一种碳点改性荧光纤维及其制备方法
1,5-二氨基戊烷溶液中,在50~80℃水浴锅中反应15~24h进行氨基化改性处理,取出氨基化改性的纤维,并用去离子水充分冲洗,放入0.5~0.8mol/l的1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、0.1~0.2mol/l的n-羟基丁二酰亚胺和0.5~1.5mol/l的脂肪酸甲酯磺酸钠缓冲液中进行交联反应,在25℃摇床中摇晃反应2~5h后用去离子水冲洗,之后置于温度约为5℃的冰箱中待用;
12.s4、荧光纤维的制备:将1~5g的表面改性后的纤维与5~40ml碳点溶液放入80ml的带有聚四氟乙烯内衬的高压反应釜中,再加入0~35ml去离子水以确保将纤维浸入液体中,随后,将高压反应釜密封并在烘箱中在60~200℃下反应4~10h,待反应釜冷却后,取出样品,抽滤,用去离子水洗涤5~10次直至滤液无荧光反应,将获得的纤维在40~60℃的真空干燥箱内干燥12~48h,最终得到长度为2~6mm的荧光纤维。
13.优选的,所述步骤s2中的透析膜的分子截留量为mwco:1000。
14.优选的,所述步骤s1中的合成碳点的碳源原料为柠檬酸、丙烯酸、葡萄糖、氨基酸、木质素、淀粉、蛋白质、聚氨酯、聚丙烯酰胺、聚乙二醇中的一种或几种混合。
15.优选的,所述步骤s1中的合成碳点的杂原子掺杂原料为苯二胺、乙二胺、聚乙烯亚胺、邻苯二胺二聚体、尿素、氨水、氨基酸、硅烷、2-氨基苯硫酚、磷酸、三聚氰胺中的一种或几种混合。
16.优选的,所述步骤s3中的纤维基材为聚酯纤维、聚丙烯腈纤维、尼龙纤维、芳纶、锦纶、涤纶、腈纶、氨纶、维纶、丙纶、氯纶、大豆纤维中的一种或几种混合。
17.一种碳点改性荧光纤维作为防伪基材在包装防伪、军事伪装、道路与高空作业的防护服、日用品、特种感光材料及信息存储材料的制备上的应用。
18.与现有技术相比,本发明的有益效果是:
19.本发明中防伪信息标记物难以被分离,采用化学载附的方式进行荧光标记得到纳米有机荧光纤维,所使用的荧光物质迁出率极低,无法采用现有的分析仪器进行采样分离与组分分析,在薄膜材料领域具有极高的防伪效能,进一步加强了技术安全性与防伪可靠性,其防伪效能是传统防伪技术所无法比拟的,市场潜力巨大。
附图说明
20.图1为傅里叶变换红外谱图;
21.图2为cds的粒径分布图;
22.图3为cds的平均粒径图;
23.图4为荧光发射光谱图。
具体实施方式
24.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。
25.一种碳点改性荧光纤维,由以下重量份的原料组成:碳点改性荧光纤维0.1~1份;相容剂:1~20份;增塑剂:1~5份。
26.碳点改性荧光纤维的碳点尺寸为5~10nm,碳点改性荧光纤维的纤维直径为22~28μm,碳点改性荧光纤维的纤维长度为2~6mm。
27.一种碳点改性荧光纤维的制备方法,包括以下步骤:
28.s1、碳点的合成:将1~5g合成碳点的碳源原料和1~5g杂原子掺杂原料在25ml去离子水中混合以形成均匀溶液,然后将溶液转移到以聚四氟乙烯为内衬的100ml的高压反应釜中,将高压反应釜封紧后放入烘箱中,反应温度设置为60~200℃后,反应时间为4~10h,反应结束后,关闭烘箱,待反应釜冷却至室温后,得到黄棕色液体;
