用于治疗帕金森病的新儿茶酚胺前药的制作方法
本发明提供了是多巴胺激动剂的前药的化合物(4ar,10ar)-1-正丙基-1,2,3,4,4a,5,10,10a-八氢-苯并[g]喹啉-6,7-二醇,及其在帕金森病和/或其他病症的治疗中的用途,对于该治疗,其中多巴胺激动剂是治疗有益的,这些病症例如但不限于下肢不宁综合征、亨廷顿病和阿尔茨海默病;以及还有神经精神性疾病和障碍,例如但不限于精神分裂症、注意缺陷多动障碍和药物成瘾。本发明还提供了包含本发明的化合物的药物组合物。
背景技术:
1、帕金森病(pd)是随着年龄变得逐渐普遍的常见神经退行性障碍,并且影响世界上估计七百万到一千万的人口。帕金森病是多方面的疾病,其特征在于运动症状和非运动症状二者。运动症状包括静止性震颤(颤动)、运动过慢/运动不能(运动的缓慢和困难)、肌僵直、姿势不稳定和步态障碍;而非运动症状包括神经精神障碍(例如抑郁、精神病症状、焦虑、情感冷漠、轻度认知损害和痴呆)连同自主神经功能障碍和睡眠障碍(poewe等人,nature review[自然评论],(2017)第3卷文章17013:1-21)。
2、帕金森病病理生理学的关键标志是致密部中色素多巴胺能神经元的损失,这些神经元提供了多巴胺能神经支配至纹状体和其他脑区。此类进行性神经变性导致多巴胺纹状体水平的降低,这最终导致基底核回路的一系列变化,最终导致帕金森病的四种主要运动特征的发生。纹状体中多巴胺的主要靶由中棘γ-氨基丁酸能神经元(msn)组成,这些神经元选择性表达等待定位投射的d1或d2受体。投射到外侧苍白球的γ-氨基丁酸能msn(也称为纹状体-苍白球‘间接途径’)表达d2受体(msn-2);而投射到黑质网状部和外侧苍白球的γ-氨基丁酸能msn(也称为纹状体-黑质‘直接途径’)表达d1受体(msn-1)。由于神经元损失的多巴胺的耗减,导致两条途径的不平衡活性,导致丘脑和皮质输出活性的显著减少,以及最终的运动障碍(gerfen等人,science[科学](1990)250:1429-32;delong,(1990)trendsin neuroscience[神经科学趋势]13:281-5;alexander et crutcher,(1990)trends inneuroscience[神经科学趋势]13:266-71;以及对于综述,poewe等人,nature review[自然评论](2017)第3卷文章17013:1-21)。
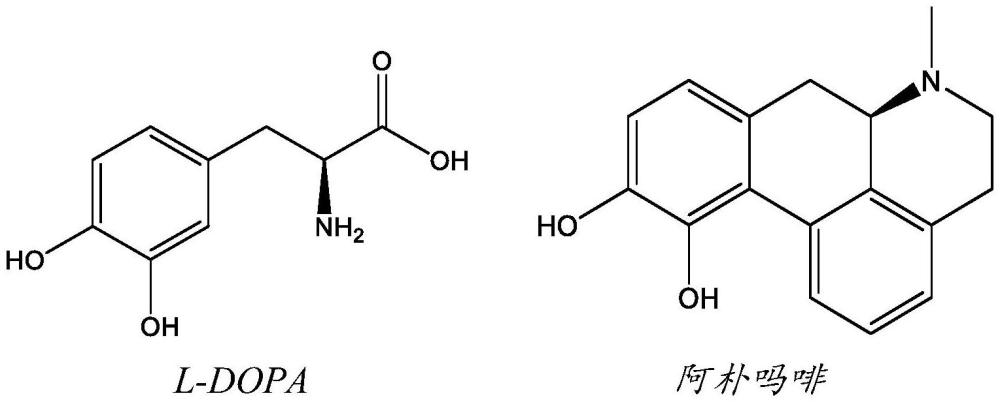
3、可供用于患有帕金森病的患者的、并且目标在于控制运动症状的最有效治疗策略主要是间接和直接的多巴胺激动剂。经典标准和黄金标准治疗方案包括l-3,4-二羟基苯丙氨酸(l-dopa)(它在大脑中脱羧基以形成多巴胺)的慢性口服摄入。其他方法在于给予多巴胺受体激动剂(例如阿朴吗啡(阿朴吗啡对d1和d2受体亚型都起作用),或主要针对d2受体亚型的普拉克索、罗匹尼罗等)。使用l-dopa和阿朴吗啡获得的最佳运动缓解是由于它们激活d1和d2受体亚型二者,并且整体重新平衡间接-直接途径(即同时d2激动剂仅逆转间接途径障碍)。
4、l-dopa和阿朴吗啡具有以下描绘的结果,并且目前是临床应用中最有效的pd药物。
5、
6、l-dopa是多巴胺的前药,并且在运动帕金森病的治疗方面仍是最有效的药物。然而,在治疗若干年(即蜜月期)后,由于疾病的固有进展(多巴胺能神经元的持续损失)连同l-dopa的差的药物代谢动力学(pk)曲线,出现并发症。那些并发症包括1)运动障碍,这是在药物的最佳‘持续时间效果’期间发生的异常不随意运动;以及2)波动,在此期间l-dopa正效应消失,并且症状重新出现或恶化(sprenger和poewe,cnsdrugs[cns药物](2013),27:259-272)。
