一种基于高通量测序动植物基因组T2T组装方法与流程
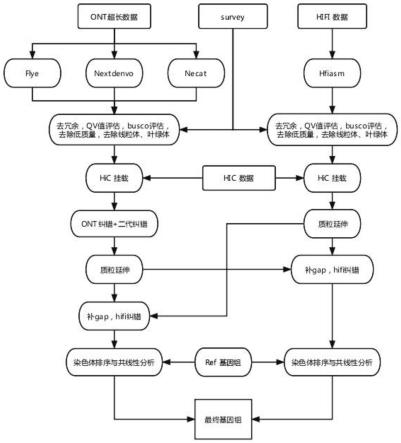
一种基于高通量测序动植物基因组t2t组装方法
技术领域
1.本发明涉及基因组装技术领域,尤其涉及一种基于高通量测序动植物基因组t2t组装方法。
背景技术:
2.高通量测序技术(high-throughputsequencing)又称下一代测序技术(next-generationsequencingtechnology)。高通量测序技术目前已经来到第四代,纳米孔测序技术(又称第四代测序技术)是最近几年兴起的新一代测序技术。目前测序长度可以达到4mb。这项技术开始于90年代,经历了三个主要的技术革新:一、单分子dna从纳米孔通过;二、纳米孔上的酶对于测序分子在单核苷酸精度的控制;三、单核苷酸的测序精度控制。随着物种基因组研究的不断发展,测序技术的不断提升,物种基因组的连续性和完整性也都得到大幅提升。物种基因组从普通二代测序组装的draftgenome(草图基因组,基因组1.0),到使用pacbio(pacificbiosciences公司)或ont(oxfordnanopore公司)(r9.4)测序技术结合hi-c(high-throughput/resolutionchromosomeconformationcapture,高通量染色体构象捕获技术)组装的high-qualitygenome(高质量基因组,基因组2.0),再到由ont(n50》50kb或r10)测序技术组装的nearcompletegenome(近完全基因组,基因组3.0),以及结合ontultra-long(牛津纳米孔超长测序技术)和pacbiohifi(highfidelityreads,高准确度读长测序技术)技术组装的t2tgenome(端粒到端粒基因组,基因组4.0)。动植物t2t基因组,指通过ontultra-longn50》100kb(测序读长n50大于1000000,n50:n50是基因组拼接之后一个评价指标,将拼接得到的所有的序列,根据序列大小从大到小进行排序,然后逐步开始累加,当加和长度超过总长一半时,加入的序列长度即为n50长度。)结合hifi和二代数据(高通量测序,如illumina hiseqtm/miseqtm)进行混合组装,得到的有一条或者多条染色体达到端粒到端粒(telomere-to-telomere,t2t)的水平基因组,t2t基因组完成图是基因组组装的终极目标。
3.t2t基因组组装的要求包括:染色体组装无空缺,qv值(基因组组装准确度评估标准)大于40,busco(基因组组装完整度评估标准)评估大于95%,端粒完整。以往单一测序手段ont普通测序或者pacbiohifi都难以达到t2t水平。普通的ont测序组装精度偏低、难以达到qv大于40的质量标准,且难以达到染色体组装无空缺;hifi测序组装虽然能带来很高的组装精度,但是无法达到染色体组装无空缺,且着丝粒区域往往组装空缺。
4.因此,有必要针对现有技术的缺陷,提出一种基于高通量测序动植物基因组t2t组装方法。
技术实现要素:
5.本发明的目的在于提供一种基于高通量测序动植物基因组t2t组装方法,通过ont超长测序技术结合hifi测序技术和高标准、高质量的组装技术能克服以往的缺点,基因组组装达到t2t水平,推动了整个物种的研究进展。
6.有鉴于此,本发明的方案如下:
7.一种基于高通量测序动植物基因组t2t组装方法,包括如下步骤:
8.s1.基于样本测序,分别进行ont超长测序数据组装及hifi测序数据组装;
9.s2.基于基因组大小评估结果对两条路线的组装结果去冗余;
10.s3.分别对两条路线组装结果进行hic挂载,得到染色体级别参考基因组其gap;
11.s4.对ont线路基因组着丝粒区域进行三代纠错,基于二代数据对ont线路基因组进行二代纠错;
12.s5.基于原始数据比对末端片段,根据端粒重复基序列的丰度大小进行重组装,再经比对进行末端端粒序列替换,对两条线路的端粒延伸;
13.s6.分别对基因组序列补gap、hifi纠错;
14.s7.结合已知物种基因组排序对染色体进行调整得到组装后的基因组。
15.在本发明的一个实施例中,所述步骤s4中三代纠错过程为:对ont基因组着丝粒区域使用k-mer锚定方法进行迭代多轮三代纠错。
