一种筛选含铋合金熔接材料的方法及含铋合金熔接材料与流程
1.本发明涉及熔接材料的筛选技术领域,具体涉及一种筛选含铋合金熔接材料的方法及含铋合金熔接材料。
背景技术:
2.对于通过封装和组装形成的电路而言,焊料合金的性能对整个电路的运行具有举足轻重的作用。在微电子连接中,对焊料合金材料的研究主要在于具有较低的熔点,优良的流动性和润湿性,足够的导电性、抗蚀性和机械强度等。对于温度敏感的材料,比如耐热性较差的电子元器件、温度传感器等设备进行封装连接时,就需要在低温下进行焊接,此时传统的焊料合金就表现出了局限性。sn-bi基钎料从工艺条件、钎料成本、机械性能、可靠性等方面都表现比较好,因此在低温无铅钎料中应用最广泛,不过sn-bi基钎料在加工过程中存在脆性大、延展性低和bi偏析等时效问题需要改进。研究表明进一步添加微量合金元素ag、cu、zn等可以细化组织,改善sn-bi基无铅钎料的力学性能,加入一定量的bi元素,可以降低熔点、改善润湿性。因此亟需寻找新型低熔点共晶含铋熔接材料,以解决上述问题,然而,研发新型焊料熔接材料是一项昂贵而耗时的工作,因此需要一种新的方法来提高研发效率,降低研发成本。
技术实现要素:
3.本发明一种筛选含铋合金熔接材料的方法,包括以下内容:
4.步骤1,获取sn-bi-tm体系中存在的二元晶体结构,tm代表sb、ag、zn、cu中的一种;
5.步骤2,采用基于密度泛函理论的第一性原理对各二元晶体结构进行低精度到高精度的结构优化,计算各二元晶体结构的基本物相信息;
6.步骤3,基于高精度优化后的各二元晶体结构,计算各二元晶体结构的声子谱;
7.步骤4,基于准简谐近似方法,计算各二元晶体结构的热学性能,得到热力学数据;
8.步骤5,采用步骤4得到的热力学数据来评估calphad模型参数,建立sn-bi-tm体系的热力学模型;
9.步骤6,基于热力学模型得到各二元晶体结构的液相投影面,基于液相投影面找到共晶点和温度,筛选出含铋合金熔接材料。
10.本发明优选的实施方式在于,所述的步骤3包括:设置参数“isif=2,ibrion=8”,利用phonopy有限位移法逐个对各二元晶体结构进行声子谱计算,筛选出声子谱无虚频的二元晶体结构。
11.本发明优选的实施方式在于,在步骤5中,calphad模型如下:
12.化合物在不同温度下的吉布斯能如下所示:
[0013][0014]
其中a,b,c,d,e和f是模型参数,由第一性原理的准简谐方法计算得到的热力学数据评估得到,h
ser
是最稳定单质在298.15k和1bar下的焓作为参考态。
[0015]
本发明优选的实施方式在于,所述步骤5进一步包括:
[0016]
液相的吉布斯能表达式为:
[0017][0018]
其中yi是组元i在液相中的摩尔分数,
xsgl
是过剩吉布斯能相,代表纯液相的吉布斯能,过剩吉布斯能
xsgl
的形式为:
[0019][0020]
其中是组元i和j之间的vth阶交互作用参数,由下式得到:
[0021][0022]
模型参数
v,liq
a和
v,liq
b由实验的热力学数据和液相相关的相边界数据评估得到。
[0023]
本发明优选的实施方式在于,在步骤5中,sn-bi-zn体系的热力学模型为:
[0024][0025]
sn-bi-ag体系的热力学模型为:
[0026][0027]
本发明优选的实施方式在于,还包括步骤7,对筛选出的含铋合金熔接材料进行结构优化,利用应力应变法进行力学性能计算,包括计算硬度,以及利用金属直流电导率公式计算电导率。
[0028]
本发明方法可以克服传统“试错法”的不足,节约资源和时间,基于高通量的第一性原理计算结合calphad模型,建立sn-bi-tm体系的热力学模型。筛选出具有低熔点共晶成分的三元合金,而且通过实验验证,误差范围很小,提高新型含铋三元合金熔接材料的研发效率,降低研发成本。