7、直接多巴胺受体激动剂能够激活位于中型多棘神经元msn-1和msn-2上的多巴胺自身受体连同突触后多巴胺受体。阿朴吗啡属于一类具有1,2-二羟基苯(儿茶酚)部分的多巴胺激动剂。当与苯乙胺基序结合时,儿茶酚胺通常拥有低的口服生物利用度或没有口服生物利用度(阿朴吗啡就属于这种情况)。尽管是以非口服递送(典型地是经由泵的间断皮下给予或白天连续肠胃外输注),但是临床上在pd治疗中使用阿朴吗啡。对于阿朴吗啡,动物研究已经显示,经皮递送或植入物可以提供可能形式的给予。然而,当在猴中研究阿朴吗啡从植入物的递送时(bibbiani等人,chase experimental neurology[实验神经学](2005),192:73-78),发现在多数情况下,必须用免疫抑制剂地塞米松治疗动物,从而防止在植入手术后的局部刺激和其他并发症。已经广泛开发了用于pd中的阿朴吗啡治疗的替代性递送策略,例如吸入和舌下配制品(参见例如grosset等人,acta neurol scand.[斯堪的纳维亚神经病学学报](2013),128:166-171和hauser等人,movement disorders[运动障碍](2016),第32卷(9):1367-1372)。然而,这些努力仍未在临床上用于治疗pd。
8、儿茶酚胺的非口服配制品的替代方案涉及使用掩蔽游离儿茶酚羟基的前药来使得能够口服给予。然而,与用于临床使用的前药的开发关联的已知问题是,在人类中,与预测转化至母体化合物关联的困难。
9、在文献中已经报道了儿茶酚胺的不同酯类前药,例如用于十二指肠递送的肠溶包衣的n-丙基-阿朴吗啡(npa)酯(参见例如wo 02/100377)以及d1样激动剂阿屈利特(a-86929的二乙酰基前药)(giardina和williams;cnsdrug reviews[cns药物综述](2001),第7卷(3):305-316)。在人类中,在口服给药后,阿屈利特经历广泛的肝脏首过代谢,并且作为结果,具有低的口服生物利用度(大约4%)。在pd患者中,静脉内(iv)阿屈利特具有与l-dopa可比较的抗帕金森功效(giardina和williams;cnsdrug reviews[cns药物综述](2001),第7卷(3):305-316)。
10、除了儿茶酚胺的酯类前药,替代性前药方法涉及将两个儿茶酚羟基掩蔽为相应的亚甲二氧基(mdo)缩醛(为衍生自除了甲醛的其他醛的缩醛),或掩蔽为衍生自不同酮类的缩酮。例如已经在campbell等人,neuropharmacology[神经药理学](1982);21(10):953-961和us 4543256、wo 2009/026934以及wo 2009/026935中描述了这一前药原理。
11、对于儿茶酚胺前药,仍另一个建议的方法是形成烯酮衍生物,如在例如wo 2001/078713和liu等人,bioorganic med.chem.[生物有机化学与医药化学](2008),16:3438-3444中建议。对于儿茶酚胺前药的另外的实例,参见例如sozio等人,exp.opin.drug disc.[关于药物发现的专家意见](2012);7(5):385-406。
