
、芳基4、杂芳基2、苯基丙-1-烯-3-基或1-甲基哌啶-4-基,r
28
为氢、1-4c-烷基、or
31
、呋喃基、吲哚基、苯基、吡啶基、被r
34
和/或r
35
取代的苯基或被r
36
和/或r
37
取代的吡啶基,r
29
为n(r
32
)r
33
,r
30
为n(r
32
)r
33
、四氢呋喃基或吡啶
基,r
31
为1-4c-烷基、r
32
为氢、1-4c-烷基、3-7c-环烷基或3-7c-环烷基甲基,r
33
为氢、1-4c-烷基、3-7c-环烷基或3-7c-环烷基甲基,或者
[0639]r32
和r
33
一起并同时包括它们所连接的氮原子共同形成4-吗啉基-、1-吡咯烷基-、1-哌啶基-或1-氮杂环庚烷基-环,
[0640]
芳基4为苯基、吡啶基、嘧啶基、被r
34
和/或r
35
取代的苯基、被r
36
和/或r
37
取代的吡啶基,r
34
为卤素、硝基、1-4c-烷基、三氟甲基或1-4c-烷氧基,r
35
为卤素或1-4c-烷基,r
36
为卤素、硝基、1-4c-烷基、三氟甲基或1-4c-烷氧基,r
37
为卤素或1-4c-烷基,
[0641]
杂芳基2为吲哚-4-基、2-甲基-喹啉-4-基、5-氯-6-氧代-1-苯基-1,6-二氢-哒嗪-4-基、3-苯基-1,2,4-噻二唑-5-基或3-邻-甲苯基-1,2,4-噻二唑-5-基,
[0642]
t为1-4的整数,u为1-4的整数,v为1-2的整数,x为-c(o)-或-s(o)
2-。
[0643]
这些化合物的制备参见us 20040127707和wo 2002/085906。
[0644]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:美国专利第6,818,651号、us20040044212和wo 2002/040450,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂或其水合物、溶剂化物、盐、盐的水合物或盐的溶剂化物具有下列结构式:
[0645][0646]
上述化合物的取代基的定义如下:
[0647]
或者r1表示氢,r2表示氟、氯、溴、氰基、三氟甲基或苯氧基,或者r1表示氢、氟、氯、溴、三氟甲基或氰基,r2表示氢,r'和r"二者均表示氢或者一起表示化学键,ar表示下列式iia、式iib或式iic的苯基:
[0648][0649]
其中r3表示氢、羟基、硝基、氨基、羧基、氨基羰基、1-4c-烷氧基、三氟甲氧基、1-4c-烷氧基羰基或一-1-4c-烷基氨基羰基或二-1-4c-烷基氨基羰基,
[0650]
r4表示1-4c-烷基、萘基、5-二甲基氨基萘-1-基、苯基乙烯-2-基、3,5-二甲基异唑-4-基、5-氯-3-甲基苯并[b]噻吩-2-基、6-氯-咪唑并[2,1b]-噻唑-5-基,或者表示被一个或多个相同或不同的选自以下基团取代的苯基或噻吩基:卤素、氰基、1-4c-烷基、三氟甲基、完全或主要被氟取代的1-4c-烷氧基、1-4c-烷氧基、1-4c-烷基羰基氨基、1-4c-烷氧基羰基、苯基磺酰基或异唑基。
[0651]
这些化合物的制备参见美国专利第6,818,651号、us 20040044212和wo 2002/040450。
[0652]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于wo 2002/040449的化合物,该专利通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂或其盐具有下列结构式:
[0653][0654]
上述化合物的取代基的定义如下:
[0655]
或者r1表示氢,r2表示氟、氯、溴、氰基、三氟甲基或苯氧基,或者
[0656]
r1表示氢、氟、氯、溴、三氟甲基或氰基,r2表示氢,
[0657]
r'和r"二者均表示氢或者一起表示化学键,
[0658]
r3表示氢、羟基、硝基、氨基、羧基、氨基羰基、1-4c-烷氧基、三氟甲氧基、1-4c-烷氧基羰基或一-1-4c-烷基氨基羰基或二-1-4c-烷基氨基羰基,r4表示c(o)-x-r5、n(h)-c(o)-r6或n(h)-c(o)-n(h)-r2,其中:
[0659]
x表示0或n(h),
[0660]
r5表示氢、1-4c-烷基、3-7c-环烷基甲基、6,6-二甲基二环[3,3,1]庚-2-基、3-7c-炔基、1-4c-烷基羰基-1-4c-烷基、氨基羰基-1-4c-烷基、呋喃-2-基甲基、2-吡啶基-2-基乙-1-基、2-吡啶基-3-基甲基、n-甲基哌啶-3-基、1-苄基哌啶-4-基、吗啉-4-基-乙-2-基、吗啉-4-基-乙-1-基、2-苯并[1,3]间二氧杂环戊烯-4-基-乙-1-基、苯并二氢呋喃-4-基、1-甲氧基羰基-2-吲哚-3-基-乙-1-基、1,3-双-甲氧基羰基丙-1-基、1-甲氧基羰基-3-甲硫基(methylsulfanyl)-乙-1-基、1-甲氧基羰基-2-噻唑-2-基-乙-1-基或4-甲基噻唑-5-基-乙-2-基,或者表示未取代或者被一个或多个选自卤素、三氟甲基和苯基取代的苄基-、苯基-乙-1-基或1-甲氧基羰基-2-苯基-乙-2-基,
[0661]
r6表示2,4-二氯苯氧基甲基、2-叔丁氧基羰基氨基-乙-1-基、1-乙酰基哌啶-4-基,ar1或ar2-ch=ch-,
[0662]
其中ar1表示3-氯苯基、4-三氟甲氧基苯基、3-苯氧基苯基、吲哚-5-基、2-甲基吡啶基-5-基、喹啉-6-基或2-苯并噻唑-6-基,ar2表示呋喃-2-基、呋喃-3-基、噻吩-2-基、吲哚-3-基、3-三氟甲基苯基、3-甲氧基苯基或吡啶基-3-基,
[0663]
r7表示1-4c-烷基、3-7c-烯基、3-7c-环烷基、1-乙氧基羰基-2-苯基-乙-1-基、噻吩-2-基乙-1-基或未取代或被一个或多个选自以下的基团取代的苯基:卤素、氰基、1-4c-烷基、三氟甲基、1-4c-烷硫基、1-4c-烷氧基、完全或主要被氟取代的1-4c-烷氧基、1-4c-烷基羰基和苯氧基。
[0664]
这些化合物的制备参见wo 2002/040449。
[0665]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于wo 2001/098274的化合物,该专利通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂及其盐、溶剂化物、水合物和n-氧化物具有下列结构式:
[0666][0667]
上述化合物的取代基的定义如下:
[0668]
w、x、y和z可相同或不同,各自表示氮原子或c(r5)基团[其中r5为氢或卤素原子或烷基、卤代烷基、烷氧基、卤代烷氧基、羟基、-no2或-cn],前提条件是w、x、y和z中的两个或更多个为c(r5)基团;
[0669]
r1、r2和r3可相同或不同,各自为原子或基团-l1(alk1)rl2(r6)s,其中l1和l2可相同或不同,各自为共价键或接头原子或基团,r为零或整数1,alk1为脂肪链或杂脂肪链(heteroaliphatic chain),s为1、2或3的整数,r6为氢或卤素原子或选自以下的基团:烷基、-or7[其中r7为氢原子或任选取代的烷基]、-sr7、nr7r8[其中r8可相同或不同且如刚才r7的定义]、-no2、cn、co2r7、so3h、s(o)r7、so2r7、oco2r7、conr7r8、oconr7r8、csnr7r8、ocr7、ocor7、n(r7)cor8、n(r7)csr8、s(o)nr7r8、so2nr7r8、n(r7)so2r8、n(r7)con(r8)(r9)[其中r9为氢原子或任选取代的烷基]、n(r7)csn(r8)(r9)、n(r7)so2n(r8)(r9)、c(r7)=no(r8)、环脂族基、杂环脂族基、芳基或杂芳基];前提条件是r1、r2或r3中的一个或多个为氢原子以外的取代基;
[0670]
r4表示任选取代的苯基、1-萘基或2-萘基、吡啶基、嘧啶基、哒嗪基或吡嗪基。
[0671]
这些化合物的制备参见wo 2001/098274。
[0672]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于wo 2001/074786的化合物,该专利通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂及其盐、溶剂化物、水合物和n-氧化物具有下列结构式:
[0673][0674]
上述化合物的取代基的定义如下:
[0675]
r1表示芳基或杂芳基;
[0676]
a、b、p和e可相同或不同,各自表示氮原子或c(r2)基团[其中r2为氢或卤素原子或烷基、卤代烷基、烷氧基、卤代烷氧基、羟基、-no2或-cn基团],前提条件是a、b、d和e中的两个或更多个为c(r2)基团;x表示氧或硫原子或n(r3)基团,其中r3为氢原子或烷基;
[0677]
q、r、s和t可相同或不同,各自表示氮原子或基团c(r4)[其中r4为一个原子或基团-l1(alk1)rl2(r5)s,其中l1和l2可相同或不同,各自为共价键或接头原子或基团,r为零或整数
1,烷基为脂肪链或杂脂肪链,s为1、2或3的整数,r5为氢或卤素原子或选自以下的基团:烷基、or6[其中r6为氢原子或任选取代的烷基]、sr6、nr6r7[其中r7可相同或不同且如刚才r6的定义]、no2、cn、co2r6、so3h、s(o)r6、so2r6、oco2r6、conr6r7、oconr6r7、csnr7r7、ocr6、ocor6、n(r6)cor7、n(r6)csr7、s(o)nr6r7、so2nr6r7、n(r6)so2r7、n(r6)con(r7)(r8)[其中r8为氢原子或任选取代的烷基]、n(r6)csn(r7)(r8)、n(r6)so2n(r7)(r8)、c(r6)=no(r7)、环脂族基、杂环脂族基、芳基或杂芳基],前提条件是q、r、s和t中的两个或更多个为c(r4)基团。
[0678]
这些化合物的制备参见wo 2001/074786。
[0679]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于wo 2000/068230的化合物,该专利通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂或其药学上可接受的盐具有下列结构式:
[0680][0681]
上述化合物的取代基的定义如下:
[0682]
x-y-z表示nr
4-c=n或n=c-nr4;
[0683]
r1表示h、烷基、环烷基、环烷基烷基、芳基烷基、杂芳基烷基或杂环烷基;
[0684]
r2表示or8、nr8r9、sr
13
、烷基或cf3;
[0685]
r3表示卤素、烷基、cf3或or8;
[0686]
r4,可与x或z连接,为选自以下的残基:
[0687][0688]
其中通过饱和环上的任何位置连接,前提是连接不发生在v的邻位上,饱和环可在任何位置上可被一个或多个r6取代;
[0689]
a、b、d和e相同或不同,各自表示clnr5、n或n-o;
[0690]
v表示o、s、nr7或c(l
1mr14
)(l
2nr14
);
[0691]
q和w相同或不同,各自表示clnr5或n;
[0692]
t表示o、s或nr7;
[0693]
l1和l2相同或不同,各自表示c(r
15
)2;
[0694]
m和n相同或不同,各自表示0、1、2、3、4或5;
[0695]
r5相同或不同,各自表示h、卤素、烷基、环烷基、or8、nr8r9、co2r
10
、conr
11r12
、conhoh、so2nr
11r12
、sonr
11r12
、cor
13
、so2r
13
、sor
13
、sr
13
、cf3、no2或cn;
[0696]
r6表示h、烷基、环烷基、or8、nr8r9、co2r
10
、conr
11r12
、so2nr
11r12
、sonr
11r12
、cor
13
、so2r
13
、sor
13
、sr
13
、cf3、cn或=o;
[0697]
r7表示h或烷基;
[0698]
r8表示h、烷基、环烷基、环烷基烷基、芳基、芳基烷基、杂芳基、杂芳基烷基、杂环基或杂环烷基;
[0699]
r9表示r8或烷基羰基、烷氧基羰基、烷基磺酰基、环烷基羰基、环烷氧基羰基、环烷基磺酰基、环烷基烷基羰基、环烷基烷氧基羰基、环烷基烷基磺酰基、芳基羰基、芳基磺酰基、杂芳基羰基、杂芳基磺酰基、杂环基羰基、杂环基磺酰基、芳基烷基羰基、芳基烷氧基羰基、芳基烷基磺酰基、杂芳基烷基羰基、杂芳基烷氧基羰基、杂芳基磺酰基、杂环烷基羰基、杂环基烷氧基羰基或杂环烷基磺酰基;或者
[0700]
nr8r9表示杂环,例如吗啉;
[0701]r10
表示h、烷基、环烷基、环烷基烷基、芳基烷基、杂芳基烷基或杂环烷基;
[0702]r11
和r
12
相同或不同,各自为r8,或者nr
11r12
表示杂环,例如吗啉;
[0703]r13
表示烷基、环烷基、环烷基烷基、芳基、芳基烷基、杂芳基、杂芳基烷基、杂环基或杂环烷基;
[0704]r14
相同或不同,各自选自h、烷基、环烷基、or8、nr8r9、co2r
10
、conr
11r12
、conhoh、so2nr
11r12
、sonr
11r12
、cor
13
、so2r
13
、sor
13
、sr
13
、cf3、no2和cn,前提条件是当m和n同时为0时,如果一个r
14
为or8、nr8r9或sr
13
,则另一个不为or8、nr8r9或sr
13
;
[0705]r15
表示h、烷基或f。
[0706]
这些化合物的制备参见wo 2000/068230,其通过引起以其整体结合到本文中。
[0707]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:us 20040106631、ep 1 400 244和wo 2004/026818,各专利通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂或其外消旋体、异构体、药学上可接受的衍生物具有下列结构式:
[0708][0709]
上述化合物的取代基的定义如下:
[0710]
m为1、2或3;r1为甲基、氯、溴或氟;r2为-q
1-q
2-q
3-q4或(c
1-c6)烷基,所述(c
1-c6)烷基被1-3个以下基团取代:or4、coor4、nr4r5、nrc(=o)r4、c(=o)nr4r5或so2nr4r5;
[0711]
r4为被1-3个以下基团取代的(c
1-c6)烷基:f、cn、s(=o)r6、so3h、so2r6、sr7、c(=o)-nh-so
2-ch3、c(=o)r7、nr'c(=o)r7、nr'so2r6、c(=o)nr7r8、o-c(=o)nr7r8或so2nr7r8;
[0712]
r5为h或被1-3个以下基团任选取代的(c
1-c6)烷基:f、cn、s(=o)r6、so3h、so2r6、sr7、c(=o)-nh-so
2-ch3、c(=o)r7、nr'c(=o)r7、nr'so2r6、c(=o)nr7r8、o-c(=o)nr7r8或so2nr7r8;或者
[0713]
所述(c
1-c6)烷基为:
[0714]
(1)被1-3个以下基团取代:oc(=o)r
4a
、sr
4a
、s(=o)r3、c(=nr9)r
4a
、c(=nr9)-nr
4ar5a
、nr-c(=nr9)-nr
4ar5a
、nrcoor
4a
、nr-c(=o)nr
4ar5a
、nr-so
2-nr
4ar5a
、nr-c(=nr9)-r
4a
或nr-so
2-r3;
[0715]
(2)被一个或两个以下基团任选取代:or
4a
、coor
4a
、c(=o)-r
4a
、nr
4ar5a
、nrc(=o)r
4a
、c(=o)nr4r
5a
或so2nr
4ar5a
;
[0716]
r9为h、cn、oh、och3、so2ch3、so2nh2或(c
1-c6)烷基;r3为被1-3个以下基团任选取代的(c
1-c6)烷基:f、cn、s(=o)r6、so3h、so2r6、c(=o)-nh-so
2-ch3、or7、sr7、coor7、c(=o)r7、o-c(=o)nr7r8、nr7r8、nr'c(=o)r7、nr'so2r6、c(=o)nr7r8或so2nr7r8;
[0717]r4a
和r
5a
相同或不同,为h或被1-3个以下基团任选取代的(c
1-c6)烷基:f、cn、s(=o)r6、so3h、so2r6、c(=o)-nh-so
2-ch3、or7、sr7、coor7、c(=o)r7、o-c(=o)nr7r8、nr7r8、nr'c(=o)r7、nr'so2r6、c(=o)nr7r8或so2nr7r8;
[0718]
q1为单键或(c
1-c6)亚烷基;q2为4-6元饱和杂环基,含有o或n中的1个或2个;q3为(c
1-c6)亚烷基;q4为4-8元芳环或非芳环杂环基,含有o、s、s(=o)、so2或n中的1-4个,所述杂环基被1-3个以下基团任选取代:or、nrr'、-cn或(c
1-c6)烷基;
[0719]
r为h或(c
1-c6)烷基;
[0720]
r6为被1个或2个or'任选取代的(c
1-c6)烷基;
[0721]
r7和r8相同或不同,为h或被1个或2个or'任选取代的(c
1-c6)烷基;
[0722]
r9为h、cn、oh、och3、so2ch3、so2nh2或(c
1-c6)烷基;
[0723]
r'为h或(c
1-c6)烷基;r"为h或(c
1-c6)烷基;
[0724]
前提条件是(1)与q1结合的q2中的原子为碳原子;(2)与q3结合的q4中的原子为碳原子。
[0725]
这些化合物的制备参见us 20040106631、ep 1 400 244和wo 2004/026818。
[0726]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献的化合物:美国专利第6,936,609号和us20040249148,所述各专利和专利申请通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂或其盐具有下列结构式:
[0727][0728][0729]
上述化合物的取代基的定义如下:
[0730]
r1表示(c
6-c
10
)-芳基,被选自以下的基团任选相同或不同地取代:卤素、甲酰基、氨基甲酰基、氰基、羟基、三氟甲基、三氟甲氧基、硝基、(c
1-c6)-烷基或(c
1-c6)-烷氧基,且被式so2nr5r6基团任选取代,其中r5和r6相互独立地表示氢或(c
1-c6)-烷基,或者nr5r6表示4-8元通过氮原子结合的杂环基,被选自以下的基团任选相同或不同地取代:氧代、卤素、
(c
1-c6)-烷基和(c
1-c6)-酰基,
[0731]
r2表示具有1-10个碳原子的饱和或部分不饱和烃基,
[0732]
r3表示甲基或乙基,
[0733]
a表示o、s或nr7,其中r7表示氢或被(c
1-c3)-烷氧基任选取代的(c
1-c6)-烷基,
[0734]
e表示化学键或(c
1-c3)-链烷二基(alkanediyl),
[0735]
r4表示(c
6-c
10
)-芳基或5-10元杂芳基,其中芳基和杂芳基被选自以下的基团任选相同或不同地取代:卤素、甲酰基、羧基、氨基甲酰基、-so3h、氨基磺酰基、氰基、羟基、三氟甲基、三氟甲氧基、硝基、(c
1-c6)-烷基、(c
1-c6)-烷氧基、1,3-二氧杂-丙-1,3-二基、(c
1-c6)-烷硫基、(c
1-c6)-烷基亚磺酰基和(c
1-c6)-烷基-磺酰基、-nr8r9和任选甲基-取代的5-6元杂芳基或苯基,
[0736]
其中r8和r9相互独立地表示氢、(c
1-c6)-烷基或(c
1-c6)-酰基。
[0737]
这些化合物的制备参见美国专利第6,936,609号和us20040249148。
[0738]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于wo 2006/092692的化合物,该专利通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0739][0740]
其中n为1-4的整数,其中存在立构中心,各中心可独立地为r或s。
[0741]
这些化合物的制备参见wo 2006/092692。
[0742]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下专利的化合物:us 2006229306和wo 2004/065391,所述各专利和专利申请通过引用以其整
体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下列结构式:
[0743][0744]
上述化合物的取代基的定义如下:
[0745]
r1和r2任一个
[0746]
(1)独立地表示:
[0747]
(a)氢原子;
[0748]
(b)选自烷基、烯基和炔基的基团,其中各个烷基、烯基和炔基独立地被一个或多个选自以下的取代基任选取代:卤素原子、羟基、烷氧基、芳氧基、烷硫基、羟基羰基、烷氧基羰基(alcoxycarbonyl)、一烷基氨基酰基和二烷基氨基酰基、氧代、氨基以及一烷基氨基和二烷基氨基;或者
[0749]
(c)式(ch2)
n-r6基团,其中n为0-4的整数,r6表示环烷基或环烯基;
[0750]
(2)r1和r2与它们所连接的氮原子一起形成含有1-4个选自氮、氧和硫的杂原子的3-8元环,该环为饱和或不饱和的且被一个或多个选自以下的取代基任选取代:卤素原子、烷基、羟基、烷氧基、酰基、羟基羰基、烷氧基羰基、亚烷基二氧基、氨基、一烷基氨基和二烷基氨基、一烷基氨基酰基和二烷基氨基酰基、硝基、氰基和三氟甲基;
[0751]
r3为式(ch2)
n-g
基团,其中n为0-4的整数,g表示含有0-4个杂原子的单环或二环芳基或杂芳基,该基团被一个或多个选自以下的取代基任选取代:
[0752]
(1)卤素原子;
[0753]
(2)烷基和亚烷基,其中各个烷基和亚烷基独立地被一个或多个选自卤素原子的取代基任选取代;
[0754]
(3)苯基、羟基、羟基烷基、烷氧基、亚烷基二氧基、芳氧基、烷硫基、氨基、一烷基氨基和二烷基氨基、酰基氨基、硝基、酰基、羟基羰基、烷氧基羰基、氰基、二氟甲氧基和三氟甲氧基;
[0755]
r4表示氢原子、烷基或芳基。
[0756]
这些化合物的制备参见us 2006229306和wo 2004/065391。
[0757]
其它用于本发明方法的化合物包括咪唑并吡啶衍生物(wo2001/34601)、二氢嘌呤衍生物(wo 2000/68203)、吡咯衍生物(wo2001/32618)、苯并硫代吡喃并咪唑酮(benzothiopyranoimidazolone)衍生物(de 19950647)、杂环化合物(wo 2002/87519)、鸟嘌呤衍生物(bioorg.med.chem.lett.11:1081-1083,2001)和苯并噻吩并噻二嗪衍生物(eur.j.med.chem.36:333,2001)。上述已发表的各专利申请和杂志文章通过引用以其整体明确结合到本文中。
[0758]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于wo 2008/130619中的那些化合物,该专利通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[0759][0760]
以上化合物的取代基定义如下:
[0761]
x为so或so2,
[0762]
r1为h,或烷基,
[0763]
r2为烷基,或卤素。
[0764]
在具体的实施方案中,r1为me。在其它的具体实施方案中,r1为f。在某些实施方案中,r2为t-bu。在具体的实施方案中,rl为甲基。在更具体的实施方案中,所述化合物选自:
[0765][0766]
在相关的实施方案中,用于本发明方法的pde7抑制剂具有下式:
[0767][0768]
其中
[0769]
r1为烷基,
[0770]
r2为芳基或杂芳基,
[0771]
r3为烷基、芳基、环烷基,或烷基芳基。
[0772]
在具体的实施方案中,r1为甲基。在某些实施方案中,r2为呋喃基或噻吩基。在其它的具体实施方案中,r2为取代的苯基或苄基。在优选的实施方案中,r3为异-丁基。在更具体的实施方案中,所述化合物选自:
[0773][0774]
在另一个相关的实施方案中,用于本发明方法的pde7抑制剂具有下式:
[0775][0776]
其中
[0777]
r1为腈,或烷基羧酸酯,
[0778]
r2为烷基、芳基,或杂芳基。
[0779]
在具体的实施方案中,r1为腈或甲基羧酸酯。在某些实施方案中,r2为5元杂芳基。在更具体的实施方案中,r2为呋喃基,或噻吩基。在其它实施方案中,r2为6元芳基。在更具体的实施方案中,r2为取代的苯基。
[0780][0781]
在另一个相关的实施方案中,用于本发明方法的pde7抑制剂具有下式:
[0782][0783]
其中
[0784]
r1为烷基、烯基,或烷基羧酸,
[0785]
r2为卤素。
[0786]
在某些实施方案中,r1为丁基。在其它实施方案中,r1为封端烯基(terminal alkenyl)。在更具体的实施方案中,r1为烯丙基或乙烯基。在其它实施方案中,r1为c1-4烷基。在具体的实施方案中,r1为甲基羧酸。在某些实施方案中,r2为cl或br。在更具体的实施方案中,所述化合物选自:
[0787][0788]
在其它相关实施方案中,用于本发明方法的pde7抑制剂具有下式:
[0789][0790]
其中
[0791]
r1为co,或烷基醇,r2为烷基,r3为烷氧基,以及c4和c9立构中心独立为(r)或(s)。
[0792]
在某些实施方案中,r1为羰基,或2-甲基丙-1-醇。在具体的实施方案中,r2为甲基。在某些实施方案中,r3为甲氧基。在更具体的实施方案中,所述化合物选自:
[0793][0794]
在另一个相关的实施方案中,用于本发明方法的pde7抑制剂具有下式:
[0795][0796]
其中
[0797]
r1为氢、羟基、羰基,或烷基醇,
[0798]
r2和r3独立选自氢、烷基、烷基羧酸酯,或羧酸,
[0799]
r4为氢,或烷基,
[0800]
r5为氢、烷基、羟基,或乙酸酯,
[0801]
r6为氢,或烷氧基,以及c4和c9立构中心独立为(r)或(s)。
[0802]
在某些实施方案中,r1为2-甲基丙-1-醇。在具体的实施方案中,r2为甲基。在某些实施方案中,r2为甲基羧酸酯。在具体的实施方案中,r2和r3均为甲基。在其它实施方案中,r2为甲基,和r3为甲基羧酸酯。在具体的实施方案中,r4为异-丙基。在具体的实施方案中,r5为甲基。在某些实施方案中,r6为甲氧基。在更具体的实施方案中所述化合物选自:
[0803][0804]
关于上述化合物,术语"烷基"、"烯基"和前缀"烷(alk)-"包括直链和支链基团二者并且还包括环状基团,即环烷基和环烯基。除非另外指明,这些基团包含1-20个碳原子,而烯基包含2-20个碳原子。优选的基团具有总共至多10个碳原子。环状基团可以是单环基或多环基,并且优选地具有3-10个环碳原子。示例性环状基团包括环丙基、环戊基、环己基、环丙基甲基、金刚烷基、降冰片烷和降冰片烯(norbornee)。这样的规定也适合于包括前缀"烷基-"的基团,例如烷基羧酸、烷基醇、烷基羧酸酯、烷基芳基等。合适的烷基羧酸基团的实例为甲基羧酸、乙基羧酸等。合适的烷基醇的实例为甲基醇、乙基醇、异丙基醇、2-甲基丙-1-醇等。合适的烷基羧酸酯的实例为甲基羧酸酯、乙基羧酸酯等。合适的烷基芳基基团的实例为苄基、苯基丙基等。
[0805]
本文所用的术语"芳基"包括碳环芳族环或环系统。芳基的实例包括苯基、萘基、联苯基、芴基和茚基。术语"杂芳基"包括包含至少一个环杂原子(如o、s、n)的芳族环或环系统。合适的杂芳基包括呋喃基、噻吩基、吡啶基、喹啉基、异喹啉基、吲哚基、异吲哚基、噻唑基、吡咯基、四唑基、咪唑基、吡唑基、唑基、噻唑基、苯并呋喃基、苯并噻吩基、咔唑基、苯并唑基、嘧啶基、苯并咪唑基、喹喔啉基、苯并噻唑基、萘啶基、异唑基、异噻唑基、嘌呤基、喹唑啉基等。
[0806]
芳基和杂芳基可以是未取代的或被一个或多个独立选自以下的取代基取代:烷基、烷氧基、亚甲二氧基、亚乙二氧基、烷硫基、卤代烷基、卤代烷氧基、卤代烷硫基、卤素,硝基、羟基、巯基、氰基、羧基、甲酰基、芳基、芳基氧基、芳基硫基、芳基烷氧基、芳基烷硫基、杂
芳基、杂芳基氧基、杂芳基烷氧基、杂芳基烷硫基、氨基、烷基氨基、二烷基氨基、杂环基、杂环烷基、烷基羰基、烯基羰基、烷氧基羰基、卤代烷基羰基、卤代烷氧基羰基、烷硫基羰基、芳基羰基、杂芳基羰基、芳基氧基羰基、杂芳基氧基羰基、芳基硫基羰基、杂芳基硫基羰基、链烷酰基氧基、链烷酰基硫基、链烷酰基氨基、芳基羰基氧基、芳基羰基硫基、烷基氨基磺酰基、烷基磺酰基、芳基磺酰基、杂芳基磺酰基、芳基二嗪基、烷基磺酰基氨基、芳基磺酰基氨基、芳基烷基磺酰基氨基、烷基羰基氨基、烯基羰基氨基、芳基羰基氨基、芳基烷基羰基氨基、芳基羰基氨基烷基、杂芳基羰基氨基(atnino),杂芳基烷基羰基氨基、烷基磺酰基氨基、烯基磺酰基氨基、芳基磺酰基氨基、芳基烷基磺酰基氨基、杂芳基磺酰基氨基、杂芳基烷基磺酰基氨基、烷基氨基羰基氨基、烯基氨基羰基氨基、芳基氨基羰基氨基、芳基烷基氨基羰基氨基、杂芳基氨基羰基氨基、杂芳基烷基氨基羰基氨基,以及在杂环基的情况下的氧代基。如果其它基团被描述为"取代的"或"任选取代的",则这些基团也可被一个或多个上文列举的取代基取代。
[0807]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献中的那些化合物:wo 2008/142550该专利通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[0808][0809]
以上化合物的取代基定义如下:
[0810]
m是0、1或2,n是0、1、2或3,
[0811]
x为o、s或n-cn,
[0812]
r1为卤素或cn,
[0813]
a为单键、ch2、o或s,
[0814]
b为单键、ch2或och2,各r2独立为卤素、(c
1-6
)烷基(任选被1-3个氟原子取代)、oh、(c
1-6
)烷硫基或cn,
[0815]
r3选自以下基团(i)-(x):
[0816][0817]
r为h或(c
1-6
)烷基(任选被1-3个氟原子取代),r'为(c
1-6
)烷基(任选被1-3个氟原子取代),或其药学上可接受的盐、溶剂化物、多晶型物或前药。
[0818]
关于上述化合物,术语"烷基"表示包含1-6个碳原子的单价、直链或支链的、饱和烃链。烷基的实例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、2-甲基丁基、3-甲基丁基、新戊基、正己基、2-甲基戊基、3-甲基戊基、4-甲基戊基、2-乙基丁基和2,2-二甲基丁基。优选的烷基特别为甲基和乙基,尤其是甲基。
[0819]
当提及时,烷基可以被1-3个氟原子取代。取代可以在烷基链上的任何位置上。优选地,这样的氟化的烷基具有1-4个碳原子,更优选1或2个碳原子。一-、二-和三氟甲基(尤其是三氟甲基),和一-、二-和三氟乙基(尤其是2,2,2-三氟乙基)是尤其优选的。
[0820]
术语"烷氧基"表示"烷基-o-",其中"烷基"如上所定义,无论是在其广义的方面,或是在其优选的方面。优选的烷氧基是尤其为甲氧基和乙氧基的基团。术语"烷硫基"表示"烷基-s-",其中"烷基"如上所定义,无论是在其广义的方面,或是在其优选的方面。优选的烷硫基基团为(c
1-4
)烷硫基,尤其是甲基硫基和乙基硫基。术语"卤素"表示氟代、氯代、溴代或碘代。优选的卤素基团为氟代和氯代。
[0821]
优选地,m是0或1,更优选1。
[0822]
优选地,n是0或1,更优选0.