[0029]
本发明还公开了含铋合金熔接材料,采用前述任一项筛选含铋合金熔接材料的方法筛选得到。
[0030]
进一步地,本发明的含铋合金熔接材料,包括sn:bi:ag的成分比为(32.51-38.54):(44.70-52.68):(13.79-16.88)的含铋合金熔接材料,sn:bi:zn的成分比为(20.76-34.09):(49.54-55.31):(10.64-14.06)的含铋合金熔接材料。
[0031]
进一步地,筛选出的sn:bi:zn的成分比为(20.76-34.09):(49.54-55.31):(10.64-14.06)的含铋合金熔接材料的共晶点温度为128.68-135.76℃;筛选出的sn:bi:ag的成分比为(32.51-38.54):(44.70-52.68):(13.79-16.88)的含铋合金熔接材料的共晶点温度为132.87-140.65℃。
[0032]
与其它合金熔接材料相比,筛选的sn-bi-zn和sn-bi-ag低熔点含铋共晶熔接材料的优势如下:
[0033]
(1)sn-bi-zn和sn-bi-ag低熔点含铋共晶熔接材料的成分属于首次发现。
[0034]
(2)sn-bi-zn和sn-bi-ag低熔点含铋共晶熔接材料的熔点很低,在106.04℃—143.85℃范围;
[0035]
sn-bi-zn和sn-bi-ag低熔点含铋共晶熔接材料是共晶合金,没有固液两相区,无
粘结现象,加工方便,造粉质量高。
附图说明
[0036]
附图1为本发明筛选含铋合金熔接材料的方法的流程图;
[0037]
附图2为sn-bi-zn体系的液相投影面;
[0038]
附图3为sn-bi-ag体系的液相投影面。
具体实施方式
[0039]
下面结合附图和具体实施方式对本发明作进一步详细说明。
[0040]
本发明基于目前开放的数据库以及相关文献报道获取sn-bi-tm体系中存在的晶体结构,共获得了78种晶体结构,其中有40种二元晶体结构。其中tm代表sb、ag、zn、cu中的一种,开放的数据库是指materials project、oqmd、springer materials、icsd和nist。sn-bi-tm体系指包含sn、bi和tm三种元素种的一种或两种的晶体结构。
[0041]
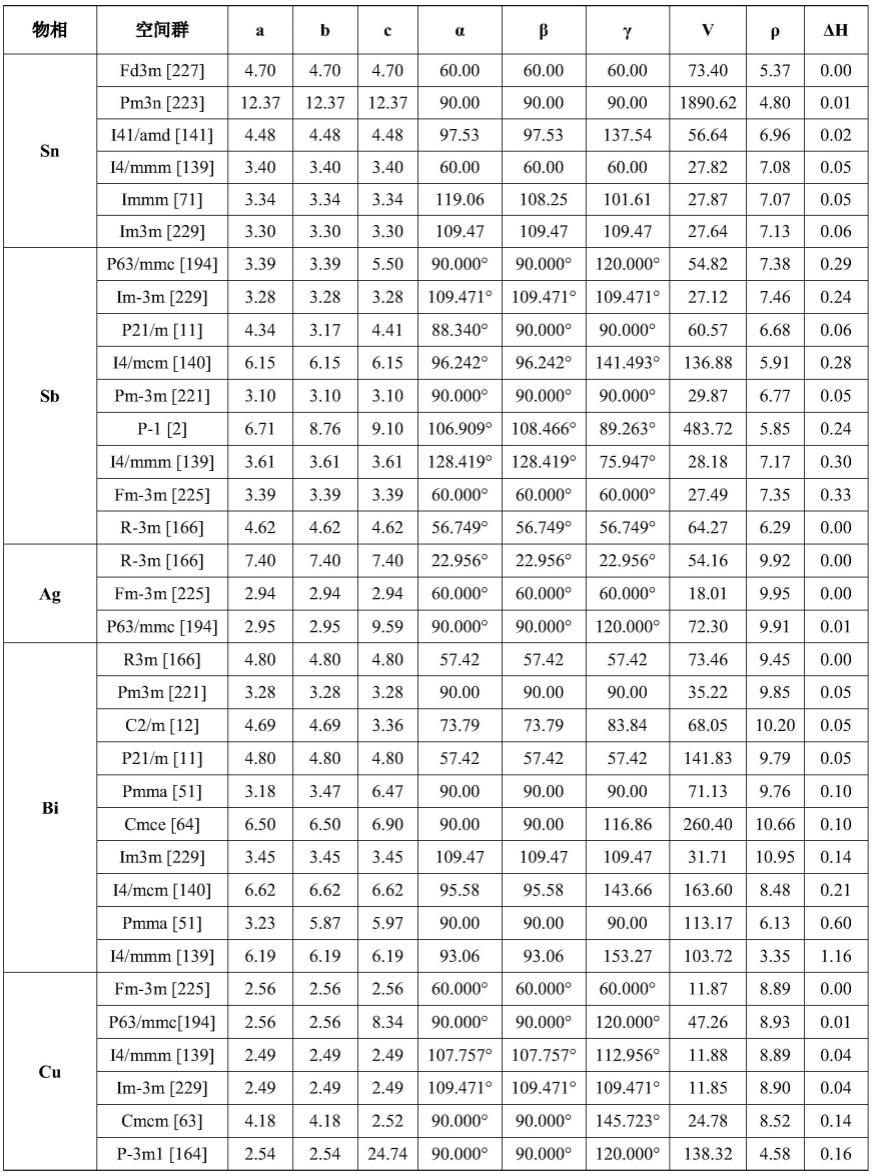
(1)采用基于密度泛函理论的第一性原理对78种晶体结构进行低精度到高精度的结构优化,通过vasp计算各晶体结构的基本物相信息,获得了sn-bi-tm体系中78种晶体结构的基本物相信息,包括晶格常数、空间群、体积、密度以及形成焓,计算结果如表1所示。表1中,a、b、c、α、β、γ代表晶格常数,δh代表形成焓。
[0042]
表1
[0043]
[0044]
[0045][0046]
(2)基于高精度优化后的40种二元晶体结构,使用phonopy软件计算这40种二元晶体结构的声子谱。
[0047]
(3)基于准简谐近似方法,即qha方法,计算各二元晶体结构的热学性能,得到热力学数据。
[0048]
(4)基于准简谐近似方法得到的热力学数据来评估calphad模型参数,最后建立sn-bi-tm体系的热力学模型,基于热力学模型得到各二元晶体结构的液相投影面,基于液相投影面找到共晶点和温度,筛选出含铋合金熔接材料。
[0049]
与其它合金熔接材料相比,筛选并实验验证的sn-bi-zn和sn-bi-ag低熔点含铋共晶熔接材料的优势如下:
[0050]
(1)sn-bi-zn和sn-bi-ag低熔点含铋共晶熔接材料的成分属于首次发现。
[0051]
(2)sn-bi-zn和sn-bi-ag低熔点含铋共晶熔接材料的熔点很低,在106.04℃—143.85℃范围,成分和熔点如表2所示。