12、描绘为以下化合物(i)的化合物(4ar,10ar)-1-正丙基-1,2,3,4,4a,5,10,10a-八氢-苯并[g]喹啉-6,7-二醇披露于wo 2009/026934中。反式异构体之前披露于liu等人,j.med.chem.[药物化学杂志](2006),49:1494-1498中,并且之后披露于liu等人,bioorganic med.chem.[生物有机化学与医药化学](2008),16:3438-3444(包括表明此化合物在大鼠中具有低的口服生物利用度的药理数据)中。外消旋物首次披露于cannon等人,j.heterocyclic chem.[杂环化学杂志](1980);17:1633-1636中。
13、
14、化合物(i)是具有混合的d1和d2活性的多巴胺受体激动剂。本领域已知化合物(i)的三种前药衍生物。
15、liu等人,j.med.chem.[药物化学杂志](2006),49:1494-1498和liu等人,bioorganic med.chem.[生物有机化学与医药化学](2008),16:3438-3444披露了以下描绘的具有式(ia)的烯酮衍生物,显示该烯酮衍生物在大鼠中被转化为活性化合物(i)。
16、
17、wo 2009/026934和wo 2009/026935披露了化合物(i)的两个类型的前药衍生物,包括具有以下式(ib)的mdo衍生物:
18、
19、已经在wo 2010/097092中证明,在大鼠和人类肝细胞中,化合物(ib)转化为化合物(i)。此外,已经在关于帕金森病的不同动物模型中,测试了化合物(ia)和(ib)连同活性“母体化合物”(i)的体内药理学(wo 2010/097092)。发现化合物(i)以及化合物(ia)和(ib)二者是有效的,表明化合物(ia)和(ib)在体内转化为化合物(i)。已经报道所有三种化合物具有与针对l-dopa和阿朴吗啡所观察到的相比,更长的作用的持续时间。
20、在wo 2009/026934和wo 2009/026935中披露的化合物(i)的其他前药是具有式(ic)的常规酯类前药:
21、
22、尽管在本领域存在长期的兴趣,但是关于开发用于治疗pd的有效的、良好耐受的并且口服活性的药物,显然仍存在未满足的需求。可以提供连续多巴胺能刺激的、给出稳定pk曲线的混合的d1/d2激动剂的前药衍生物,可以满足此类未满足的需求。
技术实现思路
1、本发明涉及用于治疗帕金森病的新化合物。更具体地,本发明涉及化合物(4ar,10ar)-1-正丙基-1,2,3,4,4a,5,10,10a-八氢-苯并[g]喹啉-6,7-二醇(化合物(i))的新前药衍生物。已经证明,对于化合物(i)的口服递送,本发明的化合物特别有用。
2、因此,本发明涉及具有式(id)的化合物
3、
4、其中
5、r1是h并且r2选自以下取代基(i)和(ii)之一;或者
6、r1选自以下取代基(i)和(ii)之一并且r2是h;或者
7、r1和r2均由以下取代基(i)表示;或者
8、r1和r2均由以下取代基(ii)表示;或者
9、r1是取代基(i)并且r2是取代基(ii);或者
10、r1是取代基(ii)并且r2是取代基(i);
11、
12、其中*指示附接点;并且
13、其中在取代基(i)上附接点处的碳原子处于s-构型;
14、或其药学上可接受的盐。
15、在一个实施例中,本发明涉及一种药物组合物,该药物组合物包括根据式(id)的化合物或其药学上可接受的盐、以及一种或多种药学上可接受的赋形剂。
16、在一个实施例中,本发明涉及用于用作药剂的根据式(id)的化合物。
17、在一个实施例中,本发明涉及根据式(id)的化合物或其药学上可接受的盐,用于治疗神经退行性疾病或障碍,例如帕金森病、亨廷顿病、下肢不宁综合征或阿尔茨海默病;或神经精神性疾病或障碍,例如精神分裂症、注意缺陷多动障碍或药物成瘾。