[0823]
优选地,x为o或n-cn,更优选o。
[0824]
优选地,r1为f或cl,更优选cl。
[0825]
优选地,a为单键或o,更优选o。
[0826]
当基团b为och2时,氧原子连接于苯环,而亚甲基连接于基团r3。
[0827]
优选地,b为单键。
[0828]
优选地,r2为f或cl,更优选f。
[0829]
优选地,r3为基团(i),(ii),(iii),(iv),(v)或(vi),更优选为基团(i)或(ii),和尤其是基团(ii)。
[0830]
在一个实施方案中,基团-b-r3存在于苯环的2-位(基团a的位置在1-位)。在其它实施方案中,基团-b-r3存在于3-位。在进一步的实施方案中,基团-b-r3存在于4-位。
[0831]
用于本发明方法的pde7抑制剂包括其中以上结构式中的每个变量选自合适的和/或优选的每个变量的组合的那些抑制剂。用于本发明方法的甚至更优选的pde7抑制剂包括其中以上结构式中的每个变量选自更优选或最优选的每个变量的组合的那些抑制剂。
3-氟-n-(甲基磺酰基)苯甲酰胺,
[0853]
n-{2-[(8'-氯-2'-氧代-2',3'-二氢-1'h-螺[环己烷-1,4'-喹唑啉]-5'-基)氧基]-3-氟代苯基}-1,1,1-三氟甲烷磺酰胺,
[0854]
{2-[(8'-氯-2'-氧代-2',3'-二氢-1'h-螺[环己烷-1,4'-喹唑啉]-5'-基)氧基]-3-氟代苯基}乙酸,
[0855]
{2-[(8'-氯-2'-氧代-2',3'-二氢-1'h-螺[环己烷-1,4'-喹唑啉]-5'-基)氧基]苯氧基}乙酸,
[0856]
{4-[(8'-氯-2'-氧代-2',3'-二氢-1'h-螺[环己烷-1,4'-喹唑啉-5'-基)氧基]苯氧基}乙酸,
[0857]
2-[(8'-氯-2'-氧代-2',3'-二氢-1'h-螺[环己烷-1,4'-喹唑啉]-5'-基)氧基]-3-氟代苯甲酸甲基酯,
[0858]
及其药学上可接受的盐、溶剂化物和前药。
[0859]
在另一个相关的实施方案中,以下pde7抑制剂用于本发明的方法中:
[0860]
8'-氯-5'-[2-氟-6-(2h-四唑-5-基)苯氧基]-1'h-螺环己烷-1,4'-喹唑啉]-2'(3'h)-酮,
[0861]
8'-氯-5'-[4-氟-2-(1h-四唑-5-基)苯氧基]-1'h-螺[环己烷-1,4'-喹唑啉]-2'(3'h)-酮,
[0862]
8'-氯-5'-[6-氟-2-(1h-四唑-5-基)苯氧基]-1'η-螺[环己烷-1,4'-喹唑啉]-2'(3'h)-酮,
[0863]
8'-氯-5'-[4-氟-2-(1h-四唑-5-基)苯氧基]-1'h-螺[环戊烷-1,4'-喹唑啉]-2'(3'h)-酮,
[0864]
8'-氯-5'-[6-氟-2-(1h-四唑-5-基)苯氧基]-1'h-螺[环戊烷-1,4'-喹唑啉]-2'(3'h)-酮,
[0865]
8'-氯-5'-[6-氯-2-(1h-四唑-5-基)苯氧基]-1'h-螺[环戊烷-1,4'-喹唑啉]-2'(3'h)-酮,
[0866]
8'-氯-5'-[2-(1h-四唑-5-基)苯氧基]-1'h-螺[环戊烷-1,4'-喹唑啉]-2'(3'h)-酮,
[0867]
8'-氯-5'-[2-(1h-四唑-5-基)苯氧基]-1'h-螺[环己烷-1,4'-喹唑啉]-2'(3'h)-酮,
[0868]
及其药学上可接受的盐、溶剂化物和前药。
[0869]
以下化合物为最优选的:
[0870]
8'-氯-5'-[2-氟-6-(2h-四唑-5-基)苯氧基]-1'h-螺[环己烷-1,4'-喹唑啉]-2'(3'h)-酮,
[0871]
8'-氯-5'-[4-氟-2-(1h-四唑-5-基)苯氧基]-1'h-螺[环己烷-1,4'-喹唑啉]-2'(3'h)-酮,
[0872]
8'-氯-5'-[6-氟-2-(1h-四唑-5-基)苯氧基]-1'h-螺[环戊烷-1,4'-喹唑啉]-2'(3'h)-酮,
[0873]
8'-氯-5'-[2-(1h-四唑-5-基)苯氧基]-1'h-螺[环己烷-1,4'-喹唑啉]-2'(3'h)-酮,
[0874]
及其药学上可接受的盐、溶剂化物和前药。
[0875]
这些化合物的制备描述于wo 2008/142550中。
[0876]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献中的那些化合物:us 7498334,us 2005/0059686和wo 2003/055882,各文献通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[0877][0878]
以上化合物的取代基定义如下:
[0879]
x为苯基或het,其各自为未取代的或被r1和/或r2单取代或多取代,r1和r2各自相互独立地为a、oh、oa、sa、soa、so2a、so2nh2、so2nha、so2aa'、cn、no2、nh2、nha、naa'、nhcoa、nhcooa、cooh、cooa、conh2、conha、conaa'或hal,r'和r2一起可选择性地为—och2o—或—och2ch2o—,r3为a、oh、oa、sa、soa、so2a、so2nh2、so2nha、so2aa'、cn、no2、nh2、nha、nhb、naa'、nhcoa、nhcooa、nhcob、nhcoob、cooh、cooa、coob、conh2、conha、conhb、conaa'或hal,r4为具有至多10个碳原子的支链或非支链烷基或烯基,其可以被1-5f和/或cl原子取代的和/或其中一个或多个ch2基团可以被o、s、so、so2、nh、na、nhco、naco、nhcoo或nacoo置换,或具有3-7个碳原子的环烷基或环烯基,其中一个或多个ch2基团可以被o、s、so、so2、so2nh、so2na、nh、nha、nhconh、naconh、nacona、nhco、naco、nhcoo或nacoo置换,r5为oh、oa、sa、soa、so2a、so2nh2、so2nha、so2aa'、cn、no2、nh2、nha、naa'、nhcoa、nhcooa、cooh、cooa、conh2、conha、conaa'或hal,r6为h、oh、oa、sa、soa、so2a、so2nh2、so2nha、so2aa'、cn、no2、nh2、nha、naa'、nhcoa、nhcooa、cooh、cooa、conh2、conha、conaa'或hal,a和a'各自相互独立地为具有至多10个碳原子的支链或非支链烷基或烯基,其可以被1-5f和/或cl原子取代和/或其中一个或多个ch2基团可以被o、s、so、so2、nh、nr7、nhco、nr7co、nhcoo或nr7coo置换。a和a'一起可选择性地为具有3-7碳原子的亚烷基,其中一个或两个ch2基团可以被chr7、chr7r8、o、s、so、so2、nh、nr7、nhco、nr7co、nhcoo或nr7coo置换。b为苯基或het,其各自为未取代的或被r1和/或r2单取代或多取代,het为具有1-3个n、o和/或s原子的芳族5-或6-元杂环基环。其为未取代的或被a
″
、hal或cf3单取代、二取代或三取代,r7和r8各自相互独立地为具有至多5碳原子的支链或非支链烷基或烯基,其可以被1-5f和/或cl原子取代和/或其中一个或多个ch2基团可以被o、s、so、so2或nh置换,a
″
为具有1-6碳原子的烷基,和hal为f、cl、br或i,及其药学上可用的衍生物、溶剂化物和立体异构体,包括其所有比例的混合物。
[0880]
在相关的实施方案中,用于本发明方法的pde7抑制剂包括以上结构式的化合物,其中r5为oh,所述化合物也可以为该结构式的其互变异构体的形式:
[0881][0882]
关于上述化合物,用于本发明方法的pde7抑制剂包括光学活性形式(立体异构体)、对映体、外消旋体、非对映体,以及这些化合物的水合物和溶剂化物。采用术语化合物的溶剂化物意指惰性溶剂分子加合到所述化合物上,所述溶剂化物是由于它们的相互吸引力而形成的。溶剂化物是例如一水合物,二水合物或醇盐。
[0883]
关于上述化合物,术语药学上可用的衍生物被用来意指,例如上述化合物的盐和所谓的前药化合物。术语前药衍生物被用来意指,例如,已被修饰的上述化合物,例如,用烷基或酰基基团、糖或低聚肽修饰,并且其在有机体内被快速裂解,因而释放出活性化合物。这些也包括上述化合物的可生物降解的聚合物衍生物,例如在int.j.pharm.115,61-67(1995)中所描述的。
[0884]
关于上述化合物,出现不只一次的所有基团的意义在每种情况下是彼此独立的。
[0885]
a和a'优选为烷基,更优选被1-5个氟原子和/或氯原子取代的烷基,还优选烯基。
[0886]
在以上结构式中,烷基优选为非支链的并具有1、2、3、4、5、6、7、8、9或10个碳原子,优选具有1、2、3、4、5或6碳原子,且优选为甲基、乙基、三氟甲基、五氟乙基或丙基,还优选异丙基、丁基、异丁基、仲丁基或叔丁基,而且还优选正戊基、新戊基、异戊基或正己基。甲基、乙基、三氟甲基、丙基、异丙基、丁基、正戊基、正己基或正癸基是特别优选的。
[0887]a″
优选地为具有1、2、3、4、5或6碳原子的烷基,例如甲基、乙基或丙基,还优选异丙基、丁基、异丁基、仲丁基或叔丁基,而且还优选正戊基、新戊基、异戊基或正己基。甲基、乙基、丙基、异丙基或丁基是特别优选的。
[0888]
环烷基优选具有3-7个碳原子且优选为环丙基或环丁基,还优选为环戊基或环己基,还进一步优选为环庚基;特别的优选给予环戊基。
[0889]
烯基优选地为乙烯基、烯丙基、2-或3-丁烯基、异丁烯基或仲丁烯基;进一步的优选给予4-戊烯基、异戊烯基或5-己烯基。
[0890]
亚烷基优选地为非支链的且优选地为亚甲基或亚乙基,还优选亚丙基或亚丁基。
[0891]
hal优选地为f、cl或br,此外也优选i。
[0892]
基团r1和r2可以是相同的或不同的且优选在苯基环的2-或4-位。它们例如彼此独立地为a或hal,或一起为亚甲二氧基。
[0893]
然而,它们优选各自为甲基、乙基、丙基、甲氧基、乙氧基、丙氧基、异丙氧基、苄氧基,而且也可为氟代-、二氟代-或三氟代-甲氧基,或1-氟代-、2-氟-、1,2-二氟代-、2,2-二氟代-、1,2,2-三氟代-或2,2,2-三氟代乙氧基,此外还可为氟或氯。
[0894]
r1特别优选地为氟、氯、甲基、乙基或丙基。
[0895]
r2特别优选地为氟、氯、甲基、乙基或丙基。
[0896]
x优选地为被r1单取代的苯基或为未取代的het。
[0897]
x特别优选地为2-氯代苯基、2-氟代苯基、4-甲基-苯基、3-氯代苯基或4-氯代苯基。
[0898]
het优选地为例如,未取代的2-或3-呋喃基、2-或3-噻吩基、1-,2-或3-吡咯基、1-,2-,4-或5-咪唑基、2-,3-或4-吡啶基、2-,4-,5-或6-嘧啶基,还优选1,2,3-三唑-1-,-4-或-5-基、1,2,4-三唑-1-,-3-或-5-基、1,2,3-二唑-4-或-5-基、1,2,4-二唑-3-或-5-基、1,3,4-噻二唑-2-或-5-基、1,2,4-噻二唑-3-或-5-基,或1,2,3-噻二唑-4-或-5-基。
[0899]
r3优选地为例如,cooa
″
或cooh。
[0900]
r4优选地为例如,具有1,2,3,4,5或6碳原子的非支链或支链烷基,其可以被1-5个f或cl原子取代,优选地为甲基、乙基、三氟甲基、五氟乙基或丙基,还优选异丙基、丁基、异丁基、仲丁基或叔丁基,而且还优选正戊基、新戊基、异戊基或正己基。特别的优选给予甲基、乙基、三氟甲基、丙基、异丙基、丁基、正戊基、正己基或正癸基。
[0901]
r5优选地为cl或oh。
[0902]
r6优选地为h。
[0903]
关于上述化合物,至少一个所述基团具有上文指定的优选含义之一。
[0904]
在相关的实施方案中,用于本发明方法的pde7抑制剂包括以下化合物,其中x为被r1单取代的苯基,或为未取代的het;r1为a或hal;r3为cooa
″
或cooh;r4为具有1、2、3、4、5或6个碳原子的非支链或支链烷基,其可以被1-5个f或cl原子取代;r5为cl或oh;和r6为h;
[0905]
在其它相关实施方案中,用于本发明方法的pde7抑制剂包括以下化合物,其中x为被r1单取代的苯基,或为未取代的het、r1为a或hal、r3为cooa
″
或cooh、r4为具有1、2、3、4、5或6碳原子的非支链或支链烷基,其可以被1-5个f或cl原子取代、r5为cl或oh,r6为h,het为呋喃基、噻吩基、吡咯基、咪唑基、吡啶基或嘧啶基、a和a
″
各自相互独立地为具有1、2、3、4、5或6碳原子的非支链或支链烷基,其可以被1-5个f或cl原子取代,hal为f、cl或br,及其药学上可用的衍生物、溶剂化物和立体异构体,包括其所有比例的混合物。
[0906]
以上化合物的制备以及用于它们的制备的起始原料描述于文献(例如在标准著作中,例如houben-weyl,methoden der organischen chemie[有机化学方法(methods oforganic chemistry)],georg-thieme-verlag,stuttgart)中,一般在对所述反应已知的和合适的反应条件下实施。也可在此使用本领域已知的但未在本文更详细提及的变量。
[0907]
在另一个相关的实施方案中,用于本发明方法的pde7抑制剂包括:
[0908]
5-异丙基-4-氧代-7-对甲苯基-4,7-二氢-3h-吡咯并[2,3-d]-嘧啶-6-羧酸乙基酯、5-甲基-4-氧代-7-(3-氯代苯基)-4,7-二氢-3h-吡咯并[2,3-d]嘧啶-6-羧酸乙基酯、5-甲基-4-氧代-7-(2-氯代苯基)-4,7-二氢-3h-吡咯并[2,3-d]嘧啶-6-羧酸乙基酯、5-甲基-4-氧代-7-(2-氟代苯基)-4,7-二氢-3h-吡咯并[2,3-d]嘧啶-6-羧酸乙基酯、5-丙基-4-氧代-7-(2-氯代苯基)-4,7-二氢-3h-吡咯并[2,3-d]嘧啶-6-羧酸乙基酯、5-甲基-4-氧代-7-(4-氯代苯基)-4,7-二氢-3h-吡咯并[2,3-d]嘧啶-6-羧酸乙基酯、5-甲基-4-氧代-7-对甲苯基-4,7-二氢-3h-吡咯并[2,3-d]-嘧啶-6-羧酸乙基酯、5-甲基-4-氧代-7-(2-氯代苯基)-4,7-二氢-3h-吡咯并[2,3-d]嘧啶-6-羧酸甲基酯、5-甲基-4-氧代-7-苯基-4,7-二氢-3h-吡咯并[2,3-d]-嘧啶-6-羧酸甲基酯、5-甲基-4-氧代-7-(2-噻吩基)-4,7-二氢-3h-吡咯并[2,3-d]-嘧啶-6-羧酸甲基酯,及其药学上可用的衍生物、溶剂化物和立体异构体,包括其所有比例的混合物。
[0909]
以上化合物的制备描述于us 7498334和wo 2003/055882中。
[0910]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下
文献中的那些化合物:u.s.pat.no.6884800和wo 01/36425,各文献通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[0911][0912]
以上化合物的取代基定义如下:
[0913]
r1和r2彼此独立地各自表示a1、oa1、sa1或hal,a1表示h、a、烯基、环烷基或亚烷基环烷基,a表示具有1-10个碳原子的烷基、hal表示f、cl、br或i,和x表示o、s、so或so2,及其生理学上可接受的盐和/或溶剂化物。
[0914]
关于上述化合物,a表示具有1-10个碳原子,即具有1、2、3、4、5、6、7、8、9或10个碳原子的烷基并优选地表示甲基、乙基或丙基,还优选异丙基、丁基、异丁基、仲丁基或叔丁基,而且还优选正戊基、新戊基、异戊基或己基。在这些基团中,1-7h原子也可以被f和/或cl置换。a因此也表示,例如,三氟甲基或五氟乙基。环烷基具有3-9个碳原子并优选地表示,例如,环戊基或环己基。烯基具有2-10个碳原子,为直链或支链的并优选表示乙烯基、丙烯基或丁烯基。亚烷基环烷基具有4-10个碳原子且表示,例如,亚甲基环戊基、亚乙基环戊基、亚甲基环己基或亚乙基环己基。r1和r2优选在每种情况下彼此独立地表示h、氟、氯、甲基、乙基、丙基、甲氧基、乙氧基、丙氧基、甲基硫基、环戊基或环己基。
[0915]
在相关的实施方案中,用于本发明方法的pde7抑制剂包括以下化合物,其中
[0916]
x为s;
[0917]
x为s,r1为h;
[0918]
x为s,r1为f或cl;
[0919]
x为s,r2为h;
[0920]
x为s,r2为f或cl;
[0921]
x为s,r1为h,r2为f或cl;
[0922]
x为s,r1为f或cl,r2为h;
[0923]
x为s;a1为h或a,a为具有1、2、3或4碳原子的烷基;
[0924]
x为s,r1和r2彼此独立地各自表示a1或hal,a1为h或a,a为具有1、2、3或4碳原子的烷基,hal为f或cl;
[0925]
及其生理学上可接受的盐和溶剂化物。
[0926]
在另一个相关的实施方案中,用于本发明方法的pde7抑制剂包括以下化合物:
[0927]
10-氯-3-咪唑-1-基-2,3-二氢-1h-吡啶并[3,2,1-kl]吩噻嗪、4-氯-3-咪唑-1-基-2,3-二氢-1h-吡啶并[3,2,1-kl]吩噻嗪、10-甲氧基-3-咪唑-1-基-2,3-二氢-1h-吡啶并[3,2,1-kl]吩噻嗪、10-丙氧基-3-咪唑-1-基-2,3-二氢-1h-吡啶并[3,2,1-kl]吩噻嗪、10-甲基硫基-3-咪唑-1-基-2,3-二氢-1h-吡啶并[3,2,1-kl]吩噻嗪、10-氟-3-咪唑-1-基-2,3-二氢-1h-吡啶并[3,2,1-kl]吩噻嗪、4,10-二氟-3-咪唑-1-基-2,3-二氢-1h-吡啶并
[3,2,1-kl]吩噻嗪、10-三氟甲基-3-咪唑-1-基-2,3-二氢-1h-吡啶并[3,2,1-kl]吩噻嗪、4-环戊氧基-3-咪唑-1-基-2,3-二氢-1h-吡啶并[3,2,1-kl]吩噻嗪、10-氯-3-咪唑-1-基-2,3-二氢-1h-7-氧杂-11b-氮杂苯并[de]-蒽,和10-氯-3-咪唑-1-基-2,3-二氢-1h-吡啶并[3,2,1-kl]吩噻嗪7,7-二氧化物。
[0928]
这些化合物的制备描述于u.s.pat.no.6,884,800和wo 01/36425中。
[0929]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献中的那些化合物:u.s.pat.no.6,531,498和wo 01/32175,各文献通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[0930][0931]
以上化合物的取代基定义如下:
[0932]
r1、r2、r3、r4各自相互独立地为hal、oa1、sa1、a、h、cooa1、cn或cona1a2,
[0933]
r5为cooa1、cn或cona1a2,
[0934]
a1、a2各自相互独立地为h、a、烯基、环烷基或亚烷基环烷基,
[0935]
a为具有1-10个c原子的烷基,
[0936]
hal为f、cl、br或i,
[0937]
及其生理学上可接受的盐和/或溶剂化物。
[0938]
关于上述化合物,a为具有1-10个c原子,即具有1、2、3、4、5、6、7、8、9或10个c原子的烷基且优选地为甲基、乙基或丙基、也优选地为异丙基、丁基、异丁基、仲丁基或叔丁基,而且还为正戊基、新戊基、异戊基或己基。基团中的1-7个h原子也可能被f和/或cl置换。a因此也为例如三氟甲基或五氟乙基。
[0939]
环烷基具有3-9个c原子且优选地为例如,环戊基或环己基。烯基具有2-10个c原子,为直链或支链的且优选地为乙烯基、丙烯基或丁烯基。
[0940]
亚烷基环烷基具有4-10个c原子且为例如亚甲基环戊基、亚乙基环戊基、亚甲基环己基或亚乙基环己基。
[0941]
在相关的实施方案中,用于本发明方法的pde7抑制剂包括所述化合物,其中
[0942]
r1为h;
[0943]
r1和r2为h;
[0944]
r1为h和r2为f或cl;
[0945]
r1、r2各自相互独立地为h或hal;
[0946]
r1、r2各自相互独立地为h或hal,a1、a2各自相互独立地为h或a;
[0947]
a1、a2各自相互独立地为h或a;
[0948]
r1、r2各自相互独立地为h或hal,a1、a2各自相互独立地为h或a,a为具有1、2、3或4c原子的烷基,hal为f或cl。
[0949]
在另一个相关的实施方案中,用于本发明方法的pde7抑制剂包括所述化合物:
[0950]
5-[2-(2-氟-4-羟基苯基氨基)乙烯基]-4-氰基-3-苯基异唑,
[0951]
5-[2-(2,4-二氟苯基氨基)乙烯基]-4-氰基-3-苯基异唑,
[0952]
5-[2-(3-甲基硫基苯基氨基)乙烯基]-4-氰基-3-苯基异唑,
[0953]
5-[2-(2,4-二甲氧基苯基氨基)乙烯基]-4-氰基-3-(2-氯代苯基)异唑,
[0954]
5-(2-氨基-2-苯基乙烯基)-4-甲基氨基羰基-3-苯基异唑,
[0955]
5-(2-苯基氨基乙烯基)-4-甲氧基羰基-3-苯基异唑,
[0956]
5-[2-(4-羧基苯基氨基)乙烯基]-4-氰基-3-苯基异唑,
[0957]
5-[2-(4-羧基苯基氨基)乙烯基]-4-甲氧基羰基-3-苯基异唑,
[0958]
5-[2-(5-氯-2-羟基苯基氨基)乙烯基]-4-氰基-3-苯基异唑,
[0959]
5-[2-(3,4-二甲基苯基氨基)乙烯基]-4-氰基-3-(2-氯代苯基)异唑,
[0960]
5-[2-(4-氯代苯基氨基)乙烯基]-4-氰基-3-(2-氯代苯基)异唑,
[0961]
5-(2-苯基氨基乙烯基)-4-氰基-3-(2-氯代苯基)异唑,
[0962]
5-[2-(4-甲氧基苯基氨基)乙烯基]-4-氰基-3-(2-氯代苯基)异唑,
[0963]
5-[2-(4-羧基苯基氨基)乙烯基]-4-氰基-3-(2-氯代苯基)异唑,
[0964]
5-[2-(2-氟-4-羟基苯基氨基)乙烯基]-4-氰基-3-(2-氯代苯基)异唑,
[0965]
5-[2-(4-氟代苯基氨基)乙烯基]-4-氰基-3-(2-氯代苯基)异唑,
[0966]
5-[2-(3,5-二氯苯基氨基)乙烯基]-4-氰基-3-(2-氯代苯基)异唑,
[0967]
5-[2-(3-氯代苯基氨基)乙烯基]-4-氰基-3-(2-氯代苯基)异唑,
[0968]
5-(2-苯基氨基乙烯基)-4-氰基-3-(2,6-二氯苯基)异唑,
[0969]
5-[2-(4-氯代苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异唑,
[0970]
5-(2-苯基氨基乙烯基)-4-甲氧基羰基-3-(2,6-二氯苯基)异唑,
[0971]
5-[2-(4-氯代苯基氨基)乙烯基]-4-甲氧基羰基-3-(2,6-二氯苯基)异唑,
[0972]
5-[2-(4-羧基苯基氨基)乙烯基]-4-甲氧基羰基-3-(2,6-二氯苯基)异唑,
[0973]
5-[2-(2,4-二氟苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异唑,
[0974]
5-[2-(2,4-二氯苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异唑,
[0975]
5-[2-(4-羧基苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异唑,
[0976]
5-[2-(3,5-二氯苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异唑,
[0977]
5-[2-(4-甲氧基苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异唑,
[0978]
5-[2-(2,4-二甲氧基苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异唑,
[0979]
5-[2-(2-苯基苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异唑,
[0980]
5-[2-(4-甲基苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异唑,
[0981]
5-(2-苯基氨基乙烯基)-4-氰基-3-(2-氯-6-氟代苯基)异唑,
[0982]
5-[2-(4-羧基苯基氨基)乙烯基]-4-氰基-3-(2-氯-6-氟代苯基)异唑,
[0983]
5-[2-(4-氯代苯基氨基)乙烯基]-4-氰基-3-(2-氯-6-氟代苯基)异唑,
[0984]
5-[2-(3-甲氧基苯基氨基)乙烯基]-4-氰基-3-(2-氯-6-氟代苯基)异唑,
[0985]
5-[2-(4-氯代苯基氨基)乙烯基]-4-甲氧基羰基-3-(2-氯-6-氟代苯基)异唑,
[0986]
5-(2-苯基氨基乙烯基)-4-甲氧基羰基-3-(2-氯-6-氟代苯基)异唑,
[0987]
5-[2-(2,4-二氯苯基氨基)乙烯基]-4-甲氧基羰基-3-(2-氯-6-氟代苯基)异唑,
[0988]
5-(2-苯基氨基乙烯基)-4-氰基-3-苯基异唑,
[0989]
5-[2-(3-三氟甲氧基苯基氨基)乙烯基]-4-氰基-3-苯基异唑,
[0990]
5-[2-(4-甲氧基苯基氨基)乙烯基]-4-氰基-3-苯基异唑,
[0991]
5-[2-(4-甲氧基苯基氨基)乙烯基]-4-甲氧基羰基-3-(2-氯-6-氟代苯基)异唑,
[0992]
5-[2-(3-甲基硫基苯基氨基)乙烯基]-4-氰基-3-苯基异唑,
[0993]
5-[2-(2,4-二氟苯基氨基)乙烯基]-4-氰基-3-苯基异唑,
[0994]
5-[2-(2-氟-4-羟基苯基氨基)乙烯基]-4-氰基-3-苯基异唑.