16.作为优选的实施例,所述ont基因组着丝粒区域基于二代数据和基因组中k-mer的频次,标记在组装中出现一次且在二代数据中出现14到46次的21-kmer。
17.在本发明的一个实施例中,所述步骤s4中二代纠错过程为:将二代原始数据进行切分并比对到基因组上,通过深度神经网络进行变异检测得到变异信息,变异信息经过滤对齐后导出一致性序列从而得到二代纠错后的基因组。
18.在本发明的一个实施例中,所述步骤s5端粒延伸过程为:
19.s51.将每条染色体分别与原始数据进行比对,收集在染色体末端定长内比对一次的所有片段,计算每条片段中出现端粒重复基序列的次数,定义出现最多次数的片段为ref,其他为query,将ref和query重新组装,得到一致性序列;
20.s52.将该一致性序列分别比对到每条染色体上,取最佳比对结果对末端端粒序列进行替换。
21.作为优选的实施例,所述步骤s52中,如果identity低于80阈值或比对上区域不在染色体末端20kbp,则不进行替换。
22.在本发明的一个实施例中,步骤s6中所述基因组序列补gap步骤为:基于补洞程序对基因组进行补洞,将补洞数据与基因组数据进行比对,按其他纠错后基因组版本、hifi数据、ont数据的先后顺序分别进行补洞。
23.作为优选的实施例,所述补gap步骤中,在数据量不够的情况下,用subreads/ont原始片段对基因组进行补洞;如果比对上的位置能刚好跨过gap两端,则选取比对上的最长长度区域的最佳比对区域用补gap数据对基因组上包含gap区域的序列进行替换。
24.在本发明的一个实施例中,步骤s6中所述基因组hifi纠错步骤为:先过滤掉低于10kbp的hifi数据,将过滤后的数据与补完洞的基因组进行比对得到合并排序后的文件;再分别过滤掉二次比对、嵌合比对的片段后进行三代纠错。
25.在本发明的一个实施例中,所述t2t组装方法还包括共线性分析步骤:对步骤s7中所述调整后的基因组与已发表的物种的基因组做共线性比对分析,分析指标包括完整度和准确度。
26.相比现有技术,本发明的有益效果包括但不限于:
27.1.本发明提出的基于高通量测序动植物基因组t2t组装方法通过ont超长测序技术结合hifi测序技术和高标准、高质量的组装技术能克服以往的缺点,基因组组装达到t2t水平,得到端粒完整、无空缺的基因组,推动了整个物种的研究进展。
28.2.本发明提出的基于高通量测序动植物基因组t2t组装方法基于多种测序手段并辅以复杂的开源软件与自研软件达到超出单一测序手段简单组装的效果,为基因组t2t组装提供新的选择。
附图说明
29.图1为本发明所述动植物基因组t2t组装方法流程图。
30.图2为本发明所述hic具体具体步骤流程图。
31.图3为本发明实施例中基于contig互作强度与位置关系得到的互作热图。
32.图4为本发明实施例中最终t2t版本hi-c互作热图。
33.图5为本发明实施例中对t2t组装方法得到的基因组共线性分析结果示意图。
具体实施方式
34.为了使本发明的目的、技术方案和有益技术效果更加清晰明白,以下结合具体实施方式,对本发明进行进一步详细说明。应当理解的是,本说明书中描述的具体实施方式仅仅是为了解释本发明,并不是为了限定本发明。
35.在一个实施例中,提出一种基于高通量测序动植物基因组t2t组装方法,流程如图1所示,步骤如下:
36.1.测序:同一份动植物生物样本需要进行二代测序(illumina)或者bgi(华大基因)、ont超长测序(oxfordnanoporetechnology平台仪器测序)、hic测序(hi-c技术源于基因组捕获技术)、pacbiohifi测序(pacificbiosciences)。
37.2.评估:通过二代测序进survey基因组分析,估计出基因组大小。
38.3.初组装:分两条线进行。
39.3.1以ont超长测序数据组装结果为骨架(后简称ont骨架),分别使用flye、nextdenovo及necat三款基因组组装软件进行组装;
40.flye:https://github.com/fenderglass/flye;
41.nextdenovo:https://github.com/nextomics/nextdenovo;
42.necat:https://github.com/xiaochuanle/necat。
43.3.2以hifi测序数据组装结果为骨架(后简称hifi骨架),使用hifiasm(https://github.com/chhylp123/hifiasm)进行组装。