18、在一个实施例中,本发明涉及用于治疗以下疾病或障碍的方法,神经退行性疾病或障碍,例如是帕金森病、亨廷顿病、下肢不宁综合征或阿尔茨海默病;或神经精神性疾病或障碍,例如精神分裂症、注意缺陷多动障碍或药物成瘾;该方法包括向有需要的患者给予治疗有效量的具有式(id)的化合物或其药学上可接受的盐。
19、在一个实施例中,本发明涉及根据式(id)的化合物或其药学上可接受的盐的用途,用于制造药剂,该药剂用于治疗神经退行性疾病或障碍,例如帕金森病、亨廷顿病、下肢不宁综合征或阿尔茨海默病;或用于治疗神经精神性疾病或障碍,例如精神分裂症、注意缺陷多动障碍或药物成瘾。
20、在本发明的上下文中,应理解,在取代基(i)上附接点处的碳原子在(i)的异头位处。
21、定义
22、本发明的化合物
23、对本发明所涵盖的化合物的提及包括本发明的化合物的游离物质(两性离子)、本发明的化合物的药学上可接受的盐,例如酸加成盐或碱加成盐、以及本发明的化合物及其药学上可接受的盐的多晶形和非结晶形式。此外,本发明的化合物及其药学上可接受的盐能潜在地以未溶剂化形式存在以及以与药学上可接受的溶剂如水、乙醇等的溶剂化形式存在。本发明涵盖了溶剂化和未溶剂化形式二者。
24、药学上可接受的盐:
25、在本上下文中的药学上可接受的盐旨在指示无毒的,即生理上可接受的盐。
26、术语“药学上可接受的盐”包括药学上可接受的酸加成盐,它们是在母体分子中的氮原子上,与无机酸和/或有机酸形成的盐。所述酸可以选自例如盐酸、氢溴酸、磷酸、亚硝酸、硫酸、苯甲酸、柠檬酸、葡糖酸、乳酸、马来酸、琥珀酸、酒石酸、乙酸、丙酸、草酸、丙二酸、富马酸、谷氨酸、焦谷氨酸、水杨酸、龙胆酸、糖精、以及磺酸,例如甲磺酸、乙磺酸、甲苯磺酸、萘-2-磺酸、2-羟基乙磺酸和苯磺酸。
27、术语“药学上可接受的盐”还包括药学上可接受的碱加成盐,它们是在具有式(id)的化合物的酸基团上,与无机碱和/或有机碱形成的盐。所述碱可以选自例如氢氧化锌,以及碱金属碱,例如氢氧化钠、氢氧化锂、氢氧化钾,以及碱土金属碱,例如氢氧化钙和氢氧化镁,以及有机碱,例如胆碱、二乙胺、三甲胺和三乙胺。
28、有用于形成药学上可接受的盐的酸和碱的另外实例可以例如在stahl和wermuth(编辑)“handbook of pharmaceutical salts.properties,selection,and use[药用盐手册:特性、选择和使用]”,威利-vch出版社(wiley-vch),2008中找到。
29、固体形式
30、在本上下文中,当本发明的化合物处于固体形式时,这表明所述化合物未溶于任何液体中,例如水性液体、有机液体及其混合物。本发明涵盖本发明的化合物的游离物质(两性离子)的固体形式,连同本发明的化合物的药学上可接受的盐的固体形式。术语“固体形式”涵盖本发明的化合物及其盐的非结晶形式,以及本发明的化合物及其盐的结晶形式二者。
31、前药
32、在本上下文中,术语“前药”或“前药衍生物”指示在给予活的受试者,例如哺乳动物,优先地人类后;化合物在体内转化为药理学上活性的部分。该转化优选地发生在哺乳动物体内,例如在小鼠、大鼠、狗、小型猪、兔、猴和/或人类体内。在本上下文中,“化合物(4ar,10ar)-1-正丙基-1,2,3,4,4a,5,10,10a-八氢-苯并[g]喹啉-6,7-二醇的前药”,或“具有式(i)的化合物的前药”,或“化合物(i)的前药”被理解为是,在给予后,在体内转化为化合物(4ar,10ar)-1-正丙基-1,2,3,4,4a,5,10,10a-八氢-苯并[g]喹啉-6,7-二醇的化合物。所述给予可以是通过本领域已知的药物组合物的常规给予途径,优选地通过口服给予。