[0995]
这些化合物的制备描述于u.s.pat.no.6531498和wo 01/32175中。
[0996]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献中的那些化合物:us 7491742和wo 2001/29049,各文献通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[0997][0998]
以上化合物的取代基定义如下:
[0999]
r1为h、a、苄基、茚满-5-基、1,2,3,4-四氢萘-5-基、二苯并噻吩-2-基,或苯基,其为未取代的或被hal、a、a—co—nh、苄氧基、烷氧基、cooh或cooa一-、二-或三取代,r2为h或a,x为o或s,hal为f、cl、br或i,a为具有1-6个c原子的烷基,及其生理学上可接受的盐和/或溶剂化物。
[1000]
关于上述化合物,a为含1-6个c原子的烷基并具有1、2、3、4、5或6个c原子且优选地为甲基、乙基或丙基,也优选地为异丙基、丁基、异丁基、仲丁基或叔丁基,而且还优选地为正戊基、新戊基、异戊基或己基。a也为环烷基例如环己基。烷氧基优选地为甲氧基、乙氧基、丙氧基或丁氧基。hal优选地为f或cl。a-co
‑‑
nh优选地为乙酰氨基。
[1001]
在相关的实施方案中,用于本发明方法的pde7抑制剂选自以下化合物:
[1002]
1-苯基-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-苄基-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-环己基-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-环戊基-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-丁基-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-异丙基-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-丙基-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-乙基-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-甲基-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、2-甲基-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-苯基-[1]苯并噻喃并[3,4-d]咪唑-4-(1h)-酮、1-苄基-[1]苯并噻喃并[3,4-d]咪唑-4-(1h)-酮、1-环己基-[1]苯并噻喃并[3,4-d]咪唑-4-(1h)-酮、1-环戊基-[1]苯并噻喃并[3,4-d]咪唑-4-(1h)-酮、1-丁基-[1]苯并噻喃并[3,4-d]咪唑-4-(1h)-酮、1-异丙基-[1]苯并噻喃并[3,4-d]咪唑-4-(1h)-酮、1-丙基-[1]苯并噻喃并[3,4-d]咪唑-4-(1h)-酮、
1-乙基-[1]苯并噻喃并[3,4-d]咪唑-4-(1h)-酮、1-甲基-[1]苯并噻喃并[3,4-d]咪唑-4-(1h)-酮、[1]苯并噻喃并[3,4-d]咪唑-4-(1h)-酮、2-甲基-[1]苯并噻喃并[3,4-d]咪唑-4-(1h)-酮、1-(2-氯代苯基-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(4-甲基-苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(4-氟代苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(2,4-二甲基-苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(3-氯代苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(2,4-二氯苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(2,5-二氯苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(4-乙酰氨基-苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(2-氟代苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(3-氟代苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(2-苄基氧基-苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(2,6-二甲基-苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(茚满-5-基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(2-甲氧基-苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(2,3-二甲基-苯基)-[1]苯并吡喃并[3,4-d]咪唑-(1h)-4-酮、1-(2,3-二氯苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(3-氯-4-甲基-苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(2,5-二甲基-苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(4-氯代苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(1,2,3,4-四氢萘-5-基)-[1]苯并吡喃并-[3,4-d]咪唑-4-(1-h)-酮、1-(二苯并噻吩-2-基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(3-甲氧基-苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮、1-(4-羧基-2-甲基-苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1h)-酮,及其生理学上可接受的盐和/或溶剂化物。
[1003]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献中的那些化合物:u.s.pat.no.6,737,436和wo 01/32618,所述文献通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[1004][1005]
以上化合物的取代基定义如下:
[1006]
r1和r2彼此独立地各自表示h、a、oa、sa或hal,
[1007]
r3表示h或a,
[1008]
r4表示a或nh2,
[1009]
r5表示h、nh2、nha或na2,
[1010]
a表示具有1-10个碳原子的烷基、烯基、环烷基或亚烷基环烷基,
[1011]
hal表示f、cl、br或i,
[1012]
及其生理学上可接受的盐和/或溶剂化物。
[1013]
关于上述化合物,a表示具有1-10个碳原子的烷基并具有1、2、3、4、5、6、7、8、9或10
个碳原子并优选表示甲基、乙基或丙基,还优选异丙基、丁基、异丁基、仲丁基或叔丁基,而且还优选正戊基、新戊基、异戊基或己基。在这些基团中,1-7h原子也可以被f和/或cl置换。a因此也表示,例如,三氟甲基或五氟乙基。
[1014]
a也表示具有3-8个碳原子的环烷基并优选表示例如环戊基或环己基。
[1015]
a也表示烯基。所述烯基具有2-10个碳原子,为直链或支链的并表示,例如,乙烯基、丙烯基或丁烯基。此外还表示亚烷基环烷基。亚烷基环烷基具有4-10个碳原子并优选表示,例如,亚甲基环戊基、亚乙基环戊基、亚甲基环己基或亚乙基环己基。
[1016]
r1和r2优选彼此独立地各自表示h、甲基、乙基、丙基、丁基、异丙基、叔丁基、甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、s-甲基、s-乙基、f或cl。
[1017]
r3优选表示h、甲基或乙基。
[1018]
r4优选表示甲基、乙基、丙基、丁基或nh2.
[1019]
r5优选表示h、氨基、甲基氨基、乙基氨基、二甲基氨基或二乙基氨基。
[1020]
在相关的实施方案中,用于本发明方法的pde7抑制剂包括以上结构式的化合物,其中r1和r2不都为h和其中当r1或r2之一为h时,则另一个不能是ch3、och3或cl。
[1021]
在另一个相关的实施方案中,用于本发明方法的pde7抑制剂包括这样的化合物,其中
[1022]
r1、r2、r3和r5为h和r4为甲基;
[1023]
r1为4-cl,r2为h,r3为乙基,r4为氨基和r5为h;
[1024]
r1和r2为h,r3为乙基,r4为甲基和r5为氨基;
[1025]
r1和r2为h,r3为乙基,r4为氨基和r5为h;
[1026]
r1和r2为h,r3为乙基,r4为h和r5为氨基;
[1027]
r1为3-cl,r2为4-o-甲基,r3为乙基,r4为氨基和r5为h;
[1028]
r1为3-cl,r2为4-o-甲基,r3为乙基,r4为甲基和r5为氨基;
[1029]
r1为4-ocf3,r2为h,r3为乙基,r4为氨基和r5为h;
[1030]
r1为3-cl,r2为4-o-甲基,r3为乙基,r4为氨基和r5为h;
[1031]
r1为3-cl,r2为4-o-甲基,r3为乙基,r4为甲基和r5为氨基;
[1032]
r1为4-ocf3,r2为h,r3为乙基,和r4为氨基和r5为h。
[1033]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献中的那些化合物:u.s.pat.no.6,613,778和wo 01/34601,所述文献通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[1034][1035]
以上化合物的取代基定义如下:
[1036]
r1表示conr4r5,
[1037]
r2表示h或a,
[1038]
r4和r5彼此独立地各自表示h或a1,
[1039]
r3表示hal,
[1040]
hal表示f、cl、br或i,
[1041]
a表示具有1-4个碳原子的烷基,
[1042]
a1表示具有1-10个碳原子的烷基,
[1043]
x表示具有1-4个碳原子的亚烷基,其中亚乙基也可被双键或三键替代,
[1044]
及其生理学上可接受的盐和/或溶剂化物。
[1045]
关于上述化合物,a表示具有1-4个碳原子的烷基并具有1、2、3或4碳原子并优选表示甲基、乙基或丙基,还优选异丙基、丁基、异丁基、仲丁基或叔丁基。基团中的1-7个h原子也可以被f和/或cl置换。a因此也表示,例如,三氟甲基或五氟乙基。
[1046]
a1表示具有1-10个碳原子的烷基并具有1、2、3、4、5、6、7、8、9或10个碳原子并优选表示甲基、乙基或丙基,还优选异丙基、丁基、异丁基、仲丁基或叔丁基,而且还表示正戊基、新戊基、异戊基或己基。基团中的1-7个h原子也可以被f和/或cl置换。a1因此也表示,例如,三氟甲基或五氟乙基。
[1047]
x表示具有1-4个碳原子的亚烷基,优选地为亚甲基、亚乙基、亚丙基或亚丁基、其中一个亚乙基也可被双键或三键替代。x因此也表示,例如,—ch2—ch
═
ch—h2—或—c≡—c—。
[1048]
在相关的实施方案中,用于本发明方法的pde7抑制剂包括以下化合物:
[1049]
2-(3-丁基-7-氯-3h-咪唑并[4,5-c]吡啶-4-基硫烷基)-n,n-二甲基乙酰胺
[1050][1051]
2-(3-丁基-7-氯-3h-咪唑并[4,5-c]吡啶-4-基硫烷基)乙酰胺,
[1052]
2-(3-丁基-7-氯-3h-咪唑并[4,5-c]吡啶-4-基硫烷基)丙酰胺,
[1053]
2-(3-丁基-7-氯-3h-咪唑并[4,5-c]吡啶-4-基硫烷基)丁酰胺,
[1054]
2-(3-丁基-7-氯-3h-咪唑并[4,5-c]吡啶-4-基硫烷基)-n-己基乙酰胺,
[1055]
2-(3-丁基-7-氯-3h-咪唑并[4,5-c]吡啶-4-基硫烷基)-n-辛基乙酰胺,
[1056]
4-(3-丁基-7-氯-3h-咪唑并[4,5-c]吡啶-4-基硫烷基)-丁-2-烯酸二甲基酰胺。
[1057]
在另一个相关的实施方案中,用于本发明方法的pde7抑制剂包括以下化合物,其中
[1058]
r3为cl;
[1059]
r3为cl,和x为具有1-4个碳原子的亚烷基;
[1060]
r3为cl,x为具有1、2、3或4碳原子的亚烷基,和a1为具有1、2、3或4碳原子的烷基。
[1061]
这些化合物的制备描述于u.s.pat.no.6,613,778和wo 01/34601中。
[1062]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献中的那些化合物:wo 2008/113881和es p200700762,各文献通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[1063][1064]
以上化合物的取代基定义如下:
[1065]
a为稠合的5、6或7元碳环基或杂环基并且可以是饱和的或不饱和的;虚线独立地表示单键或双键;x和y独立地选自烷基、氢、=o、=s、-n(烷基)、-n(芳基)、芳基、o-烷基、o-芳基、烷基-s和-s-芳基;及r1和r2独立地选自氢、卤素、烷基、卤代烷基、芳基、环烷基、(z)n-芳基、杂芳基、-or3;-c(o)or3,-(z)n-c(o)or3和-s(o),或所述化合物的药学上可接受的盐、衍生物、前药、溶剂化物或立体异构体。
[1066]
例外:当a为未取代的苯时,x=o,y=s,当a为未取代的苯时,x=o,y=o,当a为未取代的苯时,x=o,y=s-me,当a为未取代的噻吩时,x=o,y=s,和当a为未取代的苯并噻吩时,x=o,y=s。
[1067]
在相关的实施方案中,以上化合物构成有用的药用组合物,其包含治疗有效量的以上化合物,或所述化合物的混合物、所述化合物的盐、衍生物、前药、溶剂化物或药学上可接受的立体异构体,以及载体、辅助剂或用于iv给予患者的药学上可接受的溶媒。
[1068]
在其它相关实施方案中,用于本发明方法的pde7抑制剂包括以下化合物:4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉,及其选自以下基团的衍生物:
[1069][1070]
6-溴-2,3,4-四氢喹唑啉、6-溴-(2,6-二氟苯基)-4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉、6-溴-(2,3,4-三氟苯基)-4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉、6-溴-(2-溴代苯基)-4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉、3-(2,6-二氟苯基)-8-甲基-4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉、3-(2,3,4-三氟苯基)-8-甲基-4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉、和3-(2-溴代苯基)-8-甲基-4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉。
[1071]
在进一步相关的实施方案中,用于本发明方法的pde7抑制剂包括以下化合物:2-甲基硫基-4-氧代-3,4-二氢喹唑啉,及其选自以下基团的衍生物:
[1072][1073]
6-溴-(2,6-二氟苯基)-2-甲基硫基-4-氧代-3,4-二氢喹唑啉、6-溴-(2,3,4-三氟苯基)-2-甲基硫基-4-氧代-3,4-二氢喹唑啉、6-溴-(2-溴代苯基)-2-甲基硫基-4-氧代-3,4-二氢喹唑啉、3-苯基-8-甲基-2-甲基硫基-4-氧代-3,4-二氢喹唑啉、3-(2,6-二氟苯基)-8-甲基-2-甲基硫基-4-氧代-3,4-二氢喹唑啉、3-(2,3,4-三氟苯基)-8-甲基-2-甲基硫基-4-氧代-3,4-二氢喹唑啉、和3-(2-溴代苯基)-8-甲基-2-甲基硫基-4-氧代-3,4-二氢喹唑啉。
[1074]
在另一个相关的实施方案中,用于本发明方法的pde7抑制剂包括以下化合物:2,4-二硫代-1,2,3,4-四氢喹唑啉,及其选自以下基团的衍生物:
[1075][1076]
3-苯基-2,4-二硫代-1,2,3,4-四氢喹唑啉、3-(2,6-二氟苯基)-2,4-二硫代-1,2,3,4-四氢喹唑啉、和3-(2,3,4-三氟苯基)-2,4-二硫代-1,2,3,4-四氢喹唑啉。
[1077]
在另一个相关的实施方案中,用于本发明方法的pde7抑制剂包括以下化合物:(2-甲基硫基-4-硫代-3,4-二氢喹唑啉)及其选自以下基团的衍生物:
[1078][1079]
3-苯基-2-甲基硫基-4-硫代-3,4-二氢喹唑啉、3-(2,6-二氟苯基)-2-甲基硫基-4-硫代-3,4-二氢喹唑啉、3-(2,3,4-三氟苯基)-2-甲基硫基-4-硫代-3,4-二氢喹唑啉、和3-(2-溴代苯基)-2-甲基硫基-4-硫代-3,4-二氢喹唑啉。
[1080]
以上化合物的制备描述于wo 2008/113881中。
[1081]
在相关的实施方案中,用于本发明方法的pde7抑制剂描述于esp 200700762中,该文献通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、6'-(4-羧基苯基)-8'-氯-5'-甲氧基螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、6'-(3-羧基苯基)-8'-氯-5'-甲氧基螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、8'-氯-6'-[2-(4-甲基-哌嗪-1-羰基)苯基]螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、8'-氯-6'-[2-甲基-4-(4-甲基-哌嗪-1-羰基)苯基]螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、8'-氯-6'-[4-(哌嗪-1-羰基)苯基]螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、8'-氯-6'-[4-氨基甲酰基-苯基]螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、8'-氯-6'-[4-((1-甲基-哌啶-4-基)-哌嗪-1-羰基)苯基]螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、8'-氯-5'-甲氧基-6-[4-(4-甲基-哌嗪-1-羰基)苯基]螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、8'-三氟甲基螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、8'-氯-6'-氰基甲基螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、8'-氯-5'-(3-二甲基氨基-2-羟基-丙氧基)-螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、8'-氯-5'-(3-甲基氨基-2-羟基-丙氧基)-螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、8'-氯-5'-[2-(乙氧基羰基甲基-氨基)-乙氧基]-螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、8'-氯-5'-[2-(羧基甲基-氨基)-乙氧基]-螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮盐酸盐、8'-氯-5'-(2-甲烷磺酰基氨基-2-氧代-乙氧基)-螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮、8'-氯-5'-(2-[(5-甲基-异唑-3-基甲基)-氨基]乙氧基)-螺[环己烷-1-4'-(3',4'-二氢)喹唑啉]-2'(1'h)-酮。
[1085]
这些化合物的制备描述于u.s.pat.no.7,087,614、u.s.2007/0049558和wo 2002/074754中。
[1086]
在另一个实施方案中,用于本发明方法的pde7抑制剂和双重pde4/7抑制剂选自概略或详细披露于以下文献中的那些化合物:us 7,087,614、us 2003/0162802和wo 2002/102313,各文献通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[1087][1088]
用于本发明方法的pde7抑制剂包括对映体,非对映体、互变异构体,和药学上可接受的盐、前药,和上式化合物的溶剂化物。
[1089]
以上化合物的取代基定义如下:
[1090]
r1为h或烷基;
[1091]
r2为(a)杂芳基或杂环基,其中的任何一个可以被1-3个基团t1、t2、t3任选取代;(b)1-3个基团t1、t2、t3取代的芳基,前提是t1、t2、t3中的至少一个不是h;或(c)与杂芳基或杂环基稠合的芳基,其中合并的环系统可任选被1-3个基团t1、t2、t3取代;
[1092]
z为(a)-or4、-c(o)r4、-c(o)or4、-sr4、-nr3r4、-c(o)nr3r4、-nr3so2r4c、卤素、硝
基、卤代烷基;或(b)烷基、芳基、杂芳基、杂环基或环烷基,其中的任何一个可以任选被1-3个基团t1a、t2a、t3a取代;
[1093]
j为(a)氢、卤代、-or4a,或(b)烷基、烯基,或炔基,其中的任何一个可任选被1-3个基团t1b、t2b或t3b取代;
[1094]
l为(a)氢、-or4b、-c(o)r4b、-c(o)or4b、-sr4b、-nr5r6、-c(o)nr5r6、-nr5so2r4d,卤素,卤代烷基、硝基,或(b)烷基、芳基、杂芳基、杂环基,或环烷基,其中的任何一个可任选被1-3个基团t1c、t2c或t3c取代;
[1095]
r3和r4独立地为h、烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,其中的任何一个可以任选被1-3个基团t1a、t2a或t3a取代;
[1096]
或者r3和r4与它们连接的氮原子一起可结合形成任选被1-3个基团t1a、t2a或t3a取代的4-8元杂环基;
[1097]
r4a为氢、烷基、烯基、芳基、杂芳基、(芳基)烷基、(杂芳基)烷基、杂环基、(杂环基)烷基、环烷基或(环烷基)烷基,其中的任何一个可任选被1-3个基团t1b、t2b或t3取代;
[1098]
r4b为氢、烷基、烯基、芳基、杂芳基、(芳基)烷基、(杂芳基)烷基、杂环基、(杂环基)烷基、环烷基或(环烷基)烷基,其中的任何一个可任选被1-3个基团t1c、t2c或t3c取代;
[1099]
r4c和r4d独立地为烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,其中的任何一个可任选被1-3个基团t1a、t2a或t3a取代;
[1100]
r5和r6独立地为h、烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,其中的任何一个可任选独立地被1-3个基团t1c、t2c或t3c取代;
[1101]
或者r5和r6与它们连接的氮原子一起可结合形成任选被1-3个基团t1c、t2c或t3c取代的4-8-元杂环基;
[1102]
t1-1c、t2-2c和t3-3c各自独立地为(1)氢或t6,在此t6为(i)烷基、(羟基)烷基、(烷氧基)烷基、烯基、炔基、环烷基、(环烷基)烷基、环烯基、(环烯基)烷基、芳基、(芳基)烷基、杂环基、(杂环基)烷基、杂芳基、或(杂芳基)烷基;(ii)基团(i),其本身被一个或多个相同或不同的基团(i)取代;或(iii)基团(i)或(ii),其独立被t1-1c、t2-2c和t3-3c定义的一个或多个(优选1-3个)以下基团(2)-(13)取代,(2)-oh或-ot6,(3)-sh或-st6,(4)-c(o)th、-c(o)tt6,或-o-c(o)t6,在此t是1或2;(5)-so3h、-s(o)t6,或s(o)tn(t9)t6,(6)卤代,(7)氰基,(8)硝基,(9)-t4-nt7t8,(10)-t4-n(t9)-t5-nt7t8,(11)-t4-n(t10)-t5-t6,(12)-t4-n(t10)-t5-h,(13)氧代,
[1103]
t4和t5各自独立地为(1)单键,(2)-t11-s(o)t-t12-,(3)-t11-c(o)-t12-,(4)-t11-c(s)-t12-,(5)-t11-o-t12-,(6)-t11-s-t12-,(7)-t11-o-c(o)-t12-,(8)-t11-c(o)-o-t12-,(9)-t11-c(=nt9a)-t12-,或(10)-t11-c(o)-c(o)-t12,
[1104]
t7、t8、t9、t9a和t10(1)各自独立地为氢或在t6的定义中提供的基团,或(2)t7和t8可以一起为亚烷基或亚烯基,与它们连接的原子一起完成3-至8-元饱和的或不饱和的环,所述环为未取代的或由t1-1c、t2-2c和t3-3c的定义中所列的一个或多个基团取代,或(3)t7或t8,与t9一起,可以是亚烷基或亚烯基,与它们连接的氮原子一起完成3-至8-元饱
和的或不饱和的环,所述环为未取代的或由t1-1c、t2-2c和t3-3c的描述中所列的一个或多个基团取代,或(4)t7和t8或t9和t10与它们连接的氮原子一起可结合形成基团-n=ct13t14,其中t13和t14各自独立地为h或在t6的定义中提供的基团;
[1105]
以及t11和t12各自独立地为(1)单键、(2)亚烷基,(3)亚烯基,或(4)亚炔基。
[1106]
在相关的实施方案中,用于本发明方法的pde7抑制剂包括以上化合物,其中:
[1107]
z为(a)卤素、烷氧基、卤代烷基、-nr3r4、-c(o)or4、-c(o)nr3r4;(b)芳基或杂芳基,其中的任何一个可以任选由一个或多个t1a,t2a,t3a(尤其是氰基、任选取代的烷基、(羟基)烷基、-oh、-ot6、-st6、-sott6、-coth、-cott6、-t4nt7t8,或-t4n(t10)-t5-t6)取代;(c)任选取代的烷基(尤其是由一个或多个-oh、-coth、-cott6、-t4-nt7t8、-t4-n(t10)-t5-h,或-t4-n(t10)-t5-t6取代);
[1108]
j为(a)h,或(b)烷基或烯基,其中的任何一个可以任选被取代(尤其是由一个或多个-oh、-ot6、-coth,或-cott6取代);
[1109]
l为(a)h;(b)卤素、烷氧基、卤代烷基、-nr5r6、-c(o)or4b、-c(o)nr5r6;(c)芳基或杂芳基,其中的任何一个可以任选由一个或多个t1c,t2c,t3c(尤其是氰基、任选取代的烷基、(羟基)烷基、-oh、-ot6、-st6、-sott6、-coth、-cott6、-t4nt7t8,或-t4n(t10)-t5-t6)取代;或(d)任选取代的烷基(尤其是由一个或多个-oh、-coth、-cott6、-t4-nt7t8、-t4-n(t10)-t5-h,或;-t4-n(t10)-t5-t6取代);
[1110]
r1为h或烷基;
[1111]
r2为(a)杂芳基(更优选噻唑基或唑基),其任选由1-3个基团t1、t2、t3取代,优选地包括h、烷基、卤代烷基、卤代,杂芳基、氰基、c(o)tt6、ot6、-t4nt7t8;(b)芳基,其由1-3个基团t1、t2、t3(优选地包括杂芳基(优选咪唑基、唑基或噻唑基,其中的任何一个可任选被进一步取代)、氰基、c(o)tt6、s(o)tn(t9)t6、卤代、烷基和卤代烷基)取代;或(c)芳基,其与杂环稠合(如,通过芳环结合的2,3-二氢-1h-吲哚、通过芳环结合的喹啉基(尤其是喹啉-6-基)、通过芳环结合的喹唑啉基(尤其是喹唑啉-7-基)、通过芳环结合的噌啉基(尤其是噌啉-6-基)、通过芳环结合的异喹啉基(尤其是异喹啉-6-基),和通过芳环结合的酞嗪基(尤其是酞嗪-6-基)),其中合并的环系统可任选被1-3个基团t1、t2、t3(尤其是卤代、oh、ot6、烷基、-coth、-cott6,或-c(o)nt7t8)取代;
[1112]
r3为h或任选取代的烷基(尤其是被一个或多个-oh或-ot6取代);
[1113]
r4为(a)氢;(b)(芳基)烷基,其中芳基任选独立地被一个或多个基团t1a、t2a、t3a(尤其是任选取代的烷基、卤代、氰基、硝基、(羟基)烷基、-oh、-ot6、-st6,-coth、-cott6、-so3h、-sott6、-sotn(t9)(t6)、-t4nt7t8、-t4-n(t10)-t5-t6、杂环或杂芳基)取代;(c)(杂芳基)烷基,其中杂芳基任选独立地被一个或多个基团t1a、t2a、t3a(尤其是任选取代的烷基、卤代、氰基、硝基、(羟基)烷基、-oh、-ot6、-st6、-coth、-cott6、-so3h、-sott6、-sotn(t9)(t6)、-t4nt7t8、-t4-n(t10)-t5-t6、杂环基或杂芳基)取代;(d)(杂环基)烷基,其中杂环基团任选独立地被一个或多个基团t1a、t2a、t3a(尤其是任选取代的烷基、卤代、氰基、硝基、氧代、(羟基)烷基、-oh、-ot6、-st6、-coth、-cott6、-so3h、-sott6、-sotn(t9)(t6)、-t4nt7t8、-t4-n(t10)-t5-t6、杂环或杂芳基)取代;(e)烷基,其任选独立地被一个或多个基团t1a、t2a、t3a(尤其是-oh、-ot6、-coth、-cott6、-t4ntt8或-t4-n(t10)-t5-t6);(f)杂环基,其任选独立地被一个或多个基团t1a、t2a、t3a(尤其是任选取代的烷基、任选取代的芳
coth,或-cott6(在此t6为烷基)取代;
[1121]
l为(a)h;(b)卤素、烷氧基、卤代烷基、-nr5r6、-c(o)or4b、-c(o)nr5r6;(c)芳基或杂芳基,其中的任何一个可以任选被一个或多个选自氰基、任选取代的烷基(尤其是被coth或cott6取代)、(羟基)烷基、-oh、-ot6、-st6、-sott6、-coth、-cott6、-t4nt7t8,或-t4n(t10)-t5-t6(在此t4为键)或-c(o)-的t1c、t2c、t3c取代;t5为-c(o)-,或-c(o)o-;t6为烷基或卤代烷基;t7和t8独立地为h;任选被环烷基、杂芳基、羟基或-nt7t8取代的烷基;环烷基;或任选被卤素取代的芳基;或t7和t8与它们连接的氮原子一起结合形成任选被(羟基)烷基、coth或cott6取代的杂环;t10为氢;(d)烷基,其任选被一个或多个-oh、-coth、-cott6、-t4-nt7t8、-t4-n(t10)-t5-h,或-t4-n(t10)-t5-t6(其中t4为-c(o)-)取代;t5为-亚烷基-o-;t6为烷基;t7和t8独立地为h、烷基、环烷基、芳基、(芳基)烷基(任选如在r4的定义中所述的那样被取代),或杂环基(任选如在r3和r4结合形成杂环的定义中所述的那样被取代);和t10为h;
[1122]
r1为h或烷基;
[1123]
r2为(a)杂芳基(更优选噻唑基或唑基),其任选被1-3个基团t1、t2、t3(优选地包括h、烷基、卤代烷基、卤代,杂芳基、氰基、c(o)tt6、ot6、-t4nt7t8)取代;(b)芳基,其被1-3个基团t1、t2、t3(优选地包括杂芳基(优选咪唑基、唑基,或噻唑基,其中的任何一个可以任选被进一步取代)、氰基、c(o)tt6、s(o)tn(t9)t6、卤代、烷基和卤代烷基)取代;或(c)稠合于杂环的芳基(如,通过芳环连接的2,3-二氢-1h-吲哚)其中合并的环系统可任选被1-3个基团t1、t2、t3(尤其是卤代、-oh、-ot6、烷基、-coth、-cott6,或-c(o)nt7t8)取代;
[1124]
r3为h或任选取代的烷基(尤其是被一个或多个-oh,或-ot6取代);
[1125]
r4为(a)氢;(b)(芳基)烷基,其中芳基任选独立地被一个或多个选自任选取代的烷基、卤代、氰基、硝基、(羟基)烷基、-oh、-ot6、-st6、-coth、-cott6、-so3h、-sott6、-sotn(t9)(t6)、-t4nt7t8、-t4n(t10)-t5-t6、杂环基或杂芳基)的基团t1a、t2a、t3a取代,其中t4为键、-so2-,或-c(o)-;t5为-so2-,或-亚烷基-o-;t6为烷基,或环烷基;t7和t8独立地为h或烷基;及t9和t10为氢;(c)(杂芳基)烷基,其中杂芳基任选独立地被一个或多个选自任选取代的烷基、卤代、氰基、硝基、氧代、(羟基)烷基、-oh、-ot6、-st6、-coth、-cott6、-so3h、-sott6、-sotn(t9)(t6)、-t4nt7t8、-t4-n(t10)-t5-t6、杂环基,或杂芳基)的基团t1a、t2a、t3a取代,其中t4为键、-so2-,或-c(o)-;t5为-so2-,或-亚烷基-o-;t6为烷基,或环烷基;t7和t8独立地为h或烷基;及t9和t10为氢;(d)(杂环基)烷基,其中杂环基团任选独立地被一个或多个选自任选取代的烷基、卤代、氰基、硝基、(羟基)烷基、-oh、-ot6、-st6、-coth、-cott6、-so3h、-sott6、-t4nt7t8、-t4-n(t10)-t5-t6、杂环基,或杂芳基)的基团t1a、t2a、t3a取代,其中t4为键、-so2-,或-c(o)-;t5为-so2-,或-亚烷基-o-;t6为烷基,或环烷基;t7和t8独立地为h或烷基;及t9和t10为氢;(e)烷基,其任选独立地被一个或多个选自-oh、-ot6、-coth、-cott6、-t4nt7t8或-t4-n(t10)-t5-t6)的基团t1a、t2a、t3a取代,其中t4为键;t5为-c(o)-;t6为烷基;t7和t8独立地为h或烷基;和t10为氢;杂环基,其任选独立地被一个或多个选自任选取代的烷基(尤其是被-t4nt7t8取代),任选取代的芳基(尤其是被卤素或卤代烷基取代)、氰基、-oh、-ot6、-coth、-cott6、氧代、羟基(烷基)、(烷氧基)烷基、-t4-n(t10)-t5-t6,或-t4-nt7t8)的基团t1a、t2a、t3a取代,其中t4为键或-c(o)-;t5为-c(o)-、-so2-,或-亚烷基-c(o)o-;t6为烷基、烷氧基,或杂芳基;t7和t8独立地为h、烷基,或环烷基;
或t7和t8与它们连接的氮原子一起结合形成任选取代的杂环;
[1126]
或者r3和r4与它们连接的氮原子一起结合形成选自吡咯烷基、哌啶基、哌嗪基、吗啉基、二氮杂环庚烷基或1,4-二氧杂-8-氮杂螺[4.5]癸-8-基)的杂环基,其中的任何一个任选独立地被1-3个基团t1a、t2a、t3a取代,基团t1a、t2a、t3a选自任选取代的烷基(尤其是被-t4nt7t8取代),任选取代的芳基(尤其是被卤素或卤代烷基取代)、氰基、-oh、-ot6、-coth、-cott6、氧代、羟基(烷基)、(烷氧基)烷基、-t4-n(t10)-t5-t6,或-t4-nt7t8),其中t4为键或-c(o)-;t5为-c(o)-、-so2-,或-亚烷基-c(o)o-;t6为烷基、烷氧基,或杂芳基;t7和t8独立地为h、烷基,或环烷基;或t7和t8与它们连接的氮原子一起结合形成任选取代的杂环;
[1127]
r5为氢或烷基;
[1128]
r6为(a)氢;(b)(芳基)烷基,其中芳基任选独立地被一个或多个选自任选取代的烷基、卤代、氰基、硝基、(羟基)烷基、-oh、-ot6、-st6、-coth、-cott6、-so3h、-sott6、-sotn(t9)(t6)、-tnt7t8、-t4-n(t10)-t5-t6、杂环基,或杂芳基)的基团t1c、t2c、t3c取代,其中t4为键、-so2-,或-c(o)-;t5为-so2-,或-亚烷基-o-;t6为烷基,或环烷基;t7和t8独立地为h或烷基;及t9和t10为氢;(c)(杂芳基)烷基,其中杂芳基任选独立地被一个或多个选自任选取代的烷基、卤代、氰基、硝基、氧代、(羟基)烷基、-oh、-ot6、-st6、-coth、-cott6、-so3h、-sott6、-sotn(t9)(t6)、-t4nt7t8、-t4-n(t10)-t5-t6、杂环基,或杂芳基)的基团t1c、t2c、t3c取代,其中t4为键、-so2-,或-c(o)-;t5为-so2-,或-亚烷基-o-;t6为烷基,或环烷基;t7和t8独立地为h或烷基;及t9和t10为氢;(d)(杂环基)烷基,其中杂环基团任选独立地被一个或多个选自任选取代的烷基、卤代、氰基、硝基、(羟基)烷基、-oh、-ot6、-st6、-coth、-cott6、-so3h、-sott6、-t4nt7t8、-t4-n(t10)-t5-t6、杂环基,或杂芳基)的基团t1c、t2c、t3c取代,其中t4为键、-so2-,或-c(o)-;t5为-so2-,或-亚烷基-o-;t6为烷基,或环烷基;t7和t8独立地为h或烷基;及t9和t10为氢;(e)烷基,其任选独立地被一个或多个选自-oh、-ot6、-octh、-cott6、-t4nt7t8或-t4-n(t10)-t5-t6)的基团t1c、t2c、t3c取代,其中t4为键;t5为-c(o)-;t6为烷基;t7和t8独立地为h或烷基;和t10为氢;杂环基,其任选独立地被一个或多个选自任选取代的烷基(尤其是被-t4nt7t8取代),任选取代的芳基(尤其是被卤素或卤代烷基取代)、氰基、-oh、-ot6、-coth、-cott6、氧代、羟基(烷基)、(烷氧基)烷基、-t4-n(t10)-t5-t6,或-t4-nt7t8的基团t1c、t2c、t3c取代,在此t4为键或-c(o)-;t5为-c(o)-、-so2-,或-亚烷基-c(o)o-;t6为烷基、烷氧基,或杂芳基;t7和t8独立地为h、烷基,或环烷基;或t7和t8与它们连接的氮原子一起结合形成任选取代的杂环;