29.s2、碳点的分离:将产物转移至去离子水中搅拌均匀并以5000~10000rpm离心15~30min以除去较大的颗粒杂质,再用0.1~10um滤膜过滤除去不溶性物质,使用透析膜对黄色上清液透析24h,每隔12h更换一次去离子水,以除去未反应的原料,最终得到尺寸为5~10nm碳点的水溶液;
30.s3、纤维基材的表面改性:将2g长度为10~40mm的纤维基材放入无水乙醇中超声0.5~3h,之后用去离子水清洗,将清洗后的纤维基材浸入到浓度为0.5~3mol/l的2-甲基-1,5-二氨基戊烷溶液中,在50~80℃水浴锅中反应15~24h进行氨基化改性处理,取出氨基化改性的纤维,并用去离子水充分冲洗,放入0.5~0.8mol/l的1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、0.1~0.2mol/l的n-羟基丁二酰亚胺和0.5~1.5mol/l的脂肪酸甲酯磺酸钠缓冲液中进行交联反应,在25℃摇床中摇晃反应2~5h后用去离子水冲洗,之后置于温度约为5℃的冰箱中待用;
31.s4、荧光纤维的制备:将1~5g的表面改性后的纤维与5~40ml碳点溶液放入80ml的带有聚四氟乙烯内衬的高压反应釜中,再加入0~35ml去离子水以确保将纤维浸入液体中,随后,将高压反应釜密封并在烘箱中在60~200℃下反应4~10h,待反应釜冷却后,取出样品,抽滤,用去离子水洗涤5~10次直至滤液无荧光反应,将获得的纤维在40~60℃的真空干燥箱内干燥12~48h,最终得到长度为2~6mm的荧光纤维。
32.步骤s2中的透析膜的分子截留量为mwco:1000。
33.步骤s1中的合成碳点的碳源原料为柠檬酸、丙烯酸、葡萄糖、氨基酸、木质素、淀粉、蛋白质、聚氨酯、聚丙烯酰胺、聚乙二醇中的一种或几种混合。
34.步骤s1中的合成碳点的杂原子掺杂原料为苯二胺、乙二胺、聚乙烯亚胺、邻苯二胺二聚体、尿素、氨水、氨基酸、硅烷、2-氨基苯硫酚、磷酸、三聚氰胺中的一种或几种混合。
35.步骤s3中的纤维基材为聚酯纤维、聚丙烯腈纤维、尼龙纤维、芳纶、锦纶、涤纶、腈纶、氨纶、维纶、丙纶、氯纶、大豆纤维中的一种或几种混合。
36.一种碳点改性荧光纤维作为防伪基材在包装防伪、军事伪装、道路与高空作业的防护服、日用品、特种感光材料及信息存储材料的制备上的应用。
37.实施例1
38.s1、将1g柠檬酸和3g聚乙烯亚胺在25ml去离子水中混合以形成均匀溶液,然后将溶液转移到以聚四氟乙烯为内衬的100ml的高压反应釜中,将高压反应釜封紧后放入烘箱中,反应温度设置为180℃后,反应时间为8h,反应结束后,关闭烘箱,待反应釜冷却至室温后,得到黄棕色液体;
39.s2、将产物转移至超纯水中搅拌均匀并以10000rpm离心30min以除去较大的颗粒杂质,将滤液放置冰箱中冷藏12h,再用2um滤膜过滤除去不溶性物质,使用透析膜(分子截留量为mwco:1000)对黄色上清液透析24h,每隔12h更换一次超纯水,以除去未反应的原料,最终得到平均粒径为5.2nm的碳点水溶液;
40.s3、将2g长度为10mm的聚丙烯腈纤维放入无水乙醇中超声2h,之后用去离子水冲洗,将冲洗后的纤维基材浸入到浓度为2mol/l的2-甲基-1,5-二氨基戊烷溶液中,在50℃水浴锅中反应20h进行氨基化改性处理,取出氨基化改性的纤维,用去离子水冲洗后将其放入0.5mol/l的1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、0.1mol/l的n-羟基丁二酰亚胺和1mol/l的mes缓冲液中进行交联反应,在25℃摇床中摇晃反应2h后用去离子水冲洗,之后置于温度约为5℃的冰箱中待用;
