基于化学反应的分子多视图信息的PI3K抑制剂活性预测方法
本发明涉及药物性质预测,尤其涉及一种基于化学反应的分子多视图信息的pi3k抑制剂活性预测方法。
背景技术:
1、pi3k又称磷脂酰肌醇-3-激酶,可催化磷脂酰肌醇分子磷酸化,pi3k分为三类:class i、class ii和class iii,对于药物研发来说,主要针对class ipi3k。而对于classipi3k,其活性亚基p110有α、β、γ、δ四种亚型,pi3kα抑制剂对于乳腺癌适应症有着不错的效果;pi3kβ抑制剂被认为是潜在靶向pten缺失肿瘤的有效药物;pi3kγ抑制剂在控制免疫性疾病方面有着巨大潜力;δ亚型是重要的血液肿瘤靶点,因此pi3kδ抑制剂是血液肿瘤的有效药物。然而目前所上市的pi3k抑制剂药物并不多,pi3k抑制剂新药研发的一大挑战就是药物从基础科学到早期临床实验的转化成本高。专业人员需要从大量的候选化合物当中进行筛选,方式包括通过检测候选化合物的活性和毒性等理化性质。当整个流程仅使用化学方法时,时间成本和经济成本都较高,往往需要对大量的化合物做大量的实验,且伴随着较高的失败率。
2、pi3k抑制剂是一种临床上广泛被认可和应用的抗肿瘤药物,但是pi3k的毒性、选择性和耐药性等分子性质使得pi3k在临床使用上有一定局限性。使用人工智能方法寻找和筛选新的pi3k抑制剂,可以有效缩短试验周期、节约研发资金、提高临床转化率。药物性质预测是新药研发过程中的关键任务。现有的基于深度学习的药物性质预测,往往使用序列结构和图形结构给分子建模进行预测。相比序列结构,图形结构可以更恰当的表达分子的网状结构,因此往往可以取得更好的预测效果,因此成为了表达分子表征的热门方法。
3、然而,现在的许多研究只关注了分子图的建模策略,而忽略了分子的理化性质本身,因此缺少化学先验知识的介入。此外,在基于人工智能的pi3k抑制剂活性预测领域,还没有整理好的pi3k抑制剂的公共数据集。
4、因此,亟需一种新的结合反应片段和化学反应信息等化学先验知识的分子性质预测方法来和有效的pi3k数据集解决上述问题。
技术实现思路
1、针对现有技术中所存在的不足,本发明提供了一种基于化学反应的分子多视图信息的pi3k抑制剂活性预测方法,形成了有效的pi3k抑制剂数据集,并提出了局部有向通信消息传递神经网络模型ld-cmpnn,结合了化学反应来提取出分子的片段视图,通过分子的多视图信息来对pi3k抑制剂的活性进行预测,该方法预测准确率高,可以有效缩短后续临床试验失败的风险,大大提升pi3k抑制剂新药研发的成功率。
2、本发明的上述技术目的是通过以下技术方案得以实现的:
3、一种基于化学反应的分子多视图信息的pi3k抑制剂活性预测方法,包括以下步骤:
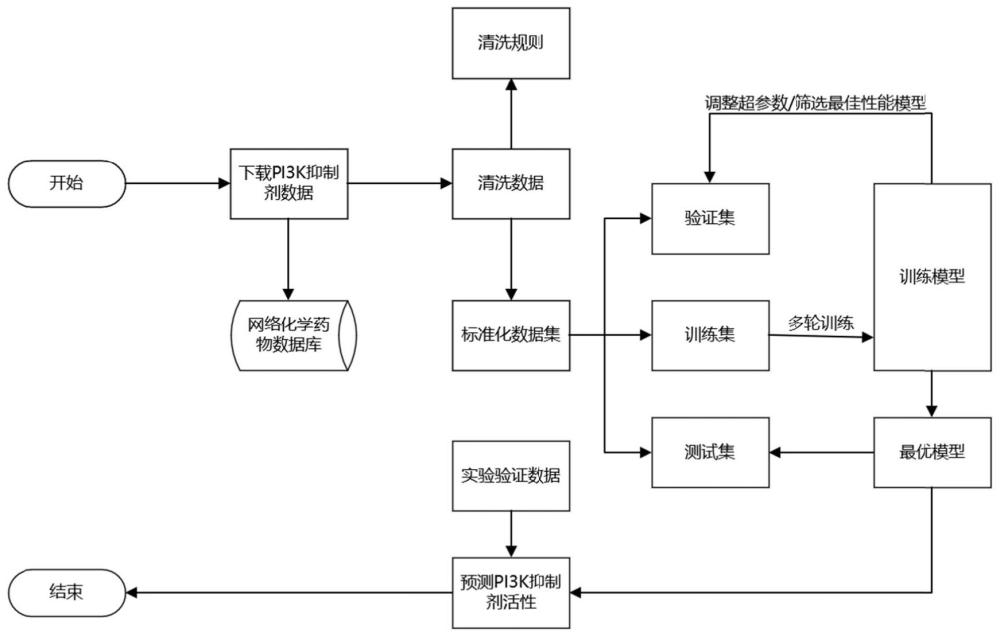
4、s1,pi3k抑制剂数据收集阶段,包括:收集pi3k抑制剂数据,对pi3k抑制剂数据进行清洗和标注并构建pi3k抑制剂数据集,将pi3k抑制剂数据集划分为训练集、验证集和测试集;
5、s2,片段视图构建阶段,包括:根据brics方法将原始分子分割成片段,并保留边上的反应信息,将各个片段当作图上的点,构建片段视图并提取各个片段的药效团信息;
6、s3,构建深度学习模型阶段,包括:将片段视图和原始分子视图分别输入到不同的图编码器来提取出两种不同的特征,最终用全连接层组合成融合型神经网络框架;
7、s4,应用预测阶段,包括:将pi3k抑制剂数据集放入模型中进行预测,得到预测结果。
8、本发明进一步设置为:在步骤s1中,具体包括以下步骤:
9、s1-1,收集pi3k抑制剂的四种亚型数据,包括pi3k-α,pi3k-β,pi3k-γ,pi3k-δ;
10、s1-2,保留具有绝对生物测定值的化合物,并丢弃没有生物活性数据的化合物;
11、s1-3,当一个分子具有多个生物活性数据点时,将这些报告的生物测定值的平均值作为最终值;