[0052]
表2
[0053]
序号体系成分/wt%计算值/℃实验值/℃1bi-sn-znbi:sn:zn=53.31:34.09:12.60131.68133.502ag-bi-snag:bi:sn=13.79:49.70:36.51136.87137.70
[0054]
(3)sn-bi-zn和sn-bi-ag的硬度和电导率计算值与实验值符合,如表3和表4所示。
[0055]
表3
[0056]
编号合金名称计算值实验值-ms/m1bi-sn-zn2.892.992ag-bi-sn2.502.44
[0057]
表4
[0058]
编号合金名称计算值实验值-hbw1bi-sn-zn14.0114.102ag-bi-sn28.5828.60
[0059]
(4)sn-bi-zn和sn-bi-ag低熔点含铋共晶熔接材料是共晶合金,没有固液两相区,无粘结现象,加工方便,造粉质量高。
[0060]
下面分别以从sn-bi-ag体系和sn-bi-zn体系中筛选含铋合金熔接材料的方法,来详细的阐述本发明筛选含铟合金熔接材料的具体步骤。从其他体系中筛选含铋合金熔接材料的方法与这两种体系的筛选方法相同。
[0061]
实施例一
[0062]
本实施例筛选含铋合金熔接材料的方法,步骤如下:
[0063]
步骤1,基于目前开放的数据库以及相关文献报道获取sn-bi-zn体系中存在的二元晶体结构,包括sn-bi二元晶体结构、bi-zn二元晶体结构和sn-zn二元晶体结构。其中,sn-bi-zn体系指包含sn、bi和zn三种元素中的一种或两种的晶体结构,sn-bi二元晶体结构指包含bi和sn两种元素的二元晶体结构,bi-zn二元晶体结构指包含bi和zn两种元素的二元晶体结构,sn-zn二元晶体结构指包含sn和zn两种元素的二元晶体结构。
[0064]
步骤2,采用基于密度泛函理论的第一性原理计算方法,通过vasp软件,对初始的sn-bi二元晶体结构、bi-zn二元晶体结构和sn-zn二元晶体结构,进行低精度的结构优化,然后分别对低精度优化后的sn-bi二元晶体结构、bi-zn二元晶体结构、sn-zn二元晶体结构进行高精度的结构优化,基于高精度优化后的sn-bi二元晶体结构、bi-zn二元晶体结构、sn-zn二元晶体结构,分别计算基本物相信息,基本物相信息包括形成焓,计算结果如表1所示。步骤2具体包括:
[0065]
步骤2.1,选取合适的关键性参数,有助于提高计算的准确性和高效性,而截断能和k点网格的密度的选取就显得至关重要。利用vaspkit功能102生成0.03精度的k点网格,截断能选用potcar文件中元素enmax的1.5倍。
[0066]
步骤2.2,首先设置参数“isif=3,ibrion=2”,分别对sn-bi二元晶体结构、bi-zn二元晶体结构、sn-zn二元晶体结构进行低精度结构优化,能量的收敛准则为10-6
ev,力的收敛准则为达到收敛标准。
[0067]
步骤2.3,低精度结构优化完成后,针对低精度结构优化后的sn-bi二元晶体结构、bi-zn二元晶体结构、sn-zn二元晶体结构再次进行高精度的结构优化,能量的收敛准则为10-8
ev,力的收敛准则为
[0068]
步骤2.4基于高精度优化后的sn-bi二元晶体结构、bi-zn二元晶体结构、sn-zn二元晶体结构,利用形成焓计算公式计算各二元晶体结构的形成焓,以作为calphad模型的参数。以sn-bi二元晶体结构为例,形成焓计算公式如公式(1)所示。
[0069][0070]
式中,δh代表形成焓,e
total
代表sn-bi二元晶体结构的总能量,e
bi
和e
sn
分别表示单质bi和sn的能量。x和y分别表示单质bi和sn在sn-bi二元晶体结构的原子数目。
[0071]