[1129]
或者r5和r6与它们连接的氮原子一起结合形成选自吡咯烷基、哌啶基、哌嗪基、吗啉基、二氮杂环庚烷基或1,4-二氧杂-8-氮杂螺[4.5]癸-8-基)的杂环,其中的任何一个任选独立地被1-3个选自任选取代的烷基(尤其是被-t4nt7t8取代),任选取代的芳基(尤其是被卤素或卤代烷基取代)、氰基、-oh、-ot6、-coth、-cott6、氧代、羟基(烷基)、(烷氧基)烷基、-t4-n(t10)-t5-t6,或-t4-nt7t8的基团t1a、t2a、t3a取代,其中t4为键或-c(o)-;5为-c(o)-、-so2-,或-亚烷基-c(o)o-;t6为烷基、烷氧基,或杂芳基;t7和t8独立地为h、烷基,或环烷基;或t7和t8与它们连接的氮原子一起结合形成任选取代的杂环。
[1130]
在进一步相关的实施方案中,用于本发明方法的pde7抑制剂包括以下化合物:
[1131]
1)2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-6-(4-甲基-1-哌嗪基)-2-嘧啶
基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1132]
2)2-[[4-[[(3,4-二甲氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯三氟乙酸盐;
[1133]
3)2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1134]
4)4-甲基-2-[[4-[[[4-(甲基磺酰基)苯基]甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-5-噻唑羧酸乙基酯;
[1135]
5)2-[[4-[[(4-甲氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1136]
6)2-[[4-[[(3-甲氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1137]
7)2-[[4-[[(2-甲氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1138]
8)4-甲基-2-[[4-(1-哌嗪基)-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑羧酸乙基酯;
[1139]
9)2-[[4-[[(2-乙氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1140]
10)2-[[4-[[(2,5-二甲氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1141]
11)2-[[4-[[(3,5-二甲氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1142]
12)2-[[4-[[(2,6-二甲基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1143]
13)2-[[4-[[[4-(甲氧基羰基)苯基]甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1144]
14)2-[[4-[[(3-溴代苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基)氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1145]
15)2-[[4-[(1,3-苯并二氧杂环戊烯-5-基甲基)氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1146]
16)4-甲基-2-[[4-[甲基(3-吡啶基甲基)氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-5-噻唑羧酸乙基酯;
[1147]
17)4-甲基-2-[[4-(1-哌嗪基)-6-[[[4-(1,2,3-噻二唑-4-基)苯基]甲基]氨基]-2-嘧啶基]氨基]-5-噻唑羧酸乙基酯;
[1148]
18)2-[[4-[[[3-(环戊基氧基)-4-甲氧基苯基]甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1149]
19)4-甲基-2-[[4-[(苯基甲基)氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-5-噻唑羧酸乙基酯;
[1150]
20)4-甲基-2-[[4-(4-甲基-1-哌嗪基)-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑羧酸乙基酯;
2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1171]
41)2-[[4-(3-羟基-1-吡咯烷基)-6-[[[4-(甲基磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1172]
42)2-[[4-[(4-羟基丁基)氨基]-6-[[[4-(甲基磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1173]
43)2-[[4-[(2,3-二羟基丙基)氨基]-6-[[[4-(甲基磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1174]
44)2-[[4-[(4-氨基-1-哌啶基)-6-[[[4-(甲基磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1175]
45)2-[[4-[4-羟基-3-(羟基甲基)-1-哌啶基]-6-[[[4-(甲基磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1176]
46)2-[[4-(4-二甲基氨基-1-哌啶基)-6-[[[4-(甲基磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1177]
47)2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-6-(甲基氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1178]
48)2-[4,6-双(4-甲基-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1179]
49)2-[4-(4-羟基-哌啶-1-基)-6-(4-甲基-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1180]
50)2-[4-(3-羟基甲基-哌啶-i-基)-6-(4-甲基-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1181]
51)4-甲基-2-[4-(4-甲基-哌嗪-1-基)-6-吗啉-4-基-嘧啶-2-基氨基]-噻唑-5-羧酸乙基酯;
[1182]
52)2-[4-(4-氨基-哌啶-1-基)-6-(4-甲基-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1183]
53)2-[4,6-双(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1184]
54)2-[4-(4-氧代-哌啶-1-基)-6-(4-甲基-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1185]
55)2-[4-(4-甲基-4-羟基-哌啶-1-基)-6-(4-甲基-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1186]
56)2-[-(4-羟基-哌啶-1-基)-6-(4-二甲基甲基-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1187]
57)2-[4-(4-羟基甲基-哌啶-1-基)-6-(4-二甲基甲基-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1188]
58)2-[4-(3-羟基甲基-哌啶-1-基)-6-(4-二甲基甲基-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1189]
59)2-[4-(4-羟基甲基-哌啶-1-基)-6-(4-羟基-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1190]
60)4-甲基-2-[4-(4-羟基-哌嗪-1-基)-6-吗啉-4-基-嘧啶-2-基氨基]-噻唑-5-羧酸乙基酯;
[1191]
61)2-[[(4-[[[4-(甲基磺酰基)苯基]甲基]氨基]-6-氯-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1192]
62)2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-6-氯-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1193]
63)2-[[4-[4-(二甲基氨基)-1-哌啶基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1194]
64)2-[[4-[1-哌嗪基(piperizinyl)]-6-甲基-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1195]
65)2-[[4-(4-氨基-1-哌啶基)-6-[[[4-(甲基磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1196]
66)2-[[4-[4-羟基-1-哌啶基]-6-甲基-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1197]
67)2-[[4-[4-(羟基甲基)-1-哌啶基]-6-甲基-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1198]
68)2-[4-(4-羟基哌啶-1-基)-6-(3-氧代-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1199]
69)2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-甲基-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1200]
70)2-[[4-[1-吗啉基]-6-甲基-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1201]
71)2-[[4-[3-氧代-1-哌嗪基]-6-[[(1,1-二氧化(dioxido)-3-氧代-1,2-苯并异噻唑-2-(3h)-基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1202]
72)2-[[4-[3-氧代-1-哌嗪基]-6-[[(4-(乙基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1203]
73)2-[[4-[3-氧代-1-哌嗪基]-6-[[(4-(羟基磺酰基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1204]
74)2-[4-(4-羟基哌啶-1-基)-6-(4-甲基-3-氧代-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1205]
75)2-[4-(4(二甲基氨基)-哌嗪-1-基)-6-(4-((1-吡咯烷基)羰基甲基)哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1206]
76)2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1207]
77)2-[[4-(4-氨基-1-哌啶基)-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1208]
78)2-[[4-[4-(羟基甲基)-1-哌啶基]-6-[4-[四唑-5-基]-4-羟基哌啶-1-基]2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1209]
79)2-[[4-[4-甲基-1-哌嗪基]-6-[n-甲基-n-[(3,4,5-三甲氧基苯基)甲基]氨
基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1210]
80)2-[[4-[4-羟基-1-哌啶基]-6-[[(4-(羟基磺酰基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1211]
81)2-[[4-[[(4-氰基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;三氟乙酸盐(1:1);
[1212]
82)2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-6-(4-吗啉基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1213]
83)2-[[4-[4-羟基-1-哌啶基]-6-[(1-氧杂-3,8-二氮杂螺[4.5]癸-2,4,二酮-8-基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1214]
84)2-[4-(2-(二甲基氨基)乙基)-哌嗪-1-基)-6-(4-甲基哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1215]
85)2-[[4-(4-羟基-1-哌啶基)-6-[甲基(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1216]
86)2-[[4-[4-羟基-3-羟基甲基哌啶-1-基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1217]
87)2-[[4-(3,4-二氢-6,7-二羟基-2(1h)-异喹啉基)-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯,三氟乙酸盐1:1);
[1218]
88)2-[[4-[4-[(甲氧基乙酰基)氨基]-1-哌啶基]-6-[[4-(甲基磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1219]
89)2-[[4-[[(3,4-二甲氧基苯基)甲基]氨基]-6-[4-(二甲基氨基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-噻唑羧酸,乙酯;
[1220]
90)2-[[4-[4-(羟基乙基)哌啶-1-基]-6-[4-(二甲基氨基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1221]
91)2-[[4-[4-(二甲基氨基)-1-哌啶基]-6-[甲基(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1222]
92)2-[[4-[4-(羟基)哌啶-1-基]-6-[4-(甲氧基羰基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1223]
93)2-[[4-[4-(羟基)哌啶-1-基]-6-[4-(甲基)-4-(羟基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1224]
94)2-[4-(3-氧代哌嗪-1-基)-6-(4-甲基哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1225]
95)2-[[4-[[(4-氰基苯基)甲基]氨基]-6-[4-二甲基氨基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1226]
96)4-甲基-2-[[4-[[(3-硝基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-5-噻唑羧酸,乙酯,三氟乙酸盐(1:1);
[1227]
97)2-[[4-(4-羟基-1-哌啶基)-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1228]
98)2-[4-(二甲基氨基)-哌嗪-1-基)-6-(4-甲基哌嗪-1-基)-嘧啶-2-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1229]
99)2-[4-(二甲基氨基)-哌啶-1-基)-6-(3-(氨基羰基)-1-哌嗪基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1230]
100)2-[4-(2-羟基乙基)-哌嗪-1-基)-6-(4-甲基-1-哌嗪基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1231]
101)2-[[4-[4-(氨基羰基)-1-哌啶基]-6-[[[4-(甲基磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1232]
102)2-[[4-[4-(羟基甲基)-1-哌啶基]-6-[n-甲基-n-(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1233]
103)2-[[4-[4-甲基哌嗪-1-基]-6-[[(3,4-二甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1234]
104)2-[[4-[哌嗪-1-基]-6-[[(4-羧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1235]
105)2-[[4-[3-羟基甲基哌啶-1-基]-6-[[n-[(3,4,5-三甲氧基苯基)甲基]]-n-(甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1236]
106)2-[4-(4-羟基哌啶-1-基)-6-(4-羧基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1237]
107)2-[[4-[哌嗪-1-基]-6-[[(3,4-二甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1238]
108)2-[[4-(4-甲酰基-1-哌嗪基)-6-[[[4-(甲基磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1239]
109)2-[4-(4-羟基哌啶-1-基)-6-(4-(羟基)-4-(4-氯代苯基)哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1240]
110)4-甲基-2-[[4-[4-二甲基氨基-1-哌啶基]-6-[[(四氢-2-呋喃基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑羧酸乙基酯;
[1241]
111)2-[[4-[哌嗪-1-基]-6-[[n-甲基-n-(5-四唑基甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1242]
112)2-[[4-[4-吗啉基]-6-[4-[四唑-5-基]-4-羟基哌啶-1-基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1243]
113)2-[[4-[4-羟基-1-哌啶基]-6-[[(1,1-二氧化-3-氧代-1,2-苯并异噻唑-2-(3h)-基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1244]
114)2-[4-(4-羟基哌啶-1-基)-6-(4-(1-甲基-1-羟基乙基)哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1245]
115)2-[[4-[3-(氨基羰基)-1-哌啶基]-6-[[n-甲基-n-(3-吡啶基甲基)]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1246]
116)2-[[4-[4-羟基甲基-1-哌啶基]-6-[[(4-(乙基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1247]
117)2-[[4-[4-羟基-1-哌啶基]-6-[4-[四唑-5-基]-4-羟基哌啶-1-基]2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1248]
118)2-[[4-[4-叔丁基氧基羰基氨基-1-哌啶基]-6-[[n-[(3,4,5-三甲氧基苯基)
甲基]]-n-(甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1249]
119)2-[[4-[[(4-氰基苯基)甲基]氨基]-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯,三氟乙酸盐(1:1);
[1250]
120)2-[[4-[4-[[(2-乙氧基-2-氧代乙基)氨基]羰基]-1-哌嗪基]-6-[甲基(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯,三氟乙酸盐(1:1);
[1251]
121)2-[4-(4-羟基哌啶-1-基)-6-(3-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1252]
122)2-[4-(4-羟基哌啶-1-基)-6-(4-羟基-4-苯基-1-哌啶基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1253]
123)4-甲基-2-[[4-[4-吗啉基]-6-[[(四氢-2-呋喃基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑羧酸乙基酯;
[1254]
124)2-[[4-[(四氢-2-呋喃基)甲基]氨基]-6-[[n-[(3,4,5-三甲氧基苯基)甲基]]-n-(甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1255]
125)2-[[4-[4-吗啉基]-6-[[(4-(羟基磺酰基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1256]
126)2-[双-4,6-(4-氰基-1-哌啶基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1257]
127)2-[[4-[4-(环戊基氨基羰基)-1-哌嗪基]-6-[n-甲基-n-(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1258]
128)2-[4-(2-甲氧基乙基)-哌嗪-1-基)-6-(4-甲基-1-哌嗪基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1259]
129)2-[4-(4-羟基哌啶-1-基)-6-(3-羧基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1260]
130)2-[[4-[4-甲基哌嗪-1-基]-6-[3-(乙酰基氨基)-1-吡咯烷基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1261]
131)2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-[[n-甲基-n-(3-吡啶基甲基)]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1262]
132)2-[[4-[2-甲基-3-oxol-哌嗪基]-6-[4-甲基-1-哌嗪基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1263]
133)2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1264]
134)2-[[4-[3-(氨基羰基)-1-哌啶基]-6-(4-二甲基氨基-1-哌啶基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1265]
135)2-[[4-[1-哌嗪基]-6-[[n-甲基-n-(2-呋喃基甲基)]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1266]
136)2-[[4-[[(4-甲氧基羰基苯基)甲基]氨基]-6-(4-二甲基-1-哌啶基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯,三氟乙酸盐(1:1);
[1267]
137)2-[[4-[3-氧代-1-哌嗪基]-6-[[(4-(甲基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1268]
138)2-[[4-[3-氧代-1-哌嗪基]-6-[[(4-(丙基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1269]
139)2-[[4-[3-(氨基羰基)-1-哌啶基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1270]
140)2-[双-4,6-(4-羟基-4-甲基-1-哌啶基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1271]
141)4-甲基-2-[[4-[4-二甲基氨基-1-哌啶基]-6-[[(2-氧代-1-吡咯烷基)丙基]氨基]-2-嘧啶基]氨基]-5-噻唑羧酸乙基酯;
[1272]
142)2-[[4-[3-氧代-1-哌嗪基]-6-[[(4-(异丙基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1273]
143)2-[4-(4-羟基哌啶-1-基)-6-(3-羟基甲基-1-哌啶基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1274]
144)4-甲基-2-[[4-[4-羟基-1-哌啶基]-6-[[(2-(4-吗啉基)乙基]氨基]-2-嘧啶基]氨基]-5-噻唑羧酸乙基酯;
[1275]
145)2-[[4-[[[4-(乙基氨基磺酰基)苯基]甲基]氨基]-6-甲氧基-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,甲酯,三氟乙酸盐(1:1);
[1276]
146)2-[[4-[4-吗啉基]-6-[(1-氧杂-3,8-二氮杂螺[4.5]癸-2,4,二酮-8-基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1277]
147)2-[[4-[4-羟基-1-哌啶基]-6-[[(4-(乙基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1278]
148)2-[[4-[叔丁基氧基羰基-1-哌嗪基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1279]
149)2-[[4-[3-(氨基羰基)-1-哌啶基]-6-[[(3,4-二甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1280]
150)2-[[4-[4-乙氧基羰基-1-哌嗪基]-6-[[n-甲基-n-(5-四唑基甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1281]
151)2-[[4-[3-氧代-1-哌嗪基]-6-[[(4-(环丙基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1282]
152)2-[[4-[4-羟基甲基-1-哌啶基]-6-[[(4-(甲基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1283]
153)2-[4-(4-二甲基氨基-1-哌嗪基)-6-(4-叔丁基氧基羰基氨基-1-哌啶基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1284]
154)2-[4-(4-羟基哌啶-1-基)-6-(4-甲氧基甲基-1-哌啶基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1285]
155)2-[4-(4-羟基哌啶-1-基)-6-(4-羟基乙基-1-哌啶基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1286]
156)2-[4-(4-羟基哌啶-1-基)-6-(4-(羟基)-4-(3-三氟甲基苯基)哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1287]
157)2-[[4-[4-吗啉基]-6-[4-[1-甲基-1-羟基乙基]-1-哌啶基]-2-嘧啶基]氨
基]-4-甲基-5-噻唑羧酸,乙酯;
[1288]
158)2-[[4-(3-氧代-1-哌嗪基]-6-[[3-吡啶基]氧基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1289]
159)2-[[4-[4-甲基-1-哌嗪基]-6-[(1,4-二氧杂螺[4.5]癸-8-基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1290]
160)2-[[4-[4-吗啉基]-6-[[(4-(甲基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1291]
161)2-[[4-[3-氧代-1-哌嗪基]-6-[(1-氧杂-3,8-二氮杂螺[4.5]癸-2,4,二酮-8-基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1292]
162)2-[[4-[4-羟基-1-哌啶基]-6-[[(4-(羧基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1293]
163)2-[4-(4-羟基哌啶-1-基)-6-(4-(羟基)-4-(4-溴代苯基)哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1294]
164)2-[[4-[4-吗啉基]-6-[[(4-乙基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基]-5-噻唑羧酸,乙酯;
[1295]
165)2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-[[(3,4-二甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1296]
166)2-[[4-[4-甲酰基-1-哌嗪基]-6-[[(3-(5-(1h)四唑基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1297]
167)2-[[4-[4-(羟基甲基)-1-哌啶基]-6-[[n-甲基-n-(5-四唑基甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1298]
168)2-[[4-[4-甲基-1-哌嗪基]-6-[[(2,5-二甲基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1299]
169)2-[[4-[3-(2-氧代-1-吡咯烷基)丙基]氨基]-6-[n-甲基-n-(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1300]
170)2-[[4-[(1-吗啉基)]-6-[[n-甲基-n-(5-四唑基甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1301]
171)2-[[4-[4-甲基-1-哌嗪基]-6-[4-[甲基磺酰基氨基]-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1302]
172)2-[[4-[4-羟基-1-哌啶基]-6-[[(2,5-二甲基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1303]
173)4-甲基-2-[[4-(4-吗啉基)-6-[[(3,4,5-三甲氧基苯基)甲基]氨基-2-嘧啶基]氨基]-5-噻唑羧酸,乙酯;
[1304]
174)2-[4-(4-羟基哌啶-1-基)-6-(3-羟基-1-哌啶基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1305]
175)4-甲基-2-[[4-(4-甲基-1-哌嗪基)-6-[甲基(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-5-噻唑羧酸,乙酯;
[1306]
176)2-[[4-[3-氧代-1-哌嗪基]-6-[[(2-(5-(1h)四唑基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-4-噻唑羧酸,乙酯;
[1307]
177)2-[[4-[(2-呋喃基甲基)氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯,三氟乙酸盐(1:1);
[1308]
178)2-[[4-[[(3,4-二甲氧基苯基)甲基]氨基]-6-(4-吗啉基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1309]
179)4-甲基-2-[[4-[甲基(3-吡啶基甲基)氨基]-6-[[(四氢-2-呋喃基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑羧酸,乙酯;
[1310]
180)2-[[4-[(4-羟基-1-哌啶基)]-6-[[n-甲基-n-(5-四唑基甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1311]
181)2-[4-(4-羟基哌啶-1-基)-6-[(4-(羟基)-4-(苯基甲基)哌啶-1-基)]-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1312]
182)2-[4-(4-二甲基氨基-1-哌嗪基)-6-[[2-(1-吗啉基)乙基]氨基]嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1313]
183)2-[[4-[4-羟基-1-哌啶基]-6-[[(3-吡啶基甲基)]氧基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1314]