44.4.去冗余与评估
45.组装后基因组大小有超过survey评估大小,为把基因组减少到评估大小,需对两条路线的组装结果进行去冗余;使用merqury(https://github.com/search?q=merqury)进行qv值计算(组装后基因组准确度评估值);使用busco(https://busco.ezlab.org/)进行组装后基因组完整度评估;使用reads回比到组装后基因组;去除低质量contig(组装后的序列);在ncbi上下载本物种线粒体、叶绿体,并使用minimap2(https://github.com/lh3/minimap2)比对组装后基因组,去除比对上的contig。
46.5.hic挂载
47.5.1ont骨架路线:选取三个组装结果中最好的数据进行hic挂载(挂载就是利用3维情况下hic互做,指导二维基因组组装的一种应用);二、hifi骨架路线:hic挂载。经过此步得出染色体数目与基因组gap(未组装出来的序列,在基因组染色体层面体现为序列空白以100个n填充)数量。hic挂载具体具体步骤如图2所示,为本领域常用的路线。
48.6.本步骤应用于ont骨架路线
49.三代纠错:先将去线粒体叶绿体低reads支持contig流程中生成的中间结果按40kbp长度过滤后的三代filterreads比对到hic挂载后的基因组上。通过meryl(https://github.com/marbl/meryl)来统计二代数据的kmer出现次数和基因组中kmer的出现次数,找到在组装中出现一次且在二代数据中出现14到46次的21-kmer,并认为是需要标记的着丝粒区域。用t2t_polish(https://github.com/arangrhie/t2t-polish)下的子程序filter_by_marker_nosplit.sh对bam文件进行标记,并用medaka consensus(https://github.com/nanoporetech/medaka)进行纠错,medaka stitch得到一致性序列,以达到避免着丝粒过度纠错的目的。
50.二代纠错:将二代原始数据进行切分,用bwa(https://github.com/lh3/bwa)分别比对到基因组上,然后sort,merge得到总的sorted.merged.bam,并建立索引文件。用deepvariant(https://github.com/google/deepvariant)进行变异检测得到vcf文件,经过过滤对齐后的vcf文件被bcftoolsconsensus(https://github.com/samtools/bcftools)导出一致性序列从而得到二代纠错后的基因组。
51.7.端粒延伸应用于两条路线
52.将每条染色体分别与原始数据用winnowmap(https://github.com/marbl/winnowmap)比对,收集在染色体末端50bp内比对一次的所有reads,计算每条read中出现端粒重复基序(
‘
ccctaaa’/
‘
tttaggg’)的次数,定义出现最多次数的read为ref,其他为query,使用medaka_consensu将ref和query重新组装,得到一致性序列。将该一致性序列分别比对到每条染色体上,取最佳比对结果对末端端粒序列进行替换。如果identity低于80阈值或比对上区域不在染色体末端20kbp,则不进行替换。
53.8.补gap
54.gap_closer流程用于对基因组上gap的填补,用winnowmap将补洞数据(不含n)与基因组数据比对(含n),流程分为三个水平对gap进行填补,其优先级为:其他纠错后基因组版本》hifi数据》ont数据。其他版本纠错后的基因组可以是nextdenovo、necat、hifiasm、canu
…
等任何版本基因组;hifi数据最好为ccs.fasta,既去环化后的hifi数据;ont数据最好为cns.fa,即ont数据经过nextdenovo自纠错后生成的一致性序列consensus.fa。在数据量不够的情况下,可以考虑选用subreads/ont原始reads对基因组进行补洞。如果比对上的位置能刚好跨过gap两端,则选取比对上的最长长度区域的最佳比对区域用补gap数据对基因组上包含gap区域的序列进行替换。从理论上说,只要所提供补洞用的hifi/ont数据量足够大,该流程就能把gap给补上。但是流程也是有一定局限性的,对于很短一片区域存在多个gap时不太适用,因此gap向两端延申的长度不建议超过两个gap间的最短距离。