步骤3,基于高精度优化后的sn-bi二元晶体结构、bi-zn二元晶体结构、sn-zn二元晶体结构,使用phonopy软件分别对sn-bi二元晶体结构、bi-zn二元晶体结构、sn-zn二元晶体结构进行声子谱计算,筛选出声子谱无虚频的二元晶体结构,具体如下:
[0072]
以sn-bi二元晶体结构为例,基于高精度优化后的sn-bi二元晶体结构,利用phonopy软件对其扩胞,生成一个spocar文件,根据sn-bi二元晶体结构的对称性,产生出不同位移的超原胞,对称性越复杂,产生的不同位移的超原胞数量就越多,计算量就越大。设置参数“isif=2,ibrion=8”,依次对不同位移的超原胞进行计算。
[0073]
步骤4,基于第一性原理的准简谐近似方法,即qha方法,分别对筛选出的声子谱无虚频的sn-bi二元晶体结构、bi-zn二元晶体结构、sn-zn二元晶体结构进行热学性能的计算。
[0074]
具体步骤如下:
[0075]
基于声子谱无虚频的sn-bi二元晶体结构、bi-zn二元晶体结构、sn-zn二元晶体结构进行变体积的能量与体积计算。具体是:
[0076]
逐个计算0.97、0.98、0.99、1.00、1.01、1.02、1.03体积下的sn-bi二元晶体结构、bi-zn二元晶体结构、sn-zn二元晶体结构的声子谱,基于准简谐近似方法得到对应的热学性能。所述热学性能指亥姆霍兹能量。
[0077]
将亥姆霍兹能量分解为三种累积贡献。如公式(2)所示:
[0078]
f(v,t)=ec(v)+f
vib
(v,t)+f
el
(v,t)
ꢀꢀꢀ
(2)
[0079]
式中f(v,t)表示亥姆霍兹能量是温度t和体积v的函数;ec为采用第一性原理的准简谐近似方法中,直接输出的0k静态总能量;f
vib
为振动自由能;f
el
是热电子对亥姆霍兹能量的贡献。
[0080]fvib
(v,t)由声子dos(p-dos)计算,如公式(3)所示:
[0081][0082]
式中,kb是玻尔兹曼常数,g(ω,v)是声子频率ω的函数声子态密度,是简约普朗克常数。热电子激发的贡献f
el
(v,t)通过mermin统计计算得到,如公式(4)所示:
[0083]fel
(v,t)=e
el
(v,t)-ts
el
(v,t)
ꢀꢀꢀ
(4)
[0084]
电子熵s
el
通过下式计算:
[0085]sel
(v,t)=-kb∫n(ε,v){f(ε,v,t)lnf(ε,v,t)+[1-f(ε,v,t)]ln[1-f(ε,v,t)]}d
ꢀꢀꢀ
(5)
[0086]
热电子能量由下式计算:
[0087][0088]
其中n(ε,v)是单电子带能量ε的函数电子态密度(e-dos),f是费米函数,εf是费米能。
[0089]
然后用修正的birch-murnaghan状态方程(eos)拟合七个给定体积下在给定温度下的亥姆霍兹能量,如公式(7)所示:
[0090]
f(v,t)=a+bv-2/3
+cv-4/3
+dv-2
+ev-8/3
ꢀꢀꢀ
(6)
[0091]
其中a,b,c,d和e为拟合参数。给定温度t下的平衡体积v
eq
(t)和最小亥姆霍兹自由能(f(v
eq
,t))由下式拟合得到:
[0092][0093]
计算的平衡体积v
eq
(t),体积热膨胀系数(β(t))由下式计算得到:
[0094][0095]
平均线膨胀系数α(t)和β(t)的关系为α(t)=β(t)/3。
[0096]
绝热体模量b
t
(v,t)由下式计算:
[0097][0098]
对于热力学建模的主要输入数据,熵推导为:
[0099][0100]
零外压下温度依赖的焓(h(v
eq
,t))由下式计算:
[0101]
h(v
eq
,t)=u(v
eq
,t)=f(v
eq
,t)+ts(v
eq
,t)
ꢀꢀꢀ
(11)
[0102]
其中u(veq,t)是内能,与等容热熔(cv(v