184)2-[[4-[3-(氨基羰基)-1-哌啶基]-6-[[(2,6-二甲基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1315]
185)2-[[4-[4-羟基-1-哌啶基]-6-[[(4-(甲基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1316]
186)2-[[4-[4-羟基-1-哌啶基]-6-[[(4-(丙基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1317]
187)2-[[4-[3-(氨基羰基)-1-哌啶基]-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1318]
188)2-[[4-(3,4-二氢-6,7-二甲氧基-2(1h)-异喹啉基)-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1319]
189)2-[[4-[4-甲酰基-1-哌嗪基]-6-[[n-甲基-n-(5-四唑基甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1320]
190)2-[[4-[[(4-羧基苯基)甲基]氨基]-6-[4-(羟基甲基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1321]
191)2-[[4-[[(4-羧基苯基)甲基]氨基]-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯,一盐酸盐;
[1322]
192)4-甲基-2-[[4-(4-甲基-1-哌嗪基)-6-[[(四氢-2-呋喃基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑羧酸,乙酯;
[1323]
193)2-[[4-[[(4-羧基苯基)甲基]氨基]-6-[3-(羟基甲基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1324]
194)2-[[4-[[[4-[[(2-甲氧基乙基)氨基]羰基]苯基]甲基]氨基]-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯,三氟乙酸盐(1:1);
[1325]
195)2-[4,6-双(1-吗啉基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1326]
196)2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-[[n-甲基-n-(5-四唑基甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1327]
197)4-甲基-2-[[4-[-甲基(3-吡啶基甲基)氨基]-6-[4-吗啉基]-2-吡啶基甲基]氨基]-5-噻唑羧酸,乙酯;
[1328]
198)2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-[[[4-(甲氧基羰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1329]
199)2-[[4-氯-6-[(1-氧杂-3,8-二氮杂螺[4.5]癸-2,4,二酮-8-基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1330]
200)2-[[4-[4-(羟基甲基)-1-哌啶基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1331]
201)2-[[4-[3-(羟基甲基)-1-哌啶基]-6-[[n-甲基-n-(5-四唑基甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1332]
202)2-[[4-[3-(羟基甲基)-1-吡咯烷基]-6-[[n-甲基-n-(5-四唑基甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1333]
203)4-甲基-2-[[4-[甲基(苯基甲基)氨基]-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-5-噻唑羧酸,乙酯;
[1334]
204)2-[[4-(二甲基氨基)-6-[[[4-(甲基磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1335]
205)2-[[4-[4-羟基-1-哌啶基]-6-[[(3-(5-(1h)四唑基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1336]
206)2-[[4-[4-羟基甲基-1-哌啶基]-6-[[(4-(丙基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1337]
207)2-[[4-[4-羟基甲基-1-哌啶基]-6-[[(4-(环丙基磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1338]
208)2-[[4-[3-(羟基甲基)-1-哌啶基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1339]
209)2-[[4-[4-四氢吡喃基]氧基-6-[[n-[(3,4,5-三甲氧基苯基)甲基]]-n-(甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1340]
210)2-[[4-[4-甲基-1-哌嗪基]-6-[(4-甲氧基苯基)氧基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1341]
211)4-甲基-2-[4-(4-甲基-哌嗪-1-基)-6-[[[4-(氨基磺酰基)苯基]甲基]氨基]嘧啶-2-基氨基]-噻唑-5-羧酸乙基酯;
[1342]
212)2-[4-异丙基-6-(4-氨磺酰基-苄基氨基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1343]
213)4-甲基-2-[4-(4-氨磺酰基-苄基氨基)-6-甲基-嘧啶-2-基氨基]-噻唑-5-羧酸乙基酯;
[1344]
214)4-甲基-2-[4-(4-氨磺酰基-苄基氨基)-6-羟基甲基-嘧啶-2-基氨基]-噻唑-5-羧酸乙基酯;
[1345]
215)4-甲基-2-[4-(4-甲基-哌嗪-1-基)-6-[4-(1h-四唑-5-基)-苄基氨基]-嘧啶-2-基氨基]-噻唑-5-羧酸乙基酯;
[1346]
216)2-[4-(4-羟基-哌啶-1-基)-6-[4-(1h-四唑-5-基)-苄基氨基]-嘧啶-2-基氨
基]-4-甲基-噻唑-5-羧酸乙基酯;
[1347]
217)4-甲基-2-[4-[(四氢-呋喃-2-基甲基)-氨基]-6-[4-(1h-四唑-5-基)-苄基氨基]-嘧啶-2-基氨基]-噻唑-5-羧酸乙基酯;
[1348]
218)4-甲基-2-[4-吗啉-4-基-6-[4-(1h-四唑-5-基)-苄基氨基]-嘧啶-2-基氨基]-噻唑-5-羧酸乙基酯;
[1349]
219)2-[4-(3-氨基甲酰基-哌啶-1-基)-6-[4-(1h-四唑-5-基)-苄基氨基]-嘧啶-2-基氨基])-4-甲基-噻唑-5-羧酸乙基酯;
[1350]
220)2-[4-(4-羟基甲基哌啶-1-基)-6-[4-(1h-四唑-5-基)-苄基氨基]-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1351]
221)2-[4-(2-羟基甲基-1-吡咯烷基)-6-[4-(1h-四唑-5-基)-苄基氨基]-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1352]
222)2-[4-(3-n,n-二乙基氨基甲酰基-1-哌啶基)-6-[4-(1h-四唑-5-基)-苄基氨基]-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1353]
223)2-[4-(3-羟基-1-吡咯烷基)-6-[4-(1h-四唑-5-基)-苄基氨基]-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1354]
224)4-甲基-2-[[[2-[4-吗啉-4-基]乙基]氨基-6-[4-(1h-四唑-5-基)-苄基氨基]嘧啶-2-基氨基]-噻唑-5-羧酸乙基酯;
[1355]
225)4-甲基-2-[[[4-羟基]丁基]氨基-6-[4-(1h-四唑-5-基)-苄基氨基]-嘧啶-2-基氨基]-噻唑-5-羧酸乙基酯;
[1356]
226)2-[4-(4-甲酰基-1-哌嗪基)-6-[4-(1h-四唑-5-基)-苄基氨基]-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1357]
227)2-[[4-[[(4-氯代苯基)甲基]氨基]-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1358]
228)2-[[4-[[(4-氨基磺酰基(sylfonyl)苯基)甲基]氨基]-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1359]
229)2-[[4-吗啉代-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1360]
230)2-[[4-[[(3,4-二甲氧基苯基)甲基]氨基]-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1361]
231)2-[[4-(1,4-二氧杂-8-氮杂-螺[4.5]癸-8-基)-6-(5-唑基(oxazoly))-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1362]
232)2-[[4-[4-羟基-4-苯基-哌啶基]-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1363]
233)2-[[4-[[(4-甲基磺酰基苯基)甲基]氨基]-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1364]
234)2-[[4-[4-羟基-哌啶基]-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1365]
235)2-[[4-[4-乙氧基羰基-哌啶基]-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1366]
236)2-[[4-哌啶基-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1367]
237)2-[[4-[n-甲基哌嗪基-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1368]
238)2-[[4-[n-(2-呋喃基羰基)哌嗪基-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1369]
239)2-[[4-[n-乙酰基-[1,4-二氮杂基]-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1370]
240)2-[[4-[n-甲基-n-(n-甲基-4-哌啶基)-氨基]-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1371]
241)2-[[4-[n-甲基-[1,4]-二氮杂基]-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1372]
242)2-[[4-n,n-二甲氧基乙基氨基-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1373]
243)2-[[4-[(1',4)-联哌啶基]-6-(5-唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1374]
244)2-[4-(4-羟基-哌啶-1-基)-6-(3,4,5-三甲氧基-苯基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1375]
245)2-[(4-(4-羟基-哌啶-1-基)-6-[4-(1h-四唑-5-基)-苯基]-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1376]
246)2-[4-(4-羟基-哌啶-1-基)-6-吡啶-3-基-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1377]
247)2-[4-(4-甲烷磺酰基-苄基氨基)-6-吡啶-3-基-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1378]
248)2-[4-(4-羟基-哌啶-1-基)-6-嘧啶-4-基-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1379]
249)2-[4-(4-氰基-苯基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1380]
250)2-[4-(4-乙酰基-苯基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1381]
251)2-[4-(4-羟基甲基-苯基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1382]
252)2-[4-(4-羟基-苯基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1383]
253)2-[4-(4-甲烷磺酰基-苄基氨基)-6-(3,4,5-三甲氧基-苯基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1384]
254)2-[4-(4-甲烷亚磺酰基苯基)-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1385]
255)2-[4-(4-(氨基)苯基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1386]
256)2-[4-(4-羧基甲基-苯基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基
噻唑-5-羧酸乙基酯;
[1387]
257)2-[4-(4-(三氟甲基羰基氨基)苯基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1388]
258)2-[4-(4-(乙氧基羰基甲基)苯基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1389]
259)2-[4-(1,2,3,6-四氢吡啶-4-基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1390]
260)2-[4-(3-(氰基)苯基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1391]
261)2-[4-(4-(甲氧基羰基)苯基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1392]
262)2-[4-(2-(甲氧基)-5-吡啶基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1393]
263)2-[4-(4-叔丁基氧基羰基-1,2,3,6-四氢吡啶-4-基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1394]
264)2-[4-(1,4-二氧杂螺[4.5]癸-7-烯-8-基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1395]
265)2-[4-(4-甲基-1-哌嗪-基)-6-(3,4,5-三甲氧基-苯基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1396]
266)2-[4-(4-吗啉基)-6-(3,4,5-三甲氧基-苯基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1397]
267)2-[4-(4-吗啉基)-6-(3-吡啶基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1398]
268)2-[4-(哌啶-4-基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1399]
269)2-[[4-[4-羟基-哌啶基]-6-(3,5-二甲基-4-异唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑-羧酸乙基酯;
[1400]
270)2-[4-(4-叔丁氧基羰基氨基-苯基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1401]
271)2-[4-(4-氰基-苯基)-6-(4-甲烷磺酰基-苄基氨基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1402]
272)2-[4-(4-甲烷磺酰基苯基)-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1403]
273)2-[4-(4-甲烷硫烷基苯基)-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1404]
274)2-[4-(4-羧基-苯基)-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-噻唑-5-羧酸乙基酯;
[1405]
275)2-[4-(4-羧基-苯基)-6-(3-氧代-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1406]
276)2-[4-(4-羧基-苯基)-6-(4-甲基-哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1407]
277)2-[4-(4-羧基-苯基)-6-吗啉-4-基-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1408]
278)2-[4-(4-羧基-苯基)-6-(4-甲基-[1,4]二氮杂环庚烷-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1409]
279)2-[4-(4-羧基-苯基)-6-(3-r-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1410]
280)2-[4-(4-羧基-苯基)-6-(3-羟基甲基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1411]
281)2-[4-(4-乙酰基-[1,4]二氮杂环庚烷-1-基)-6-(4-羧基-苯基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1412]
282)2-[4-(4-羧基-苯基)-6-[n-甲基-n-(1-n-甲基-哌啶-4-基)-氨基]-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1413]
283)2-[4-(4-羧基-苯基)-6-哌嗪-1-基-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1414]
284)2-[4-(4-羧基-苯基)-6-(4-氨磺酰基-苄基氨基)-嘧啶-2-基氨基]-4-甲基噻唑-5-羧酸乙基酯;
[1415]
285)2-[[4-[[5-烯丙基[4-(氨基磺酰基)苯基]甲基]氨基]-6-氯-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1416]
286)2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-5-甲基-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯,三氟乙酸盐(1:3);
[1417]
287)2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-5-甲基-6-(4-吗啉基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1418]
288)2-[[4-[[5-烯丙基[4-(氨基磺酰基)苯基]甲基]氨基]-6-(4-甲基哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1419]
289)2-[[4-[[5-[2-[2-甲基丙-3-烯]]-4-[4-(氨基磺酰基)苯基]甲基]氨基]-6-(4-甲基哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1420]
290)2-[[4-[[[(3,4,5-(三甲氧基)苯基]甲基]氨基]-5-甲基-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯,三氟乙酸盐;
[1421]
291)2-[[4-[[5-[2,3-丙二醇(propandiol)][4-(氨基磺酰基)苯基]甲基]氨基]-6-(4-甲基哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1422]
292)2-[[4-[[[3,4,5-(三甲氧基)苯基]甲基]氨基]-5-甲基-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯,三氟乙酸盐;
[1423]
293)2-[[4-[[5-[2-[2-甲基丙-3-烯]]-4-[4-(氨基磺酰基)苯基]甲基]氨基]-6-氯-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1424]
294)2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-5-甲基-6-(4-叔丁基氧基羰基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,乙酯;
[1425]
295)2-[[4-[n-[[3,4,5-(三甲氧基)苯基]甲基]-n-甲基氨基]-5-甲基-6-(4-甲
苄基氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1446]
316)2-[[4-[2-(4-甲基-[1,4]二氮杂环庚烷-1-基)-2-氧代-乙基]-6-(4-氨磺酰基-苄基氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1447]
317)2-[[4-[[双-(2-甲氧基-乙基)-氨基甲酰基]-甲基]-6-(4-氨磺酰基-苄基氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1448]
318)2-[[4-(2-[1,4']联哌啶-1'-基-2-氧代-乙基)-6-(4-氨磺酰基-苄基氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1449]
319)2-[[4-[2-(4-羟基-4-苯基-哌啶-1-基)-2-氧代-乙基]-6-(4-氨磺酰基-苄基氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1450]
320)2-[[4-乙氧基羰基-6-(4-氨磺酰基-苄基氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1451]
321)2-[[4-羧基-6-(4-氨磺酰基-苄基氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1452]
322)2-[[4-(羧基甲基-氨基甲酰基)-6-(4-氨磺酰基-苄基氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1453]
323)2-[4-(4-羟基-哌啶-1-基)-6-(4-甲基硫烷基-苄基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1454]
324)2-[4-(4-羟基-哌啶-1-基)-6-(4-甲烷亚磺酰基-苄基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1455]
325)2-[[4-(4-羟基-哌啶-1-基)-6-(4-甲烷磺酰基-苄基)-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1456]
326)2-[[4-[4-甲基-1-哌嗪基]-6-[n-甲基-n-[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-三氟甲基-5-噻唑羧酸,乙酯;
[1457]
327)2-[[4-[4-甲基哌嗪-1-基]-6-(n-甲基-n-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-氰基噻唑;
[1458]
328)2-[[4-[4-甲基哌嗪-1-基]-6-甲基-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,2-甲氧基乙基酯;
[1459]
329)2-[[4-[4-羟基-哌啶-1-基]-6-[n-甲基[[n-[(3,4,5-三甲氧基苯基)甲基][-n-甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,丁酯;
[1460]
330)2-[[4-[1-吗啉基]-6-[[2-[1-吗啉基]乙基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,丁基酯;
[1461]
331)2-[[4-[4-甲基-1-哌嗪基]-6-[[n-[(3,4,5-三甲氧基苯基)甲基]]-n-(甲基)氨基]-2-嘧啶基]氨基]-4-异丙基-5-噻唑羧酸,乙酯;
[1462]
332)2-[[4-[4-甲基-1-哌嗪基]-6-[[n-[(3,4,5-三甲氧基苯基)甲基]]-n-(甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑羧酸,甲基酰胺;
[1463]
333)2-[4-[4-(2-二异丙基氨基-乙基氨基甲酰基)-苯基]-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1464]
334)2-[4-[4-(3-二甲基氨基-丙基氨基甲酰基)-苯基]-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1465]
335)2-[4-[4-(环己基甲基氨基甲酰基)-苯基]-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1466]
336)2-[4-[4-(吡啶-4-基甲基氨基甲酰基)-苯基]-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1467]
337)2-[4-[4-(异丁基氨基甲酰基(carbomoyl))-苯基]-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1468]
338)2-[4-[4-(n-环己基-n-甲基氨基甲酰基)-苯基]-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1469]
339)2-[4-[4-(n-环丙基甲基-n-丙基氨基甲酰基)-苯基]-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1470]
340)2-[4-[4-(4-乙氧基羰基哌啶-1-氨基甲酰基)-苯基]-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1471]
341)2-[4-[4-(3-羟基甲基-哌啶-1-羰基)-苯基]-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1472]
342)2-[4-[4-(n-2-羟基乙基-n-乙基氨基甲酰基)-苯基]-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1473]
343)2-[4-[4-(硫代吗啉-1-羰基)-苯基]-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1474]
344)2-[4-[4-(吗啉-1-羰基)-苯基]-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;和
[1475]
345)2-[4-[4-(4-氯-苯基氨基甲酰基)-苯基]-6-(4-羟基-哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑羧酸乙基酯;
[1476]
或其立体异构体、药学上可接受的盐,或水合物。
[1477]
在另一个相关的实施方案中,用于本发明方法的pde7抑制剂包括以下化合物:
[1478]
[1479]
[1480]
[1481]
[1482]
[1483]
[1484][1485]
或其立体异构体、药学上可接受的盐,或水合物。
[1486]
这些化合物的制备描述于u.s.pat.no.7,087,614,u.s.20030162802,和wo 2002/102313中。
[1487]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献中的那些化合物:us 2007/0129388和wo2007/063391,各文献通过引用以其整体明确
结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[1488][1489]
以上化合物的取代基定义如下:
[1490]
m是0、1或2;x为o、s或n
‑‑
cn;r为f、cl或cn;a为任选被c1-4烷基取代的c3-6亚环烷基;和b为单键或c1-2亚烷基;或其药学上可接受的盐、溶剂化物、多晶型物或前药。
[1491]
关于上述化合物,术语"亚烷基"表示具有1或2碳原子的二价饱和烃链。亚烷基的实例包括亚甲基、亚乙基和甲基亚甲基,其中优选亚甲基。
[1492]
术语"亚环烷基"表示具有3-6个碳原子的二价饱和碳环。亚环烷基的实例包括亚环丙基(如,1,1-亚环丙基、顺式和反式-1,2-亚环丙基)、亚环丁基(如,1,1-亚环丁基、顺式和反式-1,2-亚环丁基,及顺式和反式-1,3-亚环丁基)、亚环戊基(如,1,1-亚环戊基、顺式和反式-1,2-亚环戊基,及顺式和反式-1,3-亚环戊基)和亚环己基(如,1,1-亚环己基、顺式和反式-1,2-亚环己基、顺式和反式-1,3-亚环己基)及顺式和反式-1,4-亚环己基)。优选的实例包括亚环丁基和亚环己基,更优选亚环丁基,甚至更优选1,3-亚环丁基,和最优选反式-1,3-亚环丁基。
[1493]
术语"烷基"表示单价、直链或支链的、包含1-4个碳原子的饱和烃链。烷基的实例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基和叔丁基。优选的实例包括甲基和乙基、尤其是甲基。
[1494]
亚环烷基任选被c1-4烷基取代。优选地,烷基取代基,如果存在的话,为甲基或乙基,更优选地为甲基。烷基取代基,如果存在的话,可以存在于所述环的任何位置上,但优选地存在于1-位(即,与羧酸基团相同的位置)。
[1495]
优选地,m是1或2,更优选1。
[1496]
优选地,x为o或n-cn,更优选o。
[1497]
优选地,r为f或c1,更优选c1。
[1498]
优选地,a为任选被甲基取代的亚环丁基或亚环己基。更优选a为亚环丁基。甚至更优选a为1,3-亚环丁基,尤其是反式-1,3-亚环丁基。
[1499]
优选地,b为单键或亚甲基。更优选b为单键。
[1500]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自以下化合物:
[1501]
顺式-3-[(8'-氯-2'-氧代-2',3'-二氢-1'h-螺[环己烷-1,4'-喹唑啉]-5'-基)氧基]环丁烷羧酸;反式-3-[(8'-氯-2'-氧代-2',3'-二氢-1'h-螺[环己烷-1,4'-喹唑啉]-5'-基)氧基]环丁烷羧酸;3-[(8'-氟-2'-氧代-2',3'-二氢-1'h-螺[环己烷-1,4'-喹唑啉]
‑‑
5'-基)氧基甲基]环丁烷羧酸;反式-3-[(8'-氰基-2'-氧代-2',3'-二氢-1'h-螺[环己烷-1,4'-喹唑啉]-5'-基)氧基]环丁烷羧酸;1-[(8'-氟-2'-氧代-2',3'-二氢-1'h-螺
[环己烷-1,4'-喹唑啉]-5'-基)氧基甲基]环丁烷羧酸;反式-3-[(8'-氯-2'-氧代-2',3'-二氢-1'h-螺[环庚基-1,4'-喹唑啉]-5'-基)氧基]环丁烷羧酸;和反式-3-[(8'-氯-2'-氧代-2',3'-二氢-1'h-螺[环戊基-1,4'-喹唑啉]-5'-基)氧基]环丁烷羧酸;或其药学上可接受的盐、溶剂化物、多晶型物或前药。
[1502]
以上化合物的制备描述于us 2007/0129388和wo 2007/063391中。
[1503]
在另一个实施方案中,用于本发明方法的pde7抑制剂包括化合物asb16165(1-环己基-n-[6-(4-羟基-1-哌啶基)-3-吡啶基]-3-甲基-1h-噻吩并[2,3-c]吡唑-5-甲酰胺一水合物),其描述于kadoshima-yamaoka,k.et al.,"asb16165,一种新的磷酸二酯酶7a(pde7a)抑制剂,通过激活的小鼠t淋巴细胞抑制il-12-诱导的ifn-g产生(a novel inhibitor forphosphodiesterase 7a(pde7a),suppresses il-12-induced ifn-g production by mouse activated t lymphocytes),"immunology letters122:193-197,2009,通过引用明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[1504][1505]
制备以上化合物的方法描述于wo 2006/004040中。
[1506]
在另一个实施方案中,用于本发明方法的pde7抑制剂包括化合物ym-393059((
±
)-n-(4,6-二甲基嘧啶-2-基)-4-[2-(4-甲氧基-3-甲基苯基)-5-(4-甲基哌嗪-1-基)-4,5,6,7-四氢-1h-吲哚-1-基]苯磺酰胺富马酸盐(difumarate),其描述于yamamoto,s.et al.,"新的磷酸二酯酶7a和-4双重抑制剂,ym-393059,在体外或体内对t细胞相关的细胞因子产生的影响(the effects ofa novel phosphodiesterase 7a and-4dual inhibitor,ym-393059,on t-cell-related cytokine production in vitro and in vivo)."european journal ofpharmacology 541:106-114,2006,通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[1507][1508]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献中的那些化合物:martinez et al.,"2,1,3-苯并-和苯并噻吩并3,2-aa噻二嗪(aathiadiazin)2,2-二氧化物的苄基衍生物:第一种磷酸二酯酶7抑制剂,"
j.med.chem.43:683-689,2000,通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂包括以下化合物:
[1509]
1-[(4-甲氧基苯基)羰基甲基]苯并噻吩并-[3,2-a]-1,2,6-噻二嗪-493h)-酮2,2-二氧化物;和1-[(3,4-二氯苯基)-甲基]-2,1,3-苯并噻二嗪-4(3h)-酮2,2二氧化物.