55.9.hifi纠错
56.上述步骤中做了填补gap的操作,为了使得我们填补上的序列可靠,以及保证基因
组质量的一致性,这一步使用hifi数据对补洞后的基因组进行纠错。过滤掉hifi数据10kbp以下的原始数据,用winnowmap将过滤后的数据与补完洞的基因组进行比对,得到比对完成的merge.sorted.bam文件后。用samtools view-f256(https://github.com/samtools/samtools)对bam文件进行过滤,并用falconcbam-filter-clipped(https://github.com/pacificbiosciences/falcon)过滤掉嵌合比对片段,最后用racon(https://github.com/isovic/racon)的liftover分支进行纠错,得到经过hifi纠错后的基因组序列。
57.10.染色体排序与共线性
58.将上述组装结果mapping到已发表的物种基因组上,并将染色体编号和方向按照已发表的基因组染色体编号和方向进行对应,然后将调整之后的基因组与已发表的物种的基因组做共线性。同样,调整位置后的基因组和ref也可以输入到共线性流程中做mummer共线性图或jcvi共线性图,这样就能得到一条直线或者是瀑布式的共线性图,可以更好的看出两版本之间共线性的情况。
59.11.基因组最终确认
60.两条线路中选择完整度高、准确度高的基因组为最终基因组。
61.实施例
62.1.使用太平洋生物公司(pacbio)高保真测序数据(hifi)进行基因组组装,生成重叠群(contig)。使用从美国国立生物技术信息中心(ncbi)下载了大约20gb水稻太平洋生物公司(pacbio)高保真测序(hifi)测序数据。使用hifiasm(https://github.com/chhylp123/hifiasm)进行组装。组装结果如表1所示。
63.表1:hifiasm初步组装结果表
64.num_seqssum_lenmin_lenavg_lenmax_lenn5014140195049891142850712.84500251431388662
65.2.使用从美国国立生物技术信息中心(ncbi)下载了大约50gb水稻ont超长数据,分别使用三款基因组组装软件进行组装flye(https://github.com/fenderglass/flye)、nextdenovo(https://github.com/nextomics/nextdenovo)、necat(https://github.com/xiaochuanle/necat)进行组装。组装结果如表2所示。
66.表2:ont数据初步组装结果表
[0067] num_seqssum_lenmin_lenavg_lenmax_lenn50nextdenovo2339848178611080417325295.04494991131426128flye7739533967414725134281.55203929424103585necat61401818451238116587187.74488457724129713
[0068]
3.对于高杂合物种,需要使用软件purge_haplotigs(v1.0.4;https://github.com/skingan/purge_haplotigs_multibam)/purge_dups(v1.2.5;https://github.com/df guan/purge_dups)对基因组进行去杂合。
[0069]
4.使用minimap2(2.17-r941)(h.li 2018)软件,比对线粒体、叶绿体,去除碱基比对超过50%的序列;通过blast refseq库去除细菌污染;去除低reads支持的contig(minimap2将ont reads(》=40kbp)与contig比对,如果一条contig上超过50%的位点的深度低于15,就进行移除),该阈值会根据数据量适当调整。
[0070]
5.利用互作关系进行辅助组装:
[0071]
1)contig聚类
[0072]