eq
,t))有如下关系:
[0103][0104]
等压热容由下列公式得到:
[0105]cp
(v
eq
,t)=cv(v
eq
,t)+v
eq
tb
t
(v
eq
,t)(β(t))2ꢀꢀꢀ
(13)
[0106]
步骤5,基于上述计算sn-bi二元晶体结构、bi-zn二元晶体结构和sn-zn二元晶体结构的热学性能,结合calphad模型,建立sn-bi-zn体系的热力学模型,采用的calphad模型如下:
[0107]
化合物在不同温度下的吉布斯能如下所示:
[0108][0109]
其中a,b,c,d,e和f是模型参数,由上面描述的第一性原理的准简谐方法计算得到的热力学数据评估得到。h
ser
是最稳定单质在298.15k和1bar下的焓作为参考态。液相的吉布斯能表达式为:
[0110][0111]
其中yi是组元i在液相中的摩尔分数,
xsgl
是过剩吉布斯能相,代表纯液相的吉布斯能,过剩吉布斯能
xsgl
的形式为:
[0112][0113]
其中是组元i和j之间的vth阶交互作用参数,由下式得到:
[0114][0115]
模型参数
v,liq
a和
v,liq
b是由实验的热力学数据和液相相关的相边界数据评估得到。
[0116]
利用calphad方法对sn-bi-zn体系相图进行热力学优化,优化后的热力学参数以及热力学模型均位于表5中。使用试错法赋值,根据热力学数据优化计算,直到与相图和热力学资料基本一致,获得sn-bi-zn体系的液相投影面,如图2所示,根据sn-bi-zn体系的液相投影面,来判断sn-bi-zn体系中是否存在共晶点和共晶点的温度范围。液相投影面的计算原理为:通过计算随第三组元的加入和成分的变化,二元零变量反应随温度变化的关系,得到三元液相面投影图,共晶点成分和比例通过热力学杠杆原理计算得到。
[0117]
表5
[0118][0119][0120]
步骤6,根据获得的sn-bi-zn体系的液相投影面,看出其存在共晶点,分析出其共晶点的成分比和共晶点温度分别为:sn:bi:zn=(20.76-34.09):(49.54-55.31):(10.64-14.06)和128.68-135.76℃。即从sn-bi-zn体系中筛选出的含铋合金熔接材料的成分比为sn:bi:zn=(20.76-34.09):(49.54-55.31):(10.64-14.06)。
[0121]
采用示差扫描量热法(dsc)对其进行实验验证,实验结果表明sn:bi:zn的共晶点温度实验值为133.50℃,误差很小。
[0122]
步骤7,对筛选出的含铋合金熔接材料进行结构优化,利用应力应变法进行力学性能计算,包括计算硬度,以及利用金属直流电导率公式计算电导率。
[0123]
步骤7.1,利用公式(18)计算筛选出的sn-bi-zn三元晶体结构的硬度,硬度的公式如下:
[0124]
hv=0.92k-1.137g0.708
ꢀꢀꢀ
(18)
[0125]
k是一个比值,k=b/g,b和g分别表示体积模量和剪切模量。b和g通过应力应变法计算sn-bi-zn三元晶体结构的弹性常数(c
11
,c
12
,c
44
)进一步得到,公式如下:
[0126][0127][0128][0129]
其中,bv和gv是voigt模型计算得到的体积模量和剪切模量,br是reuss模型计算得到的体积模量。进一步使用hill模型转化成最终需要的b和g,相应转化公式如下:
[0130][0131][0132]
获得筛选出的含铋合金熔接材料的硬度为14.01hbw。通过显微硬度计测试硬度,实验值为14.10hbw,误差范围很小。
[0133]
步骤7.2,利用金属直流电导率公式获得筛选出的含铋合金熔接材料的电导率,公式如下:
[0134][0135]