[1510]
以上化合物的制备描述于j.med.chem.43:683-689,2000中。
[1511]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献中的那些化合物:castro,a等,"codes,一种用于配体基实际筛选的新方法:作为应用实例的pde7抑制剂(codes,a novel procedure for ligand-basedvirtual screening:pde7 inhibitors as an application example),"j.med.chem.43:1349-1359,2008,通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂包括以下化合物:
[1512]
[1513][1514]
在另一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[1515][1516]
以上化合物的取代基定义如下:
[1517]
x=o或s,
[1518]
r=h、ph、4-omeph、2,6-二fph、2,3,4-三fph、2-brph、bn、萘基,或me。
[1519]
在另一个实施方案中,用于本发明方法的pde7抑制剂包括以下化合物:
[1520]
5.2.4.3-(2,3,4-三氟苯基)-2-硫代-(1h)-喹唑啉-4-酮;
[1521]
5.3.2.3-苯基-2-硫代-(1h)-噻吩并[3,2-d]嘧啶-4-酮;
[1522]
5.3.3.3-(2,6-二氟苯基)-2-硫代-(1h)-噻吩并[3,2-d]嘧啶-4-酮;和
[1523]
5.4.2.3-(2,6-二氟苯基-2-硫代-(1h)-苯并[4,5]-噻吩并[3,2-d]-嘧啶-4-酮。
[1524]
以上化合物的制备描述于j.med.chem.43:1349-1359,2008中。
[1525]
在另一个实施方案中,用于本发明方法的pde7抑制剂包括bms-586353,如在yang,g等,"磷酸二酯酶7a-缺乏小鼠具有功能性t细胞(phosphodiesterase 7a-deficient mice have functional t cells),"j.immunol 171:6414-6420,2003中所述,该文献通过引用以其整体明确结合到本文中。
[1526]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献中的那些化合物:pitts,w.j等,"磷酸二酯酶7(pde7)的嘌呤抑制剂的鉴定(identification ofpurine inhibitor of phosphodiesterase 7(pde7),"bioorg.med.chem.lett.14:2955-2958,2004,和kempson,j等,"磷酸二酯酶7(pde7)的稠合的嘧啶基抑制剂:合成和初步构效关系(fusedpyrimidine based inhibitors of phosphodiesterase 7(pde7):synthesis and initial structure-activity relationships),"bioorg.med.chem.lett.15:1829-1833,2005,各文献通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂具有下式:
[1527][1528]
以上化合物的取代基定义如下:
[1529]
r1为
[1530][1531]
r2为
[1532]
[1533][1534]
其中乙基可连接于7或9位。
[1535]
在相关的实施方案中,用于本发明方法的pde7抑制剂具有下式:
[1536][1537]
在另一个相关的实施方案中,用于本发明方法的pde7抑制剂具有下式:
[1538][1539]
其中x=ch2、ch2ch2或och2;
[1540]
ar为
[1541][1542]
和nrr'为
[1543][1544][1545]
在另一个实施方案中,用于本发明方法的pde7抑制剂选自概略或详细披露于以下文献中的那些化合物:kang,n.s等,"pde4-pde7双重抑制剂的连接和3-d qsar研究
(docking and 3-d qsar studies ofdual pde4-pde7 inhibitors),"molecularsimulation 33:1109-1117,2007,通过引用以其整体明确结合到本文中。在一个实施方案中,用于本发明方法的pde7抑制剂包括以下化合物:
[1546][1547]
制备以上化合物的方法描述于molecular simulation 33:1109-1117,2007中。
[1548]
多肽或肽抑制剂
[1549]
在一些实施方案中,pde7抑制剂包含分离的pde7多肽或肽抑制剂,包括抑制pde7活性的经分离的天然肽抑制剂和合成肽抑制剂。本文所用术语“分离的pde7多肽或肽抑制剂”是指通过结合pde7、与pde7竞争结合底物以抑制pde7依赖性切割camp,和/或与pde7直接相互作用以抑制pde7依赖性切割camp的多肽或肽,所述多肽或肽是基本纯的,基本上不含天然可与之一起存在的其它物质,以达到可用于或适用于其预期应用的程度。
[1550]
肽抑制剂已被成功用于在体内干扰蛋白质-蛋白质间的相互作用和催化部位。例如,结构上与lfa-1相关的黏着分子的肽抑制剂最近已获准用于凝血病的临床治疗(ohman,e.m.等,european heart j.16:50-55,1995)。研究发现小的线性肽(《30个氨基酸)防止或干扰整联蛋白依赖性粘附(murayama,o.等,j.biochem.120:445-51,1996)。长度范围为25-200个氨基酸残基的较长的肽已被成功用来阻断整联蛋白依赖性粘附(zhang,l.等,j.biol.chem.271(47):29953-57,1996)。一般而言,较长的肽抑制剂比短肽具有较高的亲和力和/或较慢的解离速率,因此,可以是更强效的抑制剂。研究还表明环状肽抑制剂是整联蛋白的有效抑制剂,体内用于治疗人的炎症性疾病(jackson,d.y.等,j.med.chem.40:3359-68,1997)。产生环状肽的一种方法包括合成其中肽的末端氨基酸为半胱氨酸的肽,从而通过末端氨基酸之间的二硫键结合使肽以环状形式存在,研究表明这可在体内改进亲和力和半寿期以治疗造血系统肿瘤(例如larson的美国专利第6,649,592号)。
[1551]
合成的pde7肽抑制剂
[1552]
以模拟对pde7酶活性十分重要的靶区(例如pde7的催化结构域)的氨基酸序列为例,来说明用于本发明方法的pde7抑制肽。pde7a和pde7b在催化结构域具有70%同一性(hetman,j.m.等,pnas97(1):472-476,2000)。pde7a1的催化结构域为seq id no:2的氨基酸残基185-456。pde7a2的催化结构域为seq id no:4的氨基酸残基211-424。pdeb的催化结构域为seq id no:6的氨基酸残基172-420。用于实施本发明方法的抑制肽的大小范围约为5个氨基酸至约250个氨基酸。还可采用分子建模和合理的分子设计以制备和筛选模拟pde7催化域的分子结构并抑制pde7酶活性的肽。用于建模的分子结构包括抗pde7单克隆抗体的
cdr区。与特定靶标结合的肽的鉴定方法是本领域众所周知的。例如,分子印记可用于从头构建大分子结构,例如与特定分子结合的肽。参见例如shea,k.j.,“molecular imprinting of synthetic network polymers:the de novo synthesis of macromolecular binding and catalytic sites(合成网状聚合物的分子印记:大分子结合部位和催化部位的从头合成)”,trip2(5),1994。
[1553]
举例来说,模拟pde7结合肽的一种制备方法如下。使显示抑制pde7的抗pde7抗体结合区的功能单体(模板)聚合。然后移开模板,接着在由模板余留的空处使第二类单体聚合,以提供具有一种或多种所需要的类似于模板性质的新分子。除了以这种方式制备肽以外,还可制备作为pde7抑制剂的其它pde7结合分子,例如多糖、核苷、药物、核蛋白、脂蛋白、糖、糖蛋白、类固醇、脂质和其它生物活性材料。该方法用于设计各种比其天然对应物更稳定的生物学模拟物,因为它们通常通过功能单体的自由基聚合制备,产生具有生物不能降解主链的化合物。
[1554]
pde7抑制肽可采用本领域众所周知的技术制备,例如最初由merrifield提出的固相合成技术(j.amer.chem.soc.85:2149-2154,1963)制备。可采用例如appliedbiosystems 431肽合成仪(foster city,calif.),按照生产商提供的说明书进行自动合成。其它技术可参见例如bodanszky,m.等,peptide synthesis,第二版,john wiley&sons,1976,以及本领域技术人员已知的其它参考文献。肽还可采用本领域技术人员已知的标准遗传工程技术制备。
[1555]
可以在若干实验之一(包括例如实施例1中所述的pde7磷酸二酯酶实验)中,测定候选pde7抑制肽作为pde7抑制剂所起作用的能力。
[1556]
pde7的表达抑制剂
[1557]
在本发明方法的一些实施方案中,pde7抑制剂是能够抑制pde7依赖性camp切割(pde7a、pde7b或两者)的pde7表达抑制剂。在本发明实施方案的实施中,代表性pde7表达抑制剂包括pde7反义核酸分子(例如反义mrna、反义dna或反义寡核苷酸)、pde7核酶和pde7 rnai分子。
[1558]
反义rna和dna分子通过与pde7 mrna杂交并阻止pde7蛋白翻译,从而起直接阻断pde7 mrna翻译的作用。可以按多种不同的方式构建反义核酸分子,只要能够干扰pde7表达。例如,可以使pde7a1 cdna(seq id no:1)、pde7a2 cdna(seq id no:3)或pde7b cdna(seq id no:5)的编码区(或其部分)相对于其正常转录方向逆转以便于其互补序列转录,从而来构建反义核酸分子。设计和给予反义寡核苷酸的方法是本领域众所周知的,参见例如mautino等,hum gene ther 13:1027-37,2002;及pachori等,hypertension39:969-75,2002,所述各文献均通过引起结合于此。
[1559]
反义核酸分子通常与一个或多个靶基因的至少一部分基本相同。然而,核酸无需完全相同才可抑制表达。一般可使用较高同源性以弥补较短反义核酸分子的作用。最小百分比同一性通常大于约65%,但是较高的百分比同一性可更有效地阻抑内源序列的表达。通常优选基本上大于约80%的较大百分比同一性,尽管通常最优选约95%至完全同一性。
[1560]
反义核酸分子无需与靶基因一样具有相同的内含子或外显子模式,靶基因非编码区段在实现反义抑制靶基因表达时可与编码区段同样有效。至少约8个左右核苷酸的dna序列可用作反义核酸分子,尽管优选较长的序列。在本发明中,有益的pde7抑制剂的代表性实
例为反义pde7核酸分子,该反义pde7核酸分子与由seq id no:1中所示核酸序列组成的一部分pde7a1 cdna的互补序列有至少90%同一性。有益的pde7抑制剂的另一代表性实例为反义pde7核酸分子,该反义pde7核酸分子与由seq id no:3中所示核酸序列组成的一部分pde7a2 cdna的互补序列有至少90%同一性。有益的pde7抑制剂另一代表性实例为反义pde7核酸分子,该反义pde7核酸分子与由seq id no:5中所示核酸序列组成的一部分pde7b cdna的互补序列有至少90%同一性。
[1561]
反义寡核苷酸寻靶结合pde7 mrna是可用来降低pde7蛋白质合成水平的另一种机制。例如,针对多聚半乳糖醛酸酶和毒蕈碱2型乙酰胆碱受体各自的mrna序列的反义寡核苷酸抑制其合成(cheng的美国专利第5,739,119号和shewmaker的美国专利第5,759,829号)。此外,使用核内蛋白即细胞周期蛋白、多药抗药性基因(mdg1)、icam-1、e-选择蛋白、stk-1、纹状体gabaa受体和人egf,证实了反义抑制的实例(参见例如baracchini的美国专利第5,801,154号、baker的美国专利第5,789,573号、considine的美国专利第5,718,709号和reubenstein的美国专利第5,610,288号)。
[1562]
已披露的一个系统可供普通技术人员确定哪一种寡核苷酸可用于本发明,该系统包括使用rna酶h切割探测靶mrna中合适的位点作为转录物内序列可及性的标志。scherr,m.等,nucleicacidsres.26:5079-5085,1998;lloyd等,nucleicacidsres.29:3665-3673,2001。将与pde7转录物某些区域互补的反义寡核苷酸的混合物加到表达pde7的细胞提取物中并杂交以产生rna酶h易损位点。该方法可与计算机辅助序列筛选相结合,所述计算机辅助序列筛选可根据其在宿主细胞内形成可降低或阻止与靶mrna特异性结合的二聚体、发夹结构或其它二级结构的相对能力,来预测选择反义组合物的最佳序列。可采用oligo引物分析软件(rychlik,i.,1997)和blastn 2.0.5算法软件(altschul,s.f.等,nucl.acidsres.25:3389-3402,1997)进行这些二级结构分析和靶位选择考虑事项。针对靶序列的反义化合物的长度优选包含约8至约50个核苷酸。特别优选包含约9至约35个左右核苷酸的反义寡核苷酸。本发明的发明人认为范围为9-35个核苷酸(即9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34或35左右个碱基长度的核苷酸)的所有寡核苷酸组合物对于实施基于反义寡核苷酸的本发明方法是非常优选的。非常优选的pde7 mrna的靶区为位于aug翻译起始密码子上或接近aug翻译起始密码子的靶区,以及与mrna的5’区(例如pde7基因核苷酸序列(seq id no:1、seq id no:3、seq id no:5)的0和+10区之间)基本互补的序列。
[1563]
本文所用术语“寡核苷酸”是指核糖核酸(rna)或脱氧核糖核酸(dna)或其模拟物的寡聚体或多聚体。该术语还包括由天然存在的核苷酸、糖和共价核苷酸间(主链)键以及具有非天然存在的修饰的寡核苷酸组成的寡核苷酸碱基(oligonucleobase)。这些修饰可供引入某些所需要的通过天然存在的寡核苷酸无法提供的性质,例如毒性性质的减少,对核酸酶降解的稳定性的提高,细胞摄取的增加。在示例性实施方案中,本发明的反义化合物与天然dna的区别在于:磷酸二酯主链经修饰能延长其中磷酸取代基被硫代磷酸置换的反义寡核苷酸的寿命。同样,寡核苷酸的1个或2个末端可被一个或多个间插在核酸链相邻碱基对之间的吖啶衍生物取代。
[1564]
反义的另一种替代是使用“rna干扰”(rnai)。双链rna(dsrna)可在哺乳动物体内引起基因沉默。rnai的天然功能和共抑制似乎可保护基因组免受可动遗传元件的侵袭,可
动遗传元件例如当其被激活时在宿主细胞内产生异常rna或dsrna的反转录转座子和病毒(参见例如jensen,j.等,nat.genet.21:209-12,1999)。可通过将能够形成双链rna分子的两条rna链进行合成以制备双链rna分子,每条链长约为19-25(例如19-23个核苷酸)。例如,用于本发明方法的dsrna分子可包含相应于seq id no:1、seq id no:3、seq id no:5及其互补序列中的至少一个的一部分rna。优选至少一个rna链具有1-5个核苷酸的3'突出端。经合成的rna链在形成双链分子的条件下结合起来。rna序列可包含seq id no:1、seq id no:3或seq id no:5中的至少8个核苷酸部分,总长不超过25个核苷酸。对于给定靶标的sirna序列的设计为本领域技术人员所掌握。可获得设计sirna序列并确保至少70%敲减表达的商业化服务(qiagen,valencia,ca)。示例性pde7 shrna和sirna可获自sigma-aldrich公司(产品号shdna_-nm_002603、sasi_hs01_00183420至sasi_hs01_00010490)。
[1565]
dsrna可作为药物组合物给予,并可通过已知方法进行,其中将核酸导入所需的靶细胞中。常用的基因转移方法包括磷酸钙法、deae-葡聚糖法、电穿孔法、显微注射法和病毒法。这类方法参见ausubel等,currentprotocols in molecularbiology,john wiley&sons,inc.,1993。可修饰治疗性核酸分子以穿过血脑屏障。例如,研究表明通过脑室内给药给予的针对淀粉状蛋白前体蛋白(app)aβ中央区的硫代磷酸反义寡核苷酸可以在alzheimer小鼠模型中逆转学习和记忆缺陷。banks w.a.等,journal of pharm.and exp.therapeutics,297(3):1113-1121,2001。
[1566]
核酶:
[1567]
在一些实施方案中,pde7抑制剂为特异性切割靶pde7(例如pde7a、pde7b或两者)的mrna的核酶。靶向pde7的核酶可被用作pde7抑制剂以降低pde7的量和/或生物活性。核酶是可以切割核酸分子的催化性rna分子,具有与核酶序列完全或部分同源的序列。可以设计编码与靶rna特异性配对并在特异性位置上切割磷酸二酯主链的rna核酶的核酶转基因,从而在功能上使靶rna失活。在进行这种切割时,核酶本身不发生变化,因此能够再循环切割其它分子。纳入反义rna内的核酶序列赋予其rna切割活性,从而增加反义构建体的活性。
[1568]
用于实施本发明的核酶通常包含至少约9个核苷酸的杂交区(在核苷酸序列中与至少部分靶pde7 mrna互补)和适于切割靶pde7mrna的催化区(一般参见欧洲专利号0321201;wo 88/04300;haseloff,j.等,nature 334:585-591,1988;fedor,m.j.等,proc.natl.acad.sci.usa 87:1668-1672,1990;cech,t.r.等,ann.rev.biochem.55:599-629,1986)。
[1569]
核酶可以掺入核酶序列的rna寡核苷酸的形式直接靶向细胞,或者可作为编码所需核酶rna的表达载体导入细胞。核酶还可以反义多核苷酸中所述的多种相同方式利用和应用。
[1570]
用于本发明方法的反义rna和dna、核酶和rnai分子可通过本领域已知的合成dna和rna分子的任何方法制备。这些包括本领域众所周知的用于化学合成脱氧核糖核苷酸和核糖核苷酸的技术,例如固相亚磷酰胺化学合成法。或者,rna分子可通过编码反义rna分子的dna序列的体外和体内转录来产生。可将这类dna序列掺到各种将掺入合适rna聚合酶启动子(例如t7或sp6聚合酶启动子)的载体中。或者,可将组成型或诱导型合成(取决于所用的启动子)反义rna的反义cdna构建体稳定导入细胞系。
[1571]
可提供各种众所周知的dna分子修饰作为提高稳定性和半寿期的方法。有益的修
饰包括但不限于向分子的5'端和/或3'端添加核糖核苷酸或脱氧核糖核苷酸的侧翼序列,或者在脱氧核糖核苷酸主链内使用硫代磷酸或2'o-甲基而不是磷酸二酯键。
[1572]
iv.用于治疗与神经性运动障碍病理有关的运动异常的pde7抑制剂的筛选方法
[1573]
另一方面,提供鉴定抑制pde7活性用于治疗有需要的哺乳动物受治疗者的与神经性运动障碍病理有关的运动异常的药物的方法。本发明这一方面的方法包括:(a)测定多种药物每一种的抑制pde7活性的ic
50
;(b)从抑制pde7活性的ic
50
小于约1μm的多种药物中选出药物;(c)测定抑制pde7活性的ic
50
小于约1μm的药物抑制pde4活性的ic
50
;(d)通过选出抑制pde4活性的ic
50
是抑制pde7的ic
50
的10倍以上的化合物,鉴定用于治疗运动障碍的药物;(e)在神经性运动障碍模型实验中对已鉴定出的化合物的活性进行评价,其中药物抑制pde7的ic
50
小于约1μm,抑制pde4活性的ic
50
是抑制pde7的ic
50
的10倍以上,且在模型实验中被确定为有效治疗至少一种运动异常的药物,表明是用于治疗哺乳动物受治疗者的与神经性运动障碍病理有关的运动异常的pde7抑制剂。
[1574]
可用于实施本发明这一方面方法的代表性药物包括结合pde7并抑制pde7酶活性的分子(例如结合pde7并降低酶促活性的小分子抑制剂或封闭性肽)及在转移和/或翻译水平上降低pde7表达的分子(例如pde7反义核酸分子、pde7特异性rnai分子和pde7核酶),从而阻止pde7切割camp。
[1575]
v.通用组合物描述和定义。
[1576]
一方面,本发明提供治疗与神经性运动障碍病理有关的运动异常的方法,该方法包括给予有需要的患者一定量的有效抑制pde7酶活性的pde7抑制剂,其中对pde7酶活性的这种抑制是pde7抑制剂在治疗运动异常中的主要治疗作用方式。在本方法的一些实施方案中,神经性运动障碍可用多巴胺受体激动剂或多巴胺受体激动剂前体治疗。在本方法的一些实施方案中,神经性运动障碍选自帕金森病、脑炎后帕金森综合征、多巴胺反应性肌张力障碍、夏-德雷格综合征、周期性肢体运动障碍(plmd)、睡眠中周期性肢体运动(plms)和腿多动综合征(rls)。
[1577]
在其它实施方案中,神经性运动障碍选自图雷特综合征、亨廷顿舞蹈病(即亨廷顿舞蹈症)和药物诱发的帕金森综合征。
[1578]
对于用于本发明方法的各pde7抑制化合物,所有可能的立体异构体和几何异构体都包括在内。化合物不仅包括外消旋化合物,还包括旋光异构体。当pde7抑制剂需要作为单一对映异构体时,可以通过拆分最终产物或由异构体纯的原料进行立体有择合成或使用手性助剂来获得,例如参见ma,z.等,tetrahedron:asymmetry 8(6):883-888,1997。可通过本领域已知的任何合适方法获得拆分的最终产物、中间体或原料。另外,在其中可能存在化合物互变异构体的情况下,本发明将包括化合物的所有互变异构形式。
[1579]
含有酸性部分的pde7抑制剂可与合适的阳离子形成药学上可接受的盐。合适的药学上可接受的阳离子包括碱金属(例如钠或钾)和碱土金属(例如钙或镁)阳离子。pde7抑制剂的含有碱性中心的药学上可接受的盐,是与药学上可接受的酸形成的酸加成盐。实例包括盐酸盐、氢溴酸盐、硫酸盐或硫酸氢盐、磷酸盐或磷酸氢盐、乙酸盐、苯甲酸盐、琥珀酸盐、富马酸盐、马来酸盐、乳酸盐、柠檬酸盐、酒石酸盐、葡糖酸盐、甲磺酸酸(methanefulgonate)、苯磺酸盐和对甲苯磺酸盐。根据上述情况,本文出现任何提及用于本发明方法的化合物时都包括pde7抑制剂及其药学上可接受的盐和溶剂化物。
[1580]
在治疗上可以纯的化合物给予本发明的化合物,但是优选作为药物组合物或剂型给予pde7抑制剂。因此,本发明进一步提供包含pde7抑制剂或其药学上可接受的盐连同一种或多种药学上可接受的载体以及任选其它治疗和/或预防成分的药物组合物或剂型。合适的载体可与剂型的其它成分相配伍,对其接受者无毒。本发明的化合物还可装载于递送系统以提供缓释或提高化合物的摄取或活性,例如用于注射的脂质体或水凝胶系统;用于口服或胃肠外递药的微粒、纳米粒或微团系统;或者用于口服递药的分段胶囊系统(staged capsule system)。
[1581]
具体地讲,用于本发明方法的选择性pde7抑制剂单独使用或与一种或多种另外的治疗剂联用,例如多巴胺受体激动剂、多巴胺受体激动剂前体、另一种多巴胺能药物或上述物质的组合。多巴胺受体激动剂和前体的实例包括例如左旋多巴(亦称“l-dopa”)、卡比多巴、溴隐亭、培高利特、普拉克索、罗匹尼罗、卡麦角林(cabergoline)、阿扑吗啡(apomorphine)和利舒脲(lisuride)。与选择性pde7抑制剂组合的其它药物包括抗胆碱能药,例如盐酸比哌立登(biperiden)、甲磺酸本扎托品、丙环定(procyclidine)和苯海索;单胺氧化酶b抑制剂,例如eldepryl