使用hi-c互作关系,确定有效数据中不同contig间关联的紧密程度,对contig进行聚类。对于核型为2n的基因组草图,利用allhic(v0.9.8)(zhang etal.2019)软件通过agglomerativehierarchicalclustering(自下而上的层次聚类算法),将草图的contig序列聚类为n个染色体群。
[0073]
2)contig定序和定向
[0074]
利用allhic(v0.9.8)(zhangetal.2019)软件对n个染色体群内部的contig进行定序和定向,再通过软件3d-dna(v180419)(dudchenkoetal.2017)和juicer(v1.6)(nevac.etal.2016b)将contig两两之间的互作关系转化为指定的二进制文件(即.hic文件)。再通过windows软件juicebox(v1.11.08)(nevac.etal.2016a)对已经定序和定向contig(.assembly)进行手动定序与定向(.review.assembly)。
[0075]
3)利用互作关系进行contig去冗余
[0076]
若某段序列与一相同大小的序列完全没有互作,但与其它序列互作正常,那么它极大可能为杂合序列,需要手动去除。
[0077]
将定序、定向、去冗余的contig序列,使用100个n补gap得到最终的染色体级别基因组序列。详细每条染色体及其长度统计结果如表3所示,没有互作或者有互作噪音的contig为未挂载的片段(记为chrunn)。
[0078]
4)染色体挂载结果统计:经过hi-c组装和人工调整后,共有397,785,342bp的序列长度的基因组序列被定位到12条染色体上,占比99.25%。每条染色体及其长度的详细统计结果如表3所示。
[0079]
表3:染色体长度统计表
[0080]
chromosomelength(bp)contignumberchr125,815,0086chr231,882,4771chr338,842,1042chr432,745,32713chr532,075,9785chr624,932,0231chr737,439,2563chr830,588,8192chr927,101,4762chr1031,388,6621chr1145,002,5141chr1239,971,6981chrunn3,021,77273
[0081]
5)使用软件hicexplorer(v3.6)(joachim et al.2020)基于contig互作强度与位置关系进行绘图,如图3所示。
[0082]
6.组装纠错
[0083]
6.1三代纠错:
[0084]
针对着丝粒区域使用k-mer锚定方法进行三代纠错(注意此纠错手段仅针对t2t基因组ont骨架版);
[0085]
1)使用minimap2(v2.17-r941)(h.li 2018)将ont reads(》=40kbp)比对到伪染色体上。
[0086]
2)为了避免着丝粒序列的过度纠错,找到在组装中出现一次且在illumina reads中出现14到46次的21-kmer,并使用t2t-polish
[0087]
(https://github.com/malonge/t2t-polish)的子程序filter_by_marker_nosplit.sh进行对比对好的bam进行标记。
[0088]
3)使用medakaconsensus(v1.5.0;参数:
–
modelr941_prom_hac_g507
[0089]
–
batch_size200;https://github.com/nanoporetech/medaka)进行纠错。
[0090]
4)使用medakastitch(v1.5.0;https://github.com/nanoporetech/medaka)得到三代纠错后的一致性序列。
[0091]
重复上述步骤,即t2t基因组ont骨架版需要迭代两轮三代纠错。
[0092]
6.2二代纠错:
[0093]
使用deepvariant(v1.3.0)(poplinetal.2018)进行二代纠错(注意此纠错手段仅针对t2t基因组ont骨架版);
[0094]
1)使用bwamem(v0.7.17-r1188)(hengli2013),samtools(v1.9)
[0095]
(daneceketal.2021)得到三代纠错后的基因组和二代数据比对好的bam文件。