其中n为传导电子总的电荷密度,为自由电子的有效质量,τf为弛豫时间。其中,n由下式得出:
[0136][0137]
其中n为导电电荷数,计算公式为:
[0138][0139]
自由电子有效质量的计算公式为:
[0140][0141]
获得筛选出的含铋合金熔接材料的电导率为2.89ms/m,通过高精度塞贝克系数与电阻率测试仪测试电导率,实验值为2.99ms/m,误差范围很小。
[0142]
实施例二
[0143]
在本实施例中,一种筛选含铋合金熔接材料的方法,步骤如下:
[0144]
步骤1,基于目前开放的数据库以及相关文献报道获取sn-bi-ag体系中存在的二元晶体结构,包括sn-bi二元晶体结构、bi-ag二元晶体结构和sn-ag二元晶体结构。其中,sn-bi-ag体系指包含sn、bi和ag三种元素的晶体结构,sn-bi二元晶体结构指包含bi和sn两种元素的二元晶体结构,bi-ag二元晶体结构指包含bi和ag两种元素的二元晶体结构,sn-ag二元晶体结构指包含sn和ag两种元素的二元晶体结构。
[0145]
步骤2,采用基于密度泛函理论的第一性原理计算方法,通过vasp软件,对初始的sn-bi二元晶体结构、bi-ag二元晶体结构和sn-ag二元晶体结构,进行低精度的结构优化,然后分别对低精度优化后的sn-bi二元晶体结构、bi-ag二元晶体结构、sn-ag二元晶体结构进行高精度的结构优化,基于高精度优化后的sn-bi二元晶体结构、bi-ag二元晶体结构、sn-ag二元晶体结构,分别计算基本物相信息,基本物相信息包括形成焓,计算结果如表1所示。步骤2具体包括:
[0146]
步骤2.1,选取合适的关键性参数,有助于提高计算的准确性和高效性,而截断能和k点网格的密度的选取就显得至关重要。利用vaspkit功能102生成0.03精度的k点网格,截断能选用potcar文件中元素enmax的1.5倍。
[0147]
步骤2.2,首先设置参数“isif=3,ibrion=2”,分别对sn-bi二元晶体结构、bi-ag二元晶体结构、sn-ag二元晶体结构进行低精度结构优化,能量的收敛准则为10-6
ev,力的收敛准则为达到收敛标准。
[0148]
步骤2.3,低精度结构优化完成后,针对低精度结构优化后的sn-bi二元晶体结构、bi-zn二元晶体结构、sn-zn二元晶体结构再次进行高精度的结构优化,能量的收敛准则为10-8
ev,力的收敛准则为
[0149]
步骤2.4基于高精度优化后的sn-bi二元晶体结构、bi-ag二元晶体结构、sn-ag二元晶体结构,利用形成焓计算公式计算各二元晶体结构的形成焓,以sn-bi二元晶体结构为例,形成焓计算公式如公式(1)所示。
[0150][0151]
式中,δh代表形成焓,e
total
代表sn-bi二元晶体结构的总能量,e
bi
和e
sn
分别表示单质bi和sn的能量。x和y分别表示单质bi和sn在sn-bi二元晶体结构的原子数目。
[0152]
步骤3,基于高精度优化后的sn-bi二元晶体结构、bi-ag二元晶体结构、sn-ag二元晶体结构,使用phonopy软件分别对sn-bi二元晶体结构、bi-ag二元晶体结构、sn-ag二元晶体结构进行声子谱计算。具体如下:
[0153]
以bi-ag二元晶体结构为例,基于高精度优化后的bi-ag二元晶体结构,利用phonopy软件对其扩胞,生成一个spocar文件,根据bi-ag二元晶体结构的对称性,产生出不同位移的超原胞,对称性越复杂,产生的不同位移的超原胞数量就越多,计算量就越大。设置参数“isif=2,ibrion=8”,依次对不同位移的超原胞进行计算。
[0154]
步骤4,基于准简谐近似方法,即qha方法,分别对筛选出的声子谱无虚频的sn-bi二元晶体结构、bi-ag二元晶体结构、sn-ag二元晶体结构进行热学性能的计算。具体步骤如下:
[0155]
步骤4.1,基于声子谱无虚频的sn-bi二元晶体结构、bi-ag二元晶体结构、sn-ag二元晶体结构进行变体积的能量与体积计算。具体是:
[0156]
逐个计算0.97、0.98、0.99、1.00、1.01、1.02、1.03体积下的sn-bi二元晶体结构、bi-ag二元晶体结构、sn-ag二元晶体结构的声子谱,基于准简谐近似方法得到对应的热学性能。所述热学性能指亥姆霍兹能量。
[0157]
将亥姆霍兹能量分解为三种累积贡献。如公式(2)所示:
[0158]
f(v,t)=ec(v)+f
vib
(v,t)+f
el
(v,t)
ꢀꢀꢀ
(2)
[0159]
式中f(v,t)表示亥姆霍兹能量是温度t和体积v的函数;ec为采用第一性原理的准简谐近似方法中,直接输出的0k静态总能量;f
vib
为振动自由能;f
el
是热电子对亥姆霍兹能量的贡献。
[0160]fvib
(v,t)由声子dos(p-dos)计算,如公式(3)所示:
[0161][0162]
式中,kb是玻尔兹曼常数,g(ω,v)是声子频率ω的函数声子态密度,是简约普朗克常数。热电子激发的贡献f
el
(v,t)通过mermin统计计算得到,如公式(4)所示:
[0163]fel
(v,t)=e
el
(v,t)-ts
el
(v,t)
ꢀꢀꢀ
(4)
[0164]
电子熵s
el
通过下式计算:
[0165]sel
(v,t)=-kb∫n(ε,v){f(ε,v,t)lnf(ε,v,t)+[1-f(ε,v,t)]ln[1-f(ε,v,t)]}d
ꢀꢀꢀ
(5)
[0166]
热电子能量由下式计算:
[0167][0168]
其中n(ε,v)是单电子带能量ε的函数电子态密度(e-dos),f是费米函数,εf是费米能。
[0169]
然后用修正的birch-murnaghan状态方程(eos)拟合七个给定体积下在给定温度
下的亥姆霍兹能量,如公式(7)所示:
[0170]
f(v,t)=a+bv-2/3
+cv-4/3
+dv-2
+ev-8/3
ꢀꢀꢀ
(6)
[0171]
其中a,b,c,d和e为拟合参数。给定温度t下的平衡体积veq(t)和最小亥姆霍兹自由能(f(v
eq
,t))由下式拟合得到:
[0172][0173]
从计算的平衡体积veq(t),体积热膨胀系数(β(t))由下式计算得到:
[0174][0175]
平均线膨胀系数α(t)和β(t)的关系α(t)=β(t)/3。
[0176]
绝热体模量b
t
(v,t)由下式计算:
[0177][0178]
对于热力学建模的主要输入数据,熵推导为:
[0179][0180]
零外压下温度依赖的焓(h(v
eq
,t))由下式计算:
[0181]
h(v
eq
,t)=u(v
eq
,t)=f(v
eq
,t)+ts(v
eq
,t)
ꢀꢀꢀ
(11)
[0182]
其中u(veq,t)是内能,与等容热熔(cv(v
eq
,t))有如下关系:
[0183][0184]
等压热容由下列公式得到:
[0185]cp
(v
eq
,t)=cv(v
eq
,t)+v
eq
tb
t
(v
eq
,t)(β(t))2ꢀꢀꢀ
(13)
[0186]
步骤5,基于上述计算sn-bi二元晶体结构、bi-ag二元晶体结构和sn-ag二元晶体结构的热学性能,结合calphad模型,建立sn-bi-ag体系的热力学模型,采用的calphad模型描述如下:
[0187]
化合物在不同温度下的吉布斯能如下所示:
[0188][0189]
其中a,b,c,d,e和f是模型参数,由上面描述的第一性原理的准简谐近似方法计算得到的热力学数据评估得到。h
ser
是最稳定单质在298.15k和1bar下的焓作为参考态。液相的吉布斯能表达式为:
[0190][0191]