tm
、atapryl
tm
和carbex
tm
及nmda拮抗剂金刚烷胺(amantadine)(symmetrel
tm
)。
[1582]
在一个实施方案中,选择性pde7抑制剂可与一种或多种另外的激活多巴胺d1受体和/或增加黑质纹状体神经末梢和/或黑质纹状体突触间隙中的多巴胺浓度的治疗药或治疗药的前体联用。这类药物包括左旋多巴、非选择性多巴胺受体激动剂,例如阿扑吗啡、溴隐亭和培高利特;d1选择性药物,例如abt-431、a86929和skf38393。
[1583]
在一个实施方案中,本发明提供治疗与帕金森病病理有关的运动异常的方法,该方法包括给予有需要的患者一定量的有效抑制pde7酶活性的pde7抑制剂,其中这类pde7抑制是pde7抑制剂在治疗运动异常中的主要治疗作用方式。如本文实施例5-7中所证实的一样,选择性pde7抑制剂可用于治疗与帕金森病病理有关的运动异常。在帕金森病的情况下,治疗包括减轻、减缓、掩蔽或预防至少一种选自以下运动异常症状的对症疗法:静止性震颤、强直、运动过慢或缺乏姿势反射。
[1584]
适用于本发明的化合物和药物组合物包括其中以有效量给予活性成分以达到其既定目的的化合物和药物组合物。更具体地讲,“治疗有效量”是指有效防止待治疗的受治疗者的症状发展,或者减轻待治疗的受治疗者存在的症状的用量。尤其按照本文所提供的详细公开内容,有效量的确定为本领域技术人员所掌握。要进一步了解的是,用于治疗所需要的本发明化合物的量随待治疗疾病的性质、患者年龄和健康状况而变化,最终由主治医师作出决定。然而,一般用于成人治疗的剂量范围通常为每日0.001mg/kg至约100mg/kg。
[1585]“治疗有效剂量”是指达到治疗或改善与神经性运动障碍病理有关的运动异常所需效果的化合物用量。可以在细胞培养物或实验动物中通过标准药学方法测定这类化合物的毒性和治疗功效,例如用于测定ld50(群体中50%致死的剂量)和ed50(群体中50%有效治疗的剂量)的方法。
[1586]
在一个实施方案中,治疗有效剂量是足以抑制神经元细胞内pde7酶活性的pde7抑制剂的量。在本发明方法的另一个实施方案中,治疗有效剂量是足以抑制纹状体神经元内pde7酶活性的pde7抑制剂的量。可采用pde7抑制的细胞实验(例如参见smith s.j.等,molecular pharmacology 66(6):1679-1689(2004),通过引用结合到本文中),来测定足以
穿过细胞膜并抑制细胞内pde7酶活性的pde7抑制剂的有效剂量。可采用测定pde抑制剂对纹状体camp水平的作用的实验,来测定足以抑制纹状体pde7酶活性的pde7抑制剂的有效剂量,参见siuciak j.a.等,neuropharmacology 51:386-396(2006),通过引用结合到本文中。
[1587]
毒性作用和治疗效果间的剂量比是治疗指数,用ld50与ed50之比表示。优选治疗指数高的化合物。由这类分析获得的数据可用于配制用于人的剂量范围。这类化合物的剂量优选介于包括ed50范围在内的循环浓度内,这取决于所采用的剂型和所利用的给药途径。
[1588]
可以利用实验动物模型,例如实施例5-7中所述的小鼠mptp模型,通过标准药学方法测定pde7抑制剂的毒性和治疗功效。可利用该动物模型,采用标准方法测定noael(无明显损害作用水平)和med(最小有效剂量)。noael和med作用的剂量比为治疗比率,用noael/med之比表示。最优选治疗比率或治疗指数大的pde7抑制剂。由细胞培养实验和动物研究获得的数据可用于配制各种用于人的剂量。pde7抑制剂的剂量优选介于循环浓度范围内,包括几乎无毒或无毒的med。剂量可在该范围内变化,这取决于所采用的剂型和所使用的给药途径。
[1589]
对于任何化合物剂型,可以利用动物模型对治疗有效剂量进行评价。例如,可以在动物模型配制达到包括med在内的循环血浆浓度或脑组织范围的剂量。还可测定pde7抑制剂在血浆或脑内的定量水平,例如参见本文实施例4。
[1590]
各医师根据患者情况选择确切的剂型、给药途径和剂量。可个别调整剂量和剂量间隔以提供足以维持治疗效果的活性部分的血浆水平。
[1591]
可以单剂量适当地给予所需剂量,或作为多剂量以适当间隔给予所需剂量,例如每天2次、3次、4次或以上分剂量。在实践中,医师决定最适于各个患者的实际给药方案,且剂量随具体患者的年龄、体重和反应而变化。上述剂量是平均情况的示例性剂量,不过可能有个别病例,其中较高或较低剂量是有益的,这些也都落入本发明的范围内。
[1592]
可以标准方式给予本发明的剂型用于治疗适应症,例如口服、胃肠外、经粘膜(例如舌下或通过口腔给药)、局部、经皮、直肠、通过吸入(例如经鼻或深肺部吸入)。胃肠外给药包括但不限于静脉内、动脉内、腹膜内、皮下、肌内、鞘内和关节内。还可采用高压技术,例如powderject
tm
系统实现胃肠外给药。
[1593]
另一方面,本发明提供包含pde7抑制剂用于治疗以下神经性运动障碍的药物组合物:例如帕金森病、脑炎后帕金森综合征、多巴胺反应性肌张力障碍、夏-德雷格综合征、周期性肢体运动障碍(plmd)、睡眠中周期性肢体运动(plms)、图雷特综合征或腿多动综合征(rls)。
[1594]
对于口腔或口服给药,药物组合物可以是以常规方式配制的片剂或锭剂的形式。例如,用于口服给药的片剂和胶囊剂可含有常用的赋形剂,例如粘合剂(例如糖浆、阿拉伯树胶、明胶、山梨醇、西黄蓍胶、淀粉胶浆或聚乙烯吡咯烷酮)、填充剂(例如乳糖、糖、微晶纤维素、玉米淀粉、磷酸钙或山梨醇)、润滑剂(例如硬脂酸镁、硬脂酸、滑石粉、聚乙二醇或二氧化硅)、崩解剂(例如马铃薯淀粉或羟基乙酸淀粉钠)或润湿剂(例如月桂硫酸钠)。可按照本领域众所周知的方法给片剂包衣。
[1595]
或者,可将本发明的化合物掺入口服液体药剂中,例如水性或油性混悬剂、溶液
剂、乳剂、糖浆剂或酏剂。此外,含有这些化合物的剂型可以是无水产品,用前与水或其它合适的溶媒配制。这类液体药剂可含有常用添加剂,例如悬浮剂,例如山梨醇糖浆、甲基纤维素、葡萄糖/蔗糖糖浆、明胶、羟乙基纤维素、羟丙基甲基纤维素、羧甲基纤维素、硬脂酸铝凝胶和氢化可食用脂肪;乳化剂,例如卵磷脂、失水山梨醇单油酸酯或阿拉伯树胶;非水性溶媒(可包括食用油),例如杏仁油、分馏椰子油、油性酯、丙二醇和乙醇;防腐剂,例如对羟基苯甲酸甲酯或对羟基苯甲酸丙酯和山梨酸。
[1596]
这类制剂还可配制成例如含有常用栓剂基质,例如可可脂或其它甘油脂的栓剂。用于吸入的组合物通常可以溶液剂、混悬剂或乳剂的形式提供,可作为干粉或使用常规抛射剂的气雾剂形式给予,抛射剂例如二氯二氟甲烷或三氯氟甲烷。通常局部和经皮剂型包含常用的水性或非水性溶媒,例如滴眼剂、乳膏剂、软膏剂、洗剂、糊剂或药用硬膏剂(medicatedplaster)、贴剂或膜剂的形式。
[1597]
另外,可以制备本发明的组合物用于通过注射或连续输注经胃肠外给药。用于注射的剂型可以是油性或水性溶媒中的混悬剂、溶液剂或乳剂的形式,可含有调制剂(formulation agent),例如悬浮剂、稳定剂和/或分散剂。或者,活性成分可为粉末的形式,临用前用合适的溶媒(例如无菌无热原水(hydrogen-free water))配制。
[1598]
本发明的组合物还可配制成贮库制剂。可通过植入(例如皮下或肌内)或者通过肌内注射给予这类长效剂型。因此,本发明的化合物可用合适的聚合材料或疏水材料(例如可接受油中的乳剂)、离子交换树脂配制,或者可配制成微溶性衍生物(例如微溶性盐)。
[1599]
因此,在又一方面,本发明提供包含pde7抑制剂连同其药学上可接受的稀释剂或载体的药物组合物。本发明进一步提供包含pde7抑制剂的药物组合物的制备方法,该方法包括将pde7抑制剂连同其药学上可接受的稀释剂或载体相混合。
[1600]
血脑屏障:
[1601]
在一些实施方案中,给予pde7抑制剂以便穿过或越过血脑屏障。优选治疗方法中所给予的抑制剂、化合物或组合物可以足够的量并按足够的速度穿过血脑屏障以便可治疗运动障碍。使药物穿过血脑屏障的方法为本领域所知,包括使药物大小最小化、提供有利于通过的疏水因子以及与具有相当大通透性的穿过血脑屏障的载体分子缀合。
[1602]
在一些实施方案中,pde7抑制剂的有效量是在脑组织内达到给定pde7抑制剂活性ic
50
或以上浓度的量。在一些实施方案中,以这样的方式和剂量给予pde7抑制剂,得到抑制pde7a或pde7b较大者的ic
50
浓度约1、1.5、2、2.5、5、10、20倍以上的峰值浓度。
[1603]
在一些情况下,通过外科手术植入与泵装置连接的导管给予pde7抑制剂或药物组合产品。泵装置可植入,或者可放置在体外。可以不连续脉冲方式或作为连续输注给予pde7抑制剂。注射到脑部不同部位的装置为本领域所知。在某些实施方案中,pde7抑制剂或包含pde7抑制剂的组合物通过注射局部给予脑室、黑质、纹状体、蓝斑、迈内特基底核(nucleus basalis meynert)、脚桥核(pedunculopontine nucleus)、大脑皮质和/或脊髓。
实施例
[1604]
实施例1
[1605]
本实施例描述了用于测定pde7抑制剂功效的实验,证实了用于本发明方法的若干代表性pde7抑制剂抑制pde7的功效。
[1606]
方法:
[1607]
在利用杆状病毒系统表达的重组人pde7a和pde7b酶进行的pde7磷酸二酯酶实验中,测定了表1中所列举的化合物的抑制活性。重组人pde7a酶购自bps bioscience(catalog#60070),genbank检索号nm_002603(氨基酸121至末端),带有n端gst标签,mw=66kda,在杆状病毒转染的sf9细胞表达系统中进行表达,比活为302pmol/分钟/μg。重组人pde7b酶购自bps bioscience(catalog#60071),genbank检索号nm_018945(氨基酸107至末端),带有n端gst标签,mw=66kda,在杆状病毒转染的sf9细胞表达系统中进行表达,比活为53pmol/分钟/μg。
[1608]
表1:pde7抑制化合物
[1609]
编号化合物参考号mwom691353.42om0562353.42om9553310.78om9564330.45
[1610]
pde7活性实验:
[1611]
所用实验方法为闪烁亲近测定法(spa)(得自ge healthcare,产品编码trkq7100),用[3h]-cgmp为底物(spa手册,amersham biosciences)。将纯的人pde7a和pde7b(得自bps bioscience,san diego,ca)用含有25mm tris-cl(ph 8.0)、100mmnacl、0.05%吐温20(tween 20)、50%甘油和3mm dtt的溶液稀释保存。pde7实验在下列反应混合物(终浓度)中进行:50mm tris-cl(ph 8.0)、8.3mm mgcl2、1.7mm egta、0.5mg/ml bsa、5%dmso(除对于om056为2.5%dmso外)和2ng pde7a或0.2ng pde7b重组蛋白,最终体积为0.1ml。
[1612]
为了测定针对pde7a或pde7b的ic
50
值,在单块板上以8种抑制剂浓度按一式两份进行了实验,用已知的pde7抑制剂(brl50481)作为阳性对照。抑制剂浓度的范围为0.61nm至10,000nm,只是在om056的情况下,浓度范围为0.24nm至4,000nm。通过加入酶引发反应,在30℃下温育20分钟,然后通过加入50μl含有zn2+的spa微珠终止反应。
[1613]
混合物经震荡,使之沉淀3小时,然后在平板计数器中通过闪烁计数对由底物产生的[3h]-5'amp进行定量测定。将用净cpm值表示的结果用excel与4参数逻辑模型拟合。
[1614]
结果:
[1615]
pde7磷酸二酯酶抑制实验的结果概括于下表2,并如图3a至图6b中所示。
[1616]
表2:代表性pde7抑制化合物对pde7a和pde7b抑制的ic
50
值
[1617][1618]
*本发明的发明人以前测定了om69对pde7a和pde7b活性的抑制。我们注意到有关
用om69抑制pde7b的ic
50
值,在采用不同实验方法的最初实验中,测定的ic
50
值为96.9nm。在该最初实验中,背景信号(每分钟计数)比最大信号高,希尔系数低,最初实验结果的可靠性受到质疑。
[1619]
上表2中所显示的实验结果表明,om69抑制pde7b的ic
50
值为4.8nm,我们认为这是比较准确的数值,因为如图3b所示,与4参数逻辑剂量-反应模型拟合的r2值为0.9988,正如对简单竞争性抑制剂所预期的一样。在最初的实验研究中,用om69抑制pde7a的ic
50
值为3.5nm,与表2中所显示的抑制pde7a的值1.30nm不一致。
[1620]
图3a、图4a、图5a和图6a是说明pde7a抑制活性(ic
50
)的曲线图,分别用每分钟计数(“cpm”)对代表性pde7抑制剂om69、om955、om956和om056的浓度作图来表示,浓度范围为0.61nm至10,000nm(对于om69、om955和om956),或者浓度范围为0.24nm至4,000nm(对于om056)。
[1621]
图3b、图4b、图5b和图6b是说明pde7b抑制活性(ic
50
)的曲线图,分别用每分钟计数(“cpm”)对代表性pde7抑制剂om69、om955、om956和om056的浓度作图来表示,浓度范围为0.61nm至10,000nm(对于om69、om955和om956),或者浓度范围为0.24nm至4,000nm(对于om056)。
[1622]
这些结果表明,om69和om056抑制pde7a和pde7b的功效高,而om955和om956抑制这两种酶的功效适中。om69、om056、om955和om956化合物在pde7a和pde7b之间几乎没有或者根本没有选择性。
[1623]
实施例2
[1624]
本实施例描述了用于测定pde7抑制剂选择性的一组实验,实验表明与所测定的所有其它磷酸二酯酶相比,om69(化合物1)、om056(化合物2)、om955(化合物3)和om956(化合物4)各自选择性地抑制pde7。这些实验包括了每个pde家族的代表,只是pde6除外。
[1625]
方法:
[1626]
用[3h]-camp作为pde 3a、4a、8a的底物,或者用[3h]-cgmp作为pde 1b、2a、5a、9a、10a和11a的底物,通过闪烁亲近测定法(spa,ge healthcare:产品编码trkq7100),测定了磷酸二酯酶活性。纯化的人pde1b、pde2a、pde3a、pde4a、pde5a、pde8a、pde9a和pde11a4得自bps bioscience,sandiego,ca。纯化的鼠pde10a也得自bps biosciences。要注意的是,鼠pde10a具有无法与人pde10区别开来的活动和抑制行为。因此,我们认为用鼠pde10a得到的结果可代表人pde10。
[1627]
将纯化的pde酶各自在含有25mm tris-cl(ph 8.0)、100mm nacl、0.05%吐温20、50%甘油和3mm dtt溶液中稀释保存。pde实验在下列实验混合物(终浓度)中进行:50mm tris-cl(ph 8.0)、8.3mm mgcl2、1.7mm egta、0.5mg/ml bsa、2.5%dmso和介于0.2ng和6ng之间的pde蛋白(取决于酶活性),最终体积为0.1ml。
[1628]
用pde7抑制剂om69(化合物1)、om056(化合物2)、om955(化合物3)和om956(化合物4)以4种浓度(10μm、1.25μm、0.156μm和0.019μm)按一式两份进行了实验。通过加入酶引发反应,在30℃下温育20分钟,然后通过加入50μl含有zn2+的spa微珠终止反应。
[1629]
混合物经震荡后,使之沉淀3小时。然后在平板计数器中,通过闪烁计数对由底物产生的[3h]-amp或[3h]-gmp进行了定量测定。将用净cpm值表示的结果用excel与4参数逻辑模型拟合。为了计算选择比,将用各个酶得到的ic
50
值除以实施例1
中用pde7a得到的ic
50
值。
[1630]
结果:
[1631]
表3表示用4种pde7抑制化合物针对一组受试pde所测定的选择性实验结果。表3中数值的单位用pde7a对于其它pde的选择性倍数表示,通过用针对所示pde的ic
50
值除以针对实施例1中的pde7a的ic
50
值来测定。因此,例如,对于om955与pde1b的值为342倍,是指与pde7a相比,om955作为pde1b抑制剂有效性降低342倍。括号中所表示的数字是实施例1的各种化合物针对pde7a的ic
50
值。表3中所提供的表示为“>”的选择性倍数值反映了其中最高浓度的化合物仅稍微抑制酶靶标(即小于50%)的情况,然而不能使用较高的浓度,因为化合物在实验混合物中变得不溶解。因此,仅可产生选择性的最小估计值。
[1632]
表3:代表性pde7抑制化合物的pde7a选择性
[1633][1634]
结果讨论:
[1635]
如表3所示,与pde1b、pde2a、pde3a、pde4a、pde5a、pde8a、pde9a、pde10a和pde11a相比,代表性pde7抑制剂om69、om056、om955和om956均对pde7a和pde7b有选择性。
[1636]
如表3所示,om69(化合物1)是pde7a(ic
50
=1.3nm)和pde7b(ic
50
=4.8nm)两者的强效抑制剂,与其它pde相比,抑制pde7a酶的选择性大于1000倍,且与其它pde相比,抑制pde7b酶的选择性大于250倍。
[1637]
表3进一步显示,om056(化合物2)是pde7a(ic
50
=5.7nm)和pde7b(ic
50
=9.27)两者的强效抑制剂,与其它pde相比,抑制pde7a酶的选择性大于400倍,且与其它pde相比,抑制pde7b酶的选择性大于200倍。
[1638]
表3进一步显示,om955(化合物3)是pde7a(ic
50
=51.8nm)和pde7b(ic
50
=106nm)两者的效力适中的抑制剂,与其它pde相比,抑制pde7a酶的选择性大于100倍,且与其它pde相比,抑制pde7b酶的选择性大于50倍。
[1639]
同样如表3所示,om956(化合物4)是pde7a(ic
50
=140nm)和pde7b(ic
50
=144nm)两者的效力适中的抑制剂,与其它pde相比,抑制pde7a和pde7b酶的选择性大于71倍。
[1640]
这些结果连同实施例4-7中所表示的结果一起,表明给予mptp小鼠模型这些化合物后,抑制pde7活性对于改善运动异常是必要的并且是充分的。此外,使用选择性pde7的抑制剂而不是非选择性pde抑制剂提供了较低毒性的优势,因为它们不会显著抑制已知引起
不良作用的pde(例如pde3和pde4)。
[1641]
实施例3
[1642]
本实施例描述了用于体内评价pde7抑制剂的代谢稳定性和pde7抑制剂分配到小鼠脑的能力的方法。
[1643]
动物试验方案
[1644]
小鼠品系:成年雄鼠c57bl/6j;退役种鼠,介于7-9月龄之间(jackson laboratory,bar harbor,me,usa),单独关养。
[1645]
给予化合物:将各pde7抑制剂(om69(化合物1)、om056(化合物2)、om955(化合物3)和om956(化合物4))溶于由dmso、0.03m磷酸和吐温80(5:95:0.2)组成的溶媒中,对于om69以10mg/kg,对于om56以10.0mg/kg和对于om955和om956以30.0mg/kg的剂量通过腹膜内途径注射。
[1646]
采集血样和脑样:临收获组织之前,小鼠用阿佛丁麻醉。在注射后15分钟、30分钟、60分钟、120分钟和240分钟每个时间点上,从3只动物中采集全脑或血浆样品。通过眶后放血来采集血样。制备血浆,并经离心去除红细胞。小鼠用盐水灌注去除污染血。取脑并在液氮中快速冷冻用于后续分析。将血浆和脑样品保存在-80℃下或干冰上,通过lc/ms/ms分析如下测定化合物的浓度:
[1647]
全脑组织使用mini-beaddeater-8tm(biospec products,bartlesville,ok)匀浆。血浆或均匀的脑样品用乙腈沉淀,并采用0.20微米滤板(varian corp,lake forest,ca)过滤到96孔板中。将滤液在氮气氛、50℃下蒸发至干,然后重配用于lc-ms分析。
[1648]
采用thermo ultra三重四级质谱仪(thermo fisher scientific,san jose,ca),利用电喷雾电离以数据采集多重反应监测模式,获得血浆和脑样品抑制化合物的定量测定值。将样品提取物注入2.1x 30mm xterra c18-ms高压液相色谱(hplc)充填柱(waters corp,milford,ma)中。采用thermo surveyor ms plus hplc四级泵系统(thermo fisher scientific,san jose,ca)和htc pal自动注射器(leap technologies,chapel hill,nc),供应从hplc柱洗脱化合物的流动相。
[1649]
结果:
[1650]
在每个时间点,从3小鼠中收集有关om69(化合物1)、om056(化合物2)、om955(化合物3)和om956(化合物4)的组织浓度数据,并取平均值。图7曲线图表示在240分钟时程期间化合物om69(化合物1)的血浆浓度、脑浓度和脑/血浆比率。如图7所示,在暴露的头15分钟内,在血浆(51ng/g)和脑组织(25ng/g)检测到om69。在腹膜内注射后30分钟观察到峰值水平(血浆59ng/g和脑44ng/g),之后血浆和脑水平均快速下降。在所有时间点上,当脑和血浆水平均大于lc/ms/ms实验(2ng/g)中的定量下限时,脑/血浆之比为45-75%。
[1651]
采用所述用于化合物om69的方法,对于各受试化合物,将脑内的总暴露量(曲线下面积:auc)除以血浆内的总暴露量,计算得出比率。结果见下表4。比值大于“1”表明化合物集中于脑内。表示为“》1”的值表明,在第一次测量时,脑内的化合物水平已较高。因此,在这些情况下比值为1表示最小估计值。
[1652]
表4:pde7抑制化合物的脑/血浆比
l-[3,5-3
h]酪氨酸+l-酪氨酸。溶媒对照为1%dmso。在温育缓冲液(125mm mes(ph 6.0)、0.5mg/ml过氧化氢酶、12.5mm dtt、0.5mm(6r)-5,6,7,8-四氢生物蝶呤)中,对10μm化合物om69进行了测定。将反应物在37℃下预温育15分钟。加入酪氨酸羟化酶,将反应物在37℃下温育20分钟。对[3h]-h2o进行了定量测定。在本实验中对阳性对照化合物α-甲基-l-p-酪氨酸进行了测定,其历史ic
50
值为20μm。
[1667]
4.多巴胺d1放射性配体结合实验
[1668]
按照dearry,a.等人所述方法(nature 347:72-76,1990)作下列修改后,进行了多巴胺d1放射性配体结合实验。使人重组多巴胺d1受体在cho细胞中表达。实验中所用的配体为1.4nm[3h]sch-23390。非特异性配体为10μm(+)-丁克吗。溶媒对照为1%dmso。在温育缓冲液(50mm tris-hcl(ph 7.4)、1.4mm抗坏血酸、0.001%bsa、150mm nacl)中对10μm化合物om69进行了测定。使反应物在37℃下温育2小时。对放射性配体结合进行了定量。实验中对阳性对照化合物r(+)-sch-23390进行了测定,其历史ic
50
值为1.4nm。
[1669]
5.多巴胺d
2l
放射性配体结合实验
[1670]
按照grandy,d.k.等人所述方法(pnas86:9762-9766,1989)作下列修改后,进行了多巴胺d
2l
放射性配体结合实验。使人重组多巴胺d
2l
受体在cho细胞中表达。实验中所用的配体为0.16nm[3h]-螺哌隆。非特异性配体为10μm氟哌啶醇。溶媒对照为1%dmso。在温育缓冲液(50mm tris-hcl(ph 7.4)、1.4mm抗坏血酸、0.001%bsa、150mm nacl)中对10μm化合物om69进行了测定。使反应物在25℃下温育2小时。对放射性配体结合进行了定量。本实验对阳性对照化合物螺哌隆进行了测定,其历史ic