33、在本上下文中,术语“母体化合物”和“母体分子”指示在相应前药的转化中获得的药理学上活性的部分。例如,化合物(ia)、(ib)、(ic)之一或本发明的化合物中的任一项的“母体化合物”被理解为是具有式(i)的化合物。
34、化学制造
35、在本上下文中,“由化学制造来源的”化合物指示已经通过化学工艺,例如但不限于本文实验部分中描述的工艺之一,制造的所述化合物。
36、药物代谢动力学定义和缩写
37、如本文使用的,“pk曲线”是“药物代谢动力学曲线”的缩写。本文描述的药物代谢动力学曲线和药物代谢动力学参数是基于使用非房室模型,在本发明的化合物的口服给药后,针对具有式(i)的化合物,获得的血浆浓度-时间数据。缩写的pk参数是:cmax(最大浓度);tmax(达到cmax的时间);t1/2(半衰期);auc0-∞(从给药时间直到无限的曲线下面积)。
38、治疗有效量:
39、在本上下文中,术语化合物的“治疗有效量”意指足以在包括给予所述化合物的治疗性介入中缓解、阻滞、部分阻滞、除去或延迟给定疾病及其并发症的临床表现的量。将足以实现以上的量定义为“治疗有效量”。用于各目的有效量将取决于例如疾病或损伤的严重程度以及受试者的体重及一般状态。将理解的是,确定适当剂量可以使用常规实验,通过构筑值矩阵并测试矩阵中的不同点来实现,这均在受训医师的普通技术内。
40、在本发明的上下文中,本发明的化合物的“治疗有效量”表示在给予本发明的所述化合物(优选地通过口服途径)至哺乳动物(优选地人类)时,本发明的所述化合物的量能够提供足以缓解、阻滞、部分阻滞、除去或延迟给定疾病及其并发症的临床表现的化合物(i)的量。
41、治疗(treatment和treating):
42、在本上下文中,“治疗(treatment)”或“治疗(treating)”旨在指示管理并护理患者,用于缓解、阻滞、部分阻滞、除去疾病的临床表现或延迟其进展的目的。欲治疗的患者优选是哺乳动物,特别是人类。
43、用于治疗的病症:
44、本发明的化合物旨在用于治疗神经退行性疾病和障碍,例如帕金森病和/或其他病症,对于该治疗,其中多巴胺激动剂是治疗有益的。
45、治疗适应症包括中枢神经系统障碍,其特征在于运动和/或非运动障碍,并且对此,潜在的病理生理学的一部分是纹状体介导的回路的障碍。此类功能障碍可以见于神经退行性疾病,例如但不限于帕金森病(pd)、下肢不宁综合征、亨廷顿病和阿尔茨海默病,还有神经精神性疾病,例如但不限于精神分裂症、注意缺陷多动障碍和药物成瘾。
46、除了神经退行性疾病和障碍,其他病症,其中多巴胺能转换的增加,在心智功能,包括认知的不同方面的改善中会是有益的。在抑郁患者中,它还会具有正效应,并且它还可以作为食欲抑制剂用于治疗肥胖症,以及用于治疗药物成瘾。它可以改善轻度脑机能障碍(mbd)、昏睡病、注意缺陷多动障碍以及潜在地,精神分裂症的阴性症状、阳性症状连同认知症状。
47、下肢不宁综合征(rls)和周期性肢体运动障碍(plmd)是替代的适应症,它们是临床上用多巴胺激动剂治疗的。此外,阳萎、勃起功能障碍、ssri诱导的性功能障碍、卵巢过度刺激综合征(ohss)和某些垂体肿瘤(催乳素瘤)也可能通过用多巴胺激动剂进行治疗而改善。多巴胺参与了心血管系统和肾系统的调控,并且因此肾衰竭和高血压可以被认为是针对本发明的化合物的替代的适应症。
48、本发明涵盖了本发明的化合物用于治疗以上列出的疾病和障碍的用途。
49、组合