[0096]
2)使用deepvariant(v1.3.0;参数:
–
model_type=wgs)(poplinetal.20
[0097]
18)得到vcf文件。
[0098]
3)使用bcftools(v1.15;参数:view-e
‘
type=“ref
”’
,view-i
‘
qual》1
[0099]
&&(gt=“aa”||gt=“aa”)’)(daneceketal.2021)对vcf文件进行过滤,并用bcftoolsnorm(daneceketal.2021)将vcf文件左对齐并且规范化。
[0100]
4)将vcf文件压缩并生成索引文件。
[0101]
5)bcftoolsconsensus(daneceketal.2021)导出二代纠错后的一致性序列。
[0102]
7.端粒延伸
[0103]
1)使用winnowmap(v1.11,参数:k=15,
–
md)(chiragetal.2020)将所有的ontreads比对到ref,收集在染色体末端50bp内比对一次的所有r eads。
[0104]
2)计算每条read中端粒重复基序(
‘
ccctaaa’/
‘
tttaggg’)的出现次数(端粒数据库:http://telomerase.asu.edu/sequences_telomere.html),并定义出现最多次数的read为ref,其他为query。
[0105]
3)medaka_consensu(v1.2.1,参数:-mr941_min_high_g360;https://github.
[0106]
com/nanoporetech/medaka),将ref端粒read和query端粒read进行重新组装,得到一致性序列。
[0107]
4)用nucmer(v3.1)(kurtzetal.2004)将上述端粒的一致性序列分别比对到每条染色体上,取最佳比对结果对末端端粒序列进行替换。如果identity低于80阈值或比对上区域不在染色体末端20kbp,则不进行替换。
[0108]
8.gap填补
[0109]
用于对基因组上gap的填补,用winnowmap(v1.11,参数:k=15,
–
md)
[0110]
(chiragetal.2020)将补洞数据(不含n)与基因组gap区间比对(含n),该步骤分为三个水平对gap进行填补,其优先级为:其他纠错后基因组版本》hifi数据》ont数据。其他版本纠错后的基因组可以是nextdenovo、necat、hifiasm、canu
…
等任何版本基因组;hifi数据最好为ccs.fasta,既去环化后的hifi数据;ont数据最好为cns.fa,即ont数据经过nextdenovo自纠错后生成的一致性序列consensus.fa。再数据量不够的情况下,可以考虑选用subreads/ontreads对基因组进行补洞。如果比对上的位置能刚好跨过gap两端,则选取比对上的最长长度区域的最佳比对区域用补gap数据对基因组上包含gap区域的序列进行替换。从理论上说,只要所提供补洞用的hifi/ont数据量足够大,用于补洞的基因组其他组装版本足够多,就能得到0gap基因组。
[0111]
9.hifi reads纠错
[0112]
1)使用winnowmap2(chirag et al.2020)将》=10kbp的hifi reads与填补后gap的版本基因组比对(参数:k=15greater-than distinct=0.9998
[0113]
–
md-ax map-pb);
[0114]
2)使用samtools“view”(v1.10,参数:-f 256)(danecek et al.2021)对比对片段进行过滤;
[0115]
3)使用“falconc bam-filter-clipped”来删除嵌合比对片段(-t-f 0x104);
[0116]
4)使用这些过滤后的比对信息,使用racon的特殊分支进行纠错,(v1.6.0,
[0117]-l-u,https://github.com/isovic/racon/tree/liftover)。
[0118]
10.t2t基因组评估
[0119]
1)通过基因组上gap的位置和个数来评估基因组组装的连续性,基因组gap情况如表4所示。
[0120]
表4:0gap基因组
[0121]
[0122][0123]