其中yi是组元i在液相中的摩尔分数,
xsgl
是过剩吉布斯能相,代表纯液相的吉布斯能。过剩吉布斯能
xsgl
的形式为:
[0192][0193]
其中是组元i和j之间的vth阶交互作用参数,由下式得到:
[0194][0195]
模型参数
v,liq
a和
v,liq
b是由实验的热力学数据和液相相关的相边界数据评估得到。
[0196]
利用calphad方法对sn-bi-ag体系相图进行热力学优化和计算,优化后的热力学参数和热力学模型位于表6。使用试错法赋值,根据热力学数据优化计算,直到与相图和热力学资料基本一致。获得sn-bi-ag体系的液相投影面,如图3所示,根据sn-bi-ag体系的液相投影面,来判断sn-bi-ag体系中是否存在共晶点和共晶点的温度范围。液相投影面的计算原理为:通过计算随第三组元的加入和成分的变化,二元零变量反应随温度变化的关系,得到三元液相面投影图,共晶点成分和比例通过热力学杠杆原理计算得到。
[0197]
表6
[0198][0199][0200]
步骤6,根据获得的sn-bi-ag体系的液相投影面,看出其存在共晶点,分析出其共晶点的成分比和温度分别为:sn:bi:ag=(32.51-38.54):(44.70-52.68):(13.79-16.88)和132.87-140.65℃。即从sn-bi-ag体系中筛选出的含铋合金熔接材料的成分比为sn:bi:ag=(32.51-38.54):(44.70-52.68):(13.79-16.88)。
[0201]
采用示差扫描量热法(dsc)对其进行实验验证,实验结果表明sn:bi:ag的共晶点温度实验值为137.70℃,误差很小。
[0202]
步骤7,对筛选出的含铋合金熔接材料进行结构优化,利用应力应变法进行力学性能计算,包括计算硬度,以及利用金属直流电导率公式计算电导率。
[0203]
步骤7.1,利用公式(18)计算筛选出的sn-bi-ag三元晶体结构的硬度,硬度的公式如下:
[0204]
hv=0.92k-1.137g0.708
ꢀꢀꢀ
(18)
[0205]
k是一个比值,k=b/g,b和g分别表示体积模量和剪切模量。b和g通过应力应变法计算sn-bi-ag三元晶体结构的弹性常数(c
11
,c
12
,c
44
)进一步得到,公式如下:
[0206]
[0207][0208][0209]
其中,bv和gv是voigt模型计算得到的体积模量和剪切模量,br是reuss模型计算得到的体积模量。进一步使用hill模型转化成最终需要的b和g,相应转化公式如下:
[0210][0211][0212]
获得筛选出的含铋合金熔接材料的硬度为28.58hbw。通过显微硬度计测试硬度,实验值为28.60hbw,误差范围很小。
[0213]
步骤7.2,利用金属直流电导率公式获得筛选出的含铋合金熔接材料的电导率,公式如下:
[0214][0215]
其中n为传导电子总的电荷密度,为自由电子的有效质量,τf为弛豫时间。其中,n由下式得出:
[0216][0217]
其中n为导电电荷数,计算公式为:
[0218][0219]
自由电子有效质量的计算公式为:
[0220][0221]
获得筛选出的含铋合金熔接材料的电导率为2.50ms/m,通过高精度塞贝克系数与电阻率测试仪测试电导率,实验值为2.44ms/m,误差范围很小。
[0222]
上面对本发明方法实施例结合附图进行了说明,但本发明不限于上述实施例,还可以根据本发明的发明创造的目的做出多种变化,凡依据本发明技术方案的原理下做的参数改变、或计算简化,只要符合本发明的发明目的,只要不背离本发明一种筛选含铋合金熔接材料的方法及含铋合金熔接材料的原理和构思,都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1