50、在本发明的一个实施例中,具有式(id)的化合物作为唯一活性化合物用作独立治疗(stand-alone treatment)。在本发明的另一个实施例中,具有式(id)可以与在神经退行性疾病或障碍,例如帕金森病的治疗中有用的其他药剂组合使用。如本文中在本发明的方法(包括组合给予治疗有效量的具有式(id)的化合物和另一种化合物,该化合物在治疗神经退行性疾病或障碍的治疗中有用)的上下文中使用的术语“组合使用”、“与……组合”以及“……的组合”等旨在是指同时或者顺序地(以任何顺序)与所述其他化合物一起给予具有式(id)的化合物。
51、这两种化合物可以同时给予或者在这两种化合物的给予之间有时间间隔。可以作为同一药物配制品或组合物的一部分、或在分开的药物配制品或组合物中,给予这两种化合物。可以在同一天或不同天给予这两种化合物。它们可以通过同一个途径给予,例如像通过口服给予、皮下注射、通过经皮给予、通过储库(depot)型、通过肌内注射或静脉内注射;或者通过不同途径给予,其中一种化合物例如口服给予或通过储库放置,并且例如注射另一种化合物。可以通过相同给药方案或间隔给予这两种化合物,例如每天、每周、或每月一次或两次;或可以通过不同给药方案给予这两种化合物,例如其中每天一次给予一种化合物,并且每天或每周或每月两次给予另一种化合物。
52、在一些情况下,当用具有式(id)的化合物开始治疗时,待治疗的患者可能已经用在神经退行性疾病或障碍的治疗中有用的一种或多种其他化合物进行治疗。在其他情况下,当用在神经退行性疾病或障碍的治疗中有用的一种或多种其他化合物开始治疗时,该患者可能已经用具有式(id)的化合物进行治疗。在其他情况下,用具有式(id)的化合物的治疗和用在神经退行性疾病或障碍的治疗中有用的一种或多种其他化合物的治疗同时开始。
53、用于组合治疗的化合物
54、在本发明的上下文中,与具有式(id)的化合物组合使用的化合物可以选自例如l-dopa、屈昔多巴、mao-b抑制剂,例如司来吉兰或雷沙吉兰,comt抑制剂,例如恩他卡朋或托卡朋,腺苷2a拮抗剂,例如伊曲茶碱,抗谷氨酸剂,例如金刚胺或美金刚,乙酰胆碱酯酶抑制剂,例如利凡斯的明、多奈哌齐或加兰他敏,以及抗精神病剂,例如喹硫平、氯氮平、利哌酮、匹莫范色林、奥氮平、氟哌啶醇、阿立哌唑或布瑞哌唑。
55、除了小分子,用于组合的化合物还可以包括在神经退行性疾病或障碍的治疗中,新兴的生物制剂方法,例如像靶向α-突触核蛋白、τ蛋白或a-β蛋白的抗体。
56、给药途径
57、包含具有式(id)的化合物(作为唯一活性化合物或与另一种活性化合物组合)的药物组合物可以被具体配制用于通过任何适合途径给予,例如经口、经直肠、经鼻、经颊、舌下、经肺、经皮和非经肠(例如皮下、肌内和静脉内)途径。在本发明的上下文中,口服途径是优选的给予途径。
58、将领会的是,该途径将取决于待治疗的受试者的一般状况和年龄、待治疗的病症的性质以及活性成分。
59、药物配制品和赋形剂
60、在下文中,术语“赋形剂”或“药物上可接受的赋形剂”是指药物赋形剂,包括但不限于载体、填充剂、稀释剂、抗粘合剂、粘合剂、包衣、着色剂、崩解剂、调味剂、助流剂、润滑剂、防腐剂、吸着剂、甜味剂、溶剂、运载体和佐剂。
61、本发明还提供了包括具有式(id)的化合物(例如在本文实验部分中所披露的化合物之一)的药物组合物。本发明还提供了用于制造包括具有式(id)的化合物的药物组合物的方法。根据本发明的药物组合物可以用药学上可接受的赋形剂根据常规技术进行配制,这些常规技术例如在以下中披露的那些:remington,“the science and practice ofpharmacy[药学科学与实践]”,第22版(2012),由allen,loyd v.,jr.编辑。