50
值为0.26nm。
[1671]
6.多巴胺d3放射性配体结合实验
[1672]
按照sokoloff,p.等人所述方法(nature 347:146-151,1990)作下列修改后,进行了多巴胺d3放射性配体结合实验。使人重组多巴胺d3受体在cho细胞中表达。实验中所用的配体为0.7nm[3h]-螺哌隆(spiperone)。非特异性配体为25μm s(-)舒必利(sulpiride)。溶媒对照为1%dmso。在温育缓冲液(50mm tris-hcl(ph 7.4)、1.4mm抗坏血酸、0.001%bsa、150mm nacl)中对10μm化合物om69进行了测定。使反应物在37℃下温育2小时。对放射性配体结合进行了定量。本实验对阳性对照化合物螺哌隆进行了测定,其历史ic
50
值为0.36nm。
[1673]
7.多巴胺d
4.2
放射性配体结合实验
[1674]
按照van tol,h.h.m.等人所述方法(nature 350:610-614,1991)作下列修改后,进行了多巴胺d
4.2
放射性配体结合实验。使人重组多巴胺d
4.2
受体在cho-k1细胞中表达。实验中所用的配体为0.5nm[3h]-螺哌隆。非特异性配体为10μm氟哌啶醇。溶媒对照为1%dmso。在温育缓冲液(50mm tris-hcl(ph 7.4)、1.4mm抗坏血酸、0.001%bsa、150mm nacl)中对10μm化合物om69进行了测定。使反应物在25℃下温育2小时。对放射性配体结合进行了定量。本实验对阳性对照化合物螺哌隆进行了测定,其历史ic
50
值为0.5nm。
[1675]
8.多巴胺d5放射性配体结合实验
[1676]
按照sunahara,r.k.等所述方法(nature 350:614-619,1991)作下列修改后,进行了多巴胺d5放射性配体结合实验。使人重组多巴胺d5受体在cho细胞中表达。实验中所用的配体为2nm[3h]-sch-23390。非特异性配体为10μm氟哌噻吨(flupentixol)。溶媒对照为1%dmso。在温育缓冲液(50mm tris-hcl(ph 7.4)、1.4mm抗坏血酸、0.001%bsa、150mm nacl)中对10μm化合物om69进行了测定。使反应物在37℃下温育2小时。对放射性配体结合进行了
定量。在本实验中对阳性对照化合物r(+)-sch23390进行了测定,其历史ic
50
值为1.5nm。
[1677]
9.加巴喷丁放射性配体结合实验
[1678]
按照gee,n.s.等人所述方法(j.biol.chem.271(10):5768-5776,1996)作下列修改后,进行了加巴喷丁放射性配体结合实验。从wistar大鼠脑皮质获得加巴喷丁。实验中所用的配体为0.02μm[3h]加巴喷丁。非特异性配体为100μm加巴喷丁。溶媒对照为1%dmso。在温育缓冲液(10mm hepes(ph 7.4))对10μm化合物om69进行了测定。使反应物在25℃下温育30分钟。对放射性配体结合进行了定量。在本实验中对阳性对照化合物加巴喷丁进行了测定,其历史ic
50
值为0.04μm。
[1679]
10.多巴胺转运蛋白(dat)放射性配体结合实验
[1680]
按照giros,b.等人所述方法(trendspharmacolsci 14:43-49,1993)作下列修改后,进行了多巴胺转运蛋白(dat)放射性配体结合实验。使人重组dat在cho-k1细胞中表达。实验中所用的配体为0.15nm[
125
i]-rti-55。非特异性配体为10μm诺米芬辛(nomifensine)。溶媒对照为1%dmso。在温育缓冲液(50mm tris-hcl(ph 7.4)、100mm nacl、1μm亮抑酶肽、10μm pmsf)中对10μm化合物om69进行了测定。使反应物在4℃下温育3小时。对放射性配体结合进行了定量。在实验中对阳性对照化合物gbr-12909进行了测定,其历史ic
50
值为1.7nm。
[1681]
11.腺苷a
2a
受体放射性配体结合实验
[1682]
按照varani,k.等人所述方法(br.j.pharmacol.117:1693-1701,1996)作下列修改后,进行了腺苷a
2a
受体放射性配体结合实验。使人重组a
2a
受体在hek-293细胞中表达。实验中所用的配体为0.05μm[3h]cgs-21680。非特异性配体为50μm neca。溶媒对照为1%dmso。在温育缓冲液(50mm tris-hcl(ph 7.4)、10mm mgcl2、1mm edta、2u/ml腺苷脱氨酶)中对10μm化合物om69进行了测定。使反应物在25℃下温育90分钟。对放射性配体结合进行了定量。在本实验中对阳性对照化合物cgs-21680进行了测定,其历史ic
50
值为0.13μm。
[1683]
结果:
[1684]
上述实验结果概括于下表5。在标记为“om69”一栏中的括号内标出了在实验中所用的om69的量。结果用抑制配体结合靶标或抑制靶标的酶促活性的百分比表示。阳性对照按规定包括在各实验中,作为对实验性能的检查。
[1685]
表5:用代表性pde7抑制剂om69(化合物1)对已知帕金森病靶标的抑制水平
[1686][1687]
结果讨论:
[1688]
表5中的结果表明,即使在其pde7b的ic
50
为2000倍以上的浓度下,代表性pde7抑制剂om69也基本不抑制comt、酪氨酸羟化酶、多巴胺转运蛋白、加巴喷丁受体或任何多巴胺受体亚型。
[1689]
至于腺苷a
2a
受体结果,尽管表5中的结果显示在10μm的浓度下,om69抑制与a
2a
受体结合达41%,但是至少出于下列原因,我们认为a
2a
不是om69的靶标。如实施例3所示,在1mg/kg剂量之后,om69在小鼠脑达到30-60ng/g的水平。假设为线性药代动力学特征,则如实施例5和实施例6中所表明的一样,在小鼠mptp模型中是非常有效的0.1mg/kg剂量,会产生脑内水平3-6ng/g,这相当于3-6ng/ml或8.5-17nm。在此浓度下,使用om69的ic
50
保守值10μm,a
2a
受体所占百分比可能仅约为0.17%。因此,结论是a
2a
不是om69的靶标。
[1690]
至于mao-b实验,在1μm浓度下的百分比抑制27%可表明ic
50
值约为2.7μm。因此,如果om69以17nm的浓度存在于脑内(同前述段落讨论的一样),则它可能抑制的mao-b小于0.7%。因此,结论是mao-b不是om69的靶标。
[1691]
实施例5
[1692]
mptp帕金森病模型中代表性pde7抑制化合物的药理学评价
[1693]
在本实施例中,在小鼠mptp帕金森病模型的初步研究中对代表性pde7抑制剂om69(化合物1)进行了评价。
[1694]
动物试验方案
[1695]
小鼠品系:成年雄鼠c57bl/6j;退役种鼠,介于7-9月龄之间(jackson laboratory,bar harbor,me,usa),单独关养。
[1696]
处理:注射前2周每天对动物进行处理,以便使之适应行为测试。所有小鼠在标准12小时光/暗周期下喂养,并使之任意得到实验室颗粒饲料和水。
[1697]
给予mptp:按15mg/kg(游离碱)的剂量,使小鼠接受2次甲基苯基四氢吡啶(“mptp”)皮下注射,注射间隔为10-12小时。给予对照小鼠(假损伤组)盐水而不是mptp。
[1698]
给予pde7抑制化合物:给予mptp或盐水后7天,给予小鼠om69。对于各剂量,在100%dmso中制备om69化合物的100x母液,接着将100x稀释到磷酸缓冲盐溶液(pbs)中。然后将100μl该1x溶液经腹膜内(ip)注射给药。
[1699]
之前的药代动力学研究测出,om69在12小时内清除出小鼠体内,因此可利用连续数天给予各种试验化合物,使得可从各组损伤动物获得的信息最大化。
[1700]
步长:用mptp或盐水注射前,预先训练动物不停地走过清洁纸张进入其关养笼。为了评价步长,将动物的前爪放入黑色墨水中,测量在正常行走期间前爪步伐(仅直线)的长度。在完成此项任务后,立即将动物放回其关养笼中。通过测量身体同侧每步间的距离,从第一步的中趾测到第二步的足跟,求出步长。计算至少4步清晰步伐的平均值。
[1701]
实验方案
[1702]
雄性退役种小鼠(12-15只/组,共约48-60只动物)用盐水或2x 15mg/kg mptp(12只盐水;36只mptp)处理诱发帕金森病状态(parkinsonian state)(多巴胺的神经化学性(neurochemical)丧失伴随相应的行为缺陷)。概括说明该方案的流程图见图8。
[1703]
如图8所示,在第一天,测量了基线步长,然后小鼠用mptp(n=9)或用盐水进行处理作为对照(n=10)。在第8天,再次测量步长,从盐水处理动物中得到步长的对照值。在此次测量后,在第8天给予左旋多巴(5mg/kg)或om69(0.1mg/kg)进行首次治疗,见图8。给药后20分钟,测量每只动物的步长。左旋多巴(5mg/kg)或om69(0.1mg/kg)均不改变盐水处理小鼠的步长。图8中所显示的给药治疗在连续几天内进行,每次给药后20分钟都测量了步长。如上所述,之前的药代动力学研究测出om69在给药后12小时内由小鼠体内清除出,因此认为给药后20分钟进行的步长试验所测量的只是治疗当天给予的化合物的作用。
[1704]
如图8所示,在第8天,第1组动物用左旋多巴(5mg/kg)治疗,第2组动物用om69(0.1mg/kg)治疗。在第九天,第1组动物用左旋多巴(1mg/kg)治疗,第2组动物用om69(0.01mg/kg)治疗。在第十天,将治疗组交叉,第1组动物用om69(0.01mg/kg)治疗,第2组动物用左旋多巴(1mg/kg)治疗。
[1705]
再如图8所示,对于组合药物研究,将第1组和第2组中的所有小鼠合在一起。在给予mptp后第11天,第1组和第2组的所有动物(n=9)用左旋多巴(1mg/kg)和om69(0.01mg/kg)联合治疗,在给药后20分钟进行了步长试验。在给予mptp后第12天,第1组和第2组的所有动物(n=9)用左旋多巴(1mg/kg)和om69(0.03mg/kg)联合治疗,在给药后20分钟进行了
步长试验。在第11天和第12天进行的这些组合药物研究中,无法得到9只动物中2只的平均步长数据,因为这2只动物或者是跑动而不是按正常速度行走,或者时走时停而不是连续行走。因此,对于组合药物研究,这2只动物不包括在内,仅给其余7只动物(n=7)评分。
[1706]
步长增加
[1707]
用于本实施例的mptp模型是普遍认可的小鼠pd模型,本领域技术人员认为可用来预测人类pd(tillerson,j.l.等,exp.neurol.178(1):80-90,2002;tillerson,j.l.等,j.neurosci methods123(2):189-200,2003)。
[1708]
为了评价或比较在这些剂量下om69和左旋多巴的效果,将给予同等剂量的小鼠数据合并。因此,采用由所有9只动物中得到的测量值,求出用左旋多巴(1mg/kg)治疗的动物的平均步长。同样,采用由所有9只动物中得到的测量值,求出用om69(0.01mg/kg)治疗的动物的平均步长。本实施例所述mptp研究的结果见图9-11。
[1709]
图9-11是表示墨水涂染爪步长测试结果的柱状图。这些柱状图表明,代表性pde7抑制剂om69(化合物1)加大mptp损伤小鼠的步长,且与左旋多巴相比,在显著较低剂量下加大步长。
[1710]
如图9和图11所示,与盐水对照比较起来,用mptp处理的小鼠缩短其步长,通过从墨水涂染小鼠爪留下的足迹来测量。参见图9(#1对#4)和图11(#1对#2)。
[1711]
如图9和图10所示,用左旋多巴治疗的mptp处理小鼠以剂量依赖性方式加大其步长。参见图9(#4对#5和#6)和图10(#1对#2和#6)。
[1712]
预料不到地测出,代表性pde7抑制剂om69(化合物1)当腹膜内给予0.1mg/kg时,在mptp处理小鼠中具有与50倍高剂量的左旋多巴(5mg/kg)相同的加大步长的效果。如图9所示,在上述浓度下,左旋多巴(#5)和om69(#7)两者完全恢复到未用mptp处理所观察到的步长(#1)。这些结果证实了预料不到的发现结果,即用于本发明方法的代表性pde7抑制剂om69(化合物1)在与左旋多巴相比显著较低的剂量下,有效加大mptp小鼠的步长。
[1713]
如图9-11所示,在较低的左旋多巴(1mg/kg)(参见图9,#6;图10,#2)或om69(0.01mg/kg)(参见图9,#8;图10,#3)剂量下,在mptp处理小鼠的步长方面只观察到较小作用。然而,比起两种单独的药物所观察到的步长加大合计数,当这些较低剂量的om69和左旋多巴联合给予时,步长的加大作用趋于更大。参见图9,#9和图10,#4。这些结果证实,在mptp受损小鼠中,按与左旋多巴相比显著较低的剂量给予代表性pde7抑制剂om69(化合物1)有效地加大步长,因此适用于旨在治疗与运动障碍病理有关的运动异常例如帕金森病的本发明方法。
[1714]
实施例6
[1715]
mptp帕金森病模型中代表性pde7抑制化合物的药理学评价:验证性研究
[1716]
在本实施例中,在小鼠mptp帕金森病模型中对代表性pde7抑制剂om69(化合物1)进行了评价。本研究被设计来证实实施例5的研究结果,并提供om69效果的统计依据。
[1717]
动物试验方案。
[1718]
小鼠品系:成年雄鼠c57bl/6j;退役种鼠,介于7-9月龄之间(jackson laboratory,bar harbor,me,usa),单独关养。
[1719]
处理:注射前2周每天对动物进行处理,以便使之适应行为测试。所有小鼠在标准12小时光/暗周期下喂养,并使之任意得到实验室颗粒饲料和水。
[1720]
给予mptp:按15mg/kg(游离碱)的剂量,使小鼠接受2次甲基苯基四氢吡啶(“mptp”)皮下注射,注射间隔为10-12小时。给予对照小鼠(假损伤组)盐水而不是mptp。
[1721]
给予om69(化合物1):给予mptp/盐水后7天,给予小鼠om69。对于各剂量,在100%dmso中制备om69的100x母液,接着将100x稀释到磷酸缓冲盐溶液(pbs)中。然后将100μl该1x溶液经腹膜内(ip)注射给药。
[1722]
步长:用mptp或盐水注射前,预先训练动物不停地走过清洁纸张进入其关养笼。为了评价步长,将动物的前爪放入黑色墨水中,测量在正常行走期间前爪步伐(仅直线)的长度。在完成此项任务后,立即将动物放回其关养笼中。通过测量身体同侧每步间的距离,从第一步的中趾测到第二步的足跟,求出步长。计算至少4步清晰步伐的平均值。
[1723]
统计分析
[1724]
应用prism 4.0软件对数据进行了分析。通过单向方差分析及适当时通过student-newman-keuls事后检验对各组进行了分析。达到p《0.05被视为显著。表6中有统计差异的步长图的治疗组用字母注明。有相同字母的组没有统计差异;没有相同字母的组彼此有显著差异。
[1725]
实验方案:
[1726]
雄性退役种小鼠(共28只动物)用2x 15mg/kg mptp(12只盐水;36只mptp)处理诱发帕金森病状态(多巴胺的神经化学性丧失伴随相应的行为缺陷)。概括说明该方案的流程图见图12。在第1天,测量了基线步长,然后所有小鼠用mptp或盐水处理。在第8天,再次测量步长,然后将mptp处理动物随机分成两组,每组14只。如实施例5所示,在连续几天内按图12中规定的连续疗法给药,在每种情况下,都在给药后20分钟测定了步长。流程图中框内的“n”值表示在当天获得有效数据的动物的编号(偶尔少数几只动物未能执行步行任务)。
[1727]
在第9天,第1组动物用om69(0.01mg/kg)治疗,第2组用左旋多巴(1mg/kg)治疗。在第10天,第1组动物用om69(0.03mg/kg)治疗,第2组用左旋多巴(5mg/kg)治疗。在第11天,第1组动物用om69(0.05mg/kg)治疗,第2组用om69(0.1mg/kg)治疗。在第12天,第1组用om69(0.07mg/kg)治疗。
[1728]
接着,对于组合药物研究,将两组合在一起。在第14天,所有动物均用左旋多巴(1mg/kg)和om69(0.01mg/kg)两者治疗。在第15天,所有动物用左旋多巴(1mg/kg)与较高剂量的om69(0.03mg/kg)联合治疗。
[1729]
结果:
[1730]
这些研究的结果概括于表6中,其中对用左旋多巴按两种不同剂量、用om69(化合物1)按5种不同剂量、以及左旋多巴和om69的联合治疗的mptp处理小鼠的步长与未处理对照小鼠和未接受药物的mptp处理小鼠的进行了比较。图18表明了这一子集的数据。
[1731]
表6:mptp损伤小鼠的步长
[1732]
[1733][1734]
结果讨论:
[1735]
om69与左旋多巴的比较:
[1736]
参照表6,在本实验中,mptp引起步长显著缩短(字母“b”与“a”)。用1mg/kg左旋多巴治疗不会导致步长显著加大,但是用5mg/kg左旋多巴治疗使步长回复到对照(未损伤)值(“a”与“a”)。用低剂量om69(0.01mg/kg或0.03mg/kg)治疗同样不加大步长,但用中等剂量治疗则导致统计显著性地(p《0.05)加大步长(“c”与“b”)。用较高剂量(0.1mg/kg)的om69治疗也导致统计显著性地(p《0.05)加大步长,而且,使步长回复到对照(未损伤)值(“a”与“a”)。这些结果证实,pde7抑制化合物om69在恢复mptp处理小鼠的步长中与左旋多巴一样有效,而且效价强度比左旋多巴的大约50倍。
[1737]
联合给予om69和左旋多巴:
[1738]
在本实验中还测试了om69与左旋多巴的两种剂量组合。第一组合(1mg/kg左旋多巴加上0.03mg/kg om69)引起步长统计显著性地(p《0.05)加大1.13cm,大于在该剂量下单独使用各药物时非显著性加大的总和(0.35cm)。第2组合(1mg/kg左旋多巴加上0.05mg/kg om69)同样导致步长统计显著性地(p《0.05)加大,而且,使步长回复到对照(未损伤)值(“a”与“a”)。这一加大的幅度(1.58cm)在统计上显著(p《0.0001)大于用0.05mg/kg om69的加大值加上用1mg/kg左旋多巴非显著性加大值(0.8cm)的总和,如图18的图中右手侧理论累加柱所表示。这些结果强有力的表明,pde7抑制化合物om69和左旋多巴以大于累加(greater-than-additive)的方式相互作用以纠正mptp处理小鼠的步长。
[1739]
实施例7
[1740]
mptp帕金森病模型中一组代表性pde7抑制化合物的药理学评价
[1741]
在本实施例中,在小鼠mptp帕金森病模型中对一组代表性pde7抑制剂进行了评价,以便检验这一假说,即不论所用抑制剂的具体化学结构如何,抑制pde7均可改善该模型的帕金森症状,因此,抑制pde7便足以产生所观察到的步长改进。
[1742]
动物试验方案
[1743]
小鼠品系:成年雄鼠c57bl/6j;退役种鼠,介于7-9月龄之间(charles river laboratories,wilmington,ma),单独关养。相隔一周连续对3组共35只小鼠进行了试验。
[1744]
处理:注射前2周每天对动物进行处理,以便使之适应行为测试。所有小鼠在标准12小时光/暗周期下喂养,并使之任意得到实验室颗粒饲料和水。
[1745]
给予mptp:按15mg/kg(游离碱)的剂量,使小鼠接受2次甲基苯基四氢吡啶(“mptp”)皮下注射,注射间隔为10-12小时。给予对照小鼠(假损伤组)盐水而不是mptp。
[1746]
步长:用mptp或盐水注射前,预先训练动物不停地走过清洁纸张进入其关养笼。为了评价步长,将动物的前爪放入黑色墨水中,测量在正常行走期间前爪步伐(仅直线)的长度。在完成此项任务后,立即将动物放回其关养笼中。通过测量身体同侧每步间的距离,从第一步的中趾测到第二步的足跟,求出步长。计算至少4步清晰步伐的平均值。本实验的时间表如下:
[1747]
第0周--步伐训练任务:(至少4段时间)
[1748]
天1天--采集基线步长
[1749]
第2天a--采集第二次基线步长
[1750]
第2天b--mptp注射
[1751]
第7天--采集并筛选步伐缺陷
[1752]
第8天a--采集第二次缺陷步伐
[1753]
第8天b--开始化合物试验
[1754]
第9天及以后--连续几天给予不同剂量的化合物。
[1755]
给予化合物:由于这些化合物间的水溶性在大范围内变化,所以需要许多不同的剂型。给予100μl左旋多巴的磷酸缓冲盐溶液。对于用左旋多巴治疗的动物,在左旋多巴治疗之前15分钟,给予多巴脱羧酶抑制剂苄丝肼(benserazide)(100μl的12.5mg/kg溶液)以使外周血中左旋多巴的破坏最小化。给予om955(化合物3)和om056(化合物2)200μl二甲基乙酰胺:聚乙二醇400:0.03m甲磺酸(dma:peg:msa-10%:40%:50%)溶液。给予200μl om956(化合物4)的0.03m酒石酸(ta)溶液。图13a和图13b显示,当这些溶媒的任一种单独给予时,不改变mptp处理小鼠的步长。因此,所观察到的治疗效果仅是由pde7抑制化合物本身引起的。对于各受试化合物,进行了初步实验以鉴定在mptp模型中的最有效剂量。在某些情况下,观察到高于最佳剂量的剂量不会引起步长显著加大。在使用左旋多巴的mptp处理小鼠中也观察到这种“作用过度(overshoot)”现象,其中较低剂量改善活动减退,而较高剂量诱发反常运动或活动不受控制(lundbladm.等,exp neurol.194(1):66-75(2005);pearce r.k.等,mov disord.10(6):731-40(1995);fredriksson,a.等,pharmacol-toxicol.67(4):295-301(1990))。
[1756]
实验方案:
[1757]
雄性退役种小鼠用盐水或2x 15mg/kg mptp处理以诱发帕金森病状态(多巴胺的神经化学性丧失伴随相应的行为缺陷)。在第1天,测量了基线步长,然后小鼠用mptp或用盐水作为对照处理。在第8天,再次测量步长,从盐水处理动物中得到步长的对照值。然后将动物随机分成治疗组。对0.1mg/kg、0.5mg/kg和0.1mg/kg化合物om955(化合物3)与1mg/kg左旋多巴的组合进行了测定。对0.1mg/kg和0.5mg/kg的化合物om956(化合物4)进行了测定。对0.05mg/kg、0.1mg/kg、0.5mg/kg和1mg/kg的化合物om056(化合物2)进行了测定。结果:
[1758]
本研究的结果见图14-17。图14显示在0.5mg/kg剂量下,om955引起mptp处理小鼠的步长呈统计显著性的改善(p《0.005,与mptp组相比)。在注射后20分钟和1小时,步长完全
恢复到未损伤动物的步长。图15a-c证实om955和左旋多巴的组合发挥出超过累加效应的作用以恢复步长。图15a和图15b分别显示,当单独给予低剂量左旋多巴(1mg/kg)或om955(0.1mg/kg)时不会加大步长。然而,图15c显示当同时给予这些低剂量时,步长完全恢复到未损伤动物的步长(p《0.005,与mptp组相比)。
[1759]
图16显示,剂量同样为0.5mg/kg的om956引起mptp处理动物的步长呈统计显著性的加大(p《0.01,与mptp组相比)。图17显示0.05mg/kg低剂量的om056也同样引起mptp处理动物的步长呈统计显著性的加大(p《0.05,与mptp组相比)。
[1760]
本实施例的结果表明,彼此在结构上不相关且结构上与om69不相关的3种不同的pde7抑制化合物,全都引起mptp处理动物的步长与用om69的观察结果(见实施例5和实施例6)相同的完全恢复。由于这些化合物唯一已知的共同特征是抑制pde7的能力,并且由于测试了这些化合物之一(om69)与其它已知帕金森病靶标的相互作用,并发现与这些靶标无显著的相关作用,因此得出的结论是,pde7抑制活性在用这些化合物报告的步长改进中既是必要的又是充分的。
[1761]
虽然举例说明和描述了示例性实施方案,但是要了解的是在不偏离本发明的精神和范围的情况下,可对其作各种变动。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1