2)利用软件bwa(version:0.7.17-r1188)将二代高通量测序(如illumina测序平台)得到的质控后reads(clean reads)比对(mapping)到参考基因组上。统计二代比对率和覆盖度如表5所示。
[0124]
表5:
[0125][0126]
注:sample name:样本名称;mapping rate(%):比对率;paired mapping rate(%):双端比对率;average sequencing depth:平均测序深度;coverage(%):覆盖度;coverage at least 4x(%):4x深度以上的覆盖度;coverage at least 10x(%):10x深度以上的覆盖度;coverage at least 20x(%):20x深度以上的覆盖度。
[0127]
3)使用软件busco(version:4.1.4;参数:
–
evalue 1e-05)来评估基因组组装的完整性,结果如表6所示。在基因组完整性较好时,t2t基因组中b usco评估中complete buscos(c)的占比一般大于95%。
[0128]
表6:基因组组装的完整性评估结果
[0129][0130]
[0131]
注:complete buscos为完整的busco;complete and single-copy buscos为完整且单拷贝的busco;complete and duplicated buscos为完整但多拷贝的busco;fragmented buscos为不完整的busco;missing buscos为缺失的busco;total busco groups searched为本次使用busco库中收录的保守蛋白基因的总数目。
[0132]
4)通过二代数据的kmer统计结果和基因组的kmer统计结果,结果如表7所示。比较其kmer种类数频数以及分布来评判基因组组装的质量和准确度。
[0133]
表7:基因组组装qv值统计表
[0134]
qvcompleteness(%)lengthn50readsnum45.173999.1838397,733,90132,070,99412
[0135]
注:qv为用于评估基因组质量的qv值;completeness用于评估基因组完整度;length为基因组长度;n50为contig级别的n50,即将所有的contigs按照从长到短排序后依次累加,当相加的长度达到基因组总长度的一半时,最后一个加上的contig长度即为n50;reads num为contig级别基因组reads条数。
[0136]
5)根据端粒区域碱基重复序列(5’端为ccctaaa,3’端为tttaggg),对组装结果进行端粒序列鉴定,结果如表8所示。
[0137]
表8:端粒重复序列统计结果
[0138]
[0139][0140]
6)着丝粒序列鉴定
[0141]
(i)chip-seq
[0142]
建立基于cenh3抗体的chip-seq的着丝粒分离分析方法,在获得着丝粒chip-seq数据后,通过与基因组序列比对筛选,获得在基因组上具有单一拷贝的数据,并将其锚定到对应物种的基因组上,即可获得chip-seq富集的峰图,该峰图则指示着丝粒区。这一方法目前已被用于着丝粒的基因组精确定位。
[0143]
(ii)fish验证
[0144]
结合细胞学的荧光原位杂交技术(fish)验证,最终可鉴定出着丝粒区域特异的重复序列。
[0145]
11.最终t2t版本hi-c互作热图
[0146]
在经过端粒延申,补gap,纠错后,以最终t2t版本基因组,再一次通过hi-c数据来得到新的染色体级别的hi-c互作热图,如图4所示,通过热图中互作信号的异常与否来鉴定我们前面的操作是否正确。
[0147]
12.共线性分析
[0148]
本次组装的基因组与目标物种历史基因组版本进行比较;该分析仅限有历史基因组版本的物种,共线性分析结果如图5所示。该历史基因组版本为另一种t2t组装方法得到的基因组。
[0149]
本发明并不仅仅限于说明书和实施方式中所描述,因此对于熟悉领域的人员而言
可容易地实现另外的优点和修改,故在不背离权利要求及等同范围所限定的一般概念的精神和范围的情况下,本发明并不限于特定的细节、代表性的方案和这里描述的示例。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1