62、优选地,包含本发明的化合物的药物组合物是用于口服给予的药物组合物。用于口服给予的药物组合物包括固体口服剂型,例如片剂、胶囊、粉剂以及颗粒剂;和液体口服剂型,例如溶液、乳剂、悬浮液和糖浆剂以及待溶解或悬浮在合适液体中的粉剂和颗粒剂。
63、固体口服剂型可以离散单位形式呈现(例如片剂或硬胶囊或者软胶囊),各自包含预定量的活性成分,并且优选地包括一种或多种适合的赋形剂。适当时,根据本领域中熟知的方法,这些固体剂型可以制备为具有包衣,例如肠溶包衣,或者它们可以被配制以提供活性成分的改变的释放,例如延迟或延长释放。适当时,该固体剂型可以是在唾液中崩解的剂型,例如口腔分散片剂。
64、适于固体口服配制品的赋形剂的实例包括但不限于:微晶纤维素、玉米淀粉、乳糖、甘露醇、聚维酮、交联羧甲纤维素钠、蔗糖、环糊精、滑石、明胶、果胶、硬脂酸镁、硬脂酸和纤维素的低级烷基醚。类似地,固体配制品剂可包括本领域已知的、延迟或延长释放配制品的赋形剂,例如单硬脂酸甘油酯或羟丙甲纤维素。如果将固体材料用于口服给予,则该配制品可以例如通过将活性成分与固体赋形剂混合,并且随后在常规压片机中压缩该混合物来制备;或可以例如将该配制品以例如粉剂、丸剂或微型片剂形式置于硬胶囊中。固体赋形剂的量将广泛变化,但将典型地在每剂量单位从约25mg至约1g的范围。
65、液体口服剂型能以例如酏剂、糖浆剂、口服滴剂或充液胶囊呈现。液体口服剂型还能以用于在水性或非水性液体中的溶液或悬浮液的粉剂呈现。适合于液体口服配制品的赋形剂的实例包括,但不限于乙醇、丙二醇、甘油、聚乙二醇、泊洛沙姆、山梨醇、聚山梨醇酯、甘油单酯和甘油二酯、环糊精、椰子油、棕榈油和水。液体口服剂型可以例如通过将活性成分溶解或悬浮在水性或非水性液体中,或通过将活性成分掺入水包油或油包水液体乳液中来制备。
66、可以将另外的赋形剂(例如,着色剂、调味剂和防腐剂等)用于固体和液体口服配制品中。
67、用于非经肠给药的药物组合物包括:用于注射或输注的无菌水性及非水性溶液、分散液、悬浮液或乳液、用于注射或输注的浓缩物以及欲在使用之前在用于注射或输注的无菌溶液或分散液中复水的无菌粉剂。适合于非经肠配制品的赋形剂的实例包括,但不限于水、椰子油、棕榈油和环糊精溶液。必要时应该适当缓冲水性配制品,并且用足够盐水或葡萄糖使水性配制品变得等张。
68、其他类型的药物组合物包括栓剂、吸入剂、乳膏剂、凝胶剂、皮肤贴片、植入物和用于经颊或舌下给药的配制品。
69、用于任何药物配制品的赋形剂必须符合预期的给药途径并且与活性成分相容。
70、剂量:
71、在一个实施例中,每天以从约0.0001mg/kg体重至约5mg/kg体重的量给予本发明的化合物。具体而言,每日剂量可以处于每天0.001mg/kg体重至约1mg/kg体重的范围内。精确剂量将取决于给予频率及模式,待治疗的受试者的性别、年龄、体重及一般状况,待治疗的病症、任何待治疗的伴随疾病的性质及严重程度,所希望的治疗效果以及本领域的普通技术人员已知的其他因素。
72、针对成人的典型口服剂量在以下范围内:0.01-100mg/天的本发明的化合物,例如0.05-50mg/天,例如0.1-10mg/天或0.1-5mg/天。方便地,本发明的化合物是以一种单位剂型给予,该单位剂型以如下的量包含所述化合物:约0.01至50mg,例如0.05mg、0.1mg、0.2mg、0.5mg、1mg、5mg、10mg、15mg、20mg或多达50mg的本发明化合物。
- 还没有人留言评论。精彩留言会获得点赞!