12、s1-4,去除重复分子;
13、s1-5,将数据集中的每种化合物标准化为简化分子线性输入规范格式smiles,包括去除氢离子、溶剂成分和盐,以及通过添加或减去原子来中和电荷;最终生物活性值≤1um的分子被标记为活性分子,作为正样本;生物活性值>1um的分子被标记为非活性分子,作为负样本。
14、本发明进一步设置为:在步骤s2中,具体包括以下步骤:
15、s2-1,使用基本化学反应模板的brics方法将分子分割成片段;
16、s2-2,将各个片段视作一个整体,映射成分子图中节点,片段之间连接的化学键映射成分子图中的边,由此构建片段视图;
17、s2-3,根据输入片段的smiles格式生成两种不同类型的分子指纹:基于子结构的分子指纹maccs和基于药效团的分子指纹;将两种指纹拼接得到片段的初始特征向量;根据brics方法分割时得到的化学反应信息作为片段之间连接的化学键的初始特征向量。
18、本发明进一步设置为:在步骤s3中,具体包括以下步骤:
19、s3-1,对于原始的分子视图,将原子映射成分子图的节点,将化学键映射成分子图中的边;计算原子的理化性质,作为节点的初始特征向量;根据键的类型,作为边的初始特征向量;
20、s3-2,将片段视图和原始分子视图分别输入到不同的图编码器得到出两种不同的特征向量,将这两个特征向量拼接,然后输入到多层全连接层,以用来预测pi3k抑制剂的活性;
21、s3-3,原始分子视图所输入的图编码器为局部有向通信消息传递神经网络模型ld-cmpnn,片段视图所输入的图编码器为通信消息传递神经网络模型cmpnn。
22、本发明进一步设置为:在步骤s4中,具体包括以下步骤:
23、s4-1,模型可以自定义拆分比例、拆分方式、训练迭代轮数和交叉验证折数;
24、s4-2,将pi3k抑制剂数据集以smiles格式输入到模型,得到pi3k抑制剂活性的预测结果。
25、本发明进一步设置为:在步骤s1-1中,从bindingdb,pubchem和chembl三个数据库里抽取pi3k抑制剂的四种亚型数据。
26、本发明进一步设置为:在步骤s3-1中,原子的理化性质包括原子序数、带电荷数、原子的度、手性碳信息、相连氢原子数、杂化轨道类型、原子质量、是否是芳香原子。
27、本发明进一步设置为:在步骤s3-1中,键的类型包括单键、双键、三键和芳香键。
28、本发明进一步设置为:在步骤s3-3中,所述局部有向通信消息传递神经网络模型ld-cmpnn在消息传递时的计算流程包括:
29、k代表消息传递的深度,k代表消息传递的当前深度,其中k=1…k;设定每个节点v聚合其入边,创建一个中间消息向量mk(v),在获得消息向量后,将节点的当前隐藏状态hk-1(v)与消息向量连接,并通过通信函数将该连接的特征向量馈送,以将节点的隐藏状态更新为hk(v);其中,
30、
31、hk(v)=communicate(mk(v),hk-1(v))#
32、若节点到节点w之间的边为非切割边,那么该边为无向消息传递,计算公式如下:
33、
34、若节点v到节点w之间的边为切割边,那么该边为有向消息传递,由于之前已经在hk(v)中获得了邻边信息,因此可以通过hk(v)中减去其逆键信息来获得mk(ev,w),计算公式如下:
35、mk(ev,w)=hk(v)-hk-1(ew,v)#。
36、本发明具有以下有益效果:
37、本发明基于化学反应划分了分子片段,融合了分子的片段视图中的药效团信息和反应信息,解决了深度学习在小规模数据集上无法有效提取到重要特征的缺陷。本发明还收集了大量可靠的pi3k抑制剂数据,为以后基于机器学习的pi3k抑制剂活性预测工作提供了数据支撑。传统的pi3k抑制剂药物研发,若要去寻找候选药物,其搜索空间是巨大的,通过本发明所提供的方法能够及时发现大量物理化学性质不达标的化合物,为后续临床试验大大降低了成本和失败的风险,从而能够提高药物研发的成功率。
- 还没有人留言评论。精彩留言会获得点赞!