41.s4、将表面改性后的聚丙烯腈纤维与30ml碳点溶液放入80ml的带有聚四氟乙烯内衬的高压反应釜中,再加入10ml去离子水以确保将纤维浸入液体中,随后,将高压反应釜密封并在烘箱中在180℃下反应8h,待反应釜冷却后,取出样品,抽滤,用去离子水洗涤8次直至滤液无荧光反应,将获得的纤维在50℃的真空干燥箱内干燥12h,最终得到长度为7mm的荧光纤维。
42.实施例2
43.s1、将5g丙烯酸和2g尿素在25ml去离子水中混合以形成均匀溶液,然后将溶液转移到以聚四氟乙烯为内衬的100ml高压反应釜中,将高压反应釜封紧后放入烘箱中,反应温度设置为160℃后,反应时间为10h,反应结束后,关闭烘箱,待反应釜冷却至室温后,得到黄棕色液体;
44.s2、将产物转移至超纯水中搅拌均匀并以8000rpm离心30min以除去较大的颗粒杂质,将滤液放置冰箱中冷藏12h,再用2um滤膜过滤除去不溶性物质,使用透析膜(分子截留量为mwco:1000)对黄色上清液透析24h,每隔12h更换一次超纯水,以除去未反应的原料,最终得到平均粒径为5.5nm的碳点水溶液;
45.s3、将2g长度为20mm的聚酯纤维放入无水乙醇中超声3h,之后用去离子水冲洗,将冲洗后的纤维基材浸入到浓度为1.5mol/l的2-甲基-1,5-二氨基戊烷溶液中,在60℃水浴锅中反应15h进行氨基化改性处理。取出氨基化改性的纤维,用去离子水冲洗后将其放入20ml15%(体积分数)的戊二醛溶液中,在30℃摇床中摇晃反应4h后用去离子水冲洗,之后置于温度约为5℃的冰箱中待用;
46.s4、将表面改性后的聚酯纤维与25ml碳点溶液放入80ml的带有聚四氟乙烯内衬的高压反应釜中,再加入15ml去离子水以确保将纤维浸入液体中,随后,将高压反应釜密封并在烘箱中在160℃下反应10h,冷却后,抽滤,洗涤直至呈中性,将获得的纤维在80℃条件下干燥12h,最终得到长度为8mm的荧光纤维。
47.实施例3
48.s1、将2g葡萄糖和5g乙二胺在30ml去离子水中混合以形成均匀溶液,然后将溶液转移到以聚四氟乙烯为内衬的100ml高压反应釜中,将高压反应釜封紧后放入烘箱中,反应温度设置为140℃后,反应时间为6h,反应结束后,关闭烘箱,待反应釜冷却至室温后,得到黄棕色液体;
49.s2、将产物转移至超纯水中搅拌均匀并以10000rpm离心25min以除去较大的颗粒杂质,将滤液放置冰箱中冷藏12h,再用1um滤膜过滤除去不溶性物质,使用透析膜(分子截留量为mwco:1000)对黄色上清液透析24h,每隔12h更换一次超纯水,以除去未反应的原料,最终得到平均粒径为5.4nm的碳点水溶液;
50.s3、将2g长度为20mm的大豆纤维放入无水乙醇中超声1h,之后用去离子水冲洗,将
冲洗后的纤维基材浸入到浓度为1.5mol/l的2-甲基-1,5-二氨基戊烷溶液中,在60℃水浴锅中反应15h进行氨基化改性处理,取出氨基化改性的纤维,用去离子水冲洗后将其放入20ml25%(体积分数)的戊二醛溶液中,在25℃摇床中摇晃反应5h后用去离子水冲洗,之后置于温度约为5℃的冰箱中待用;
51.s4、将表面改性后的大豆纤维与20ml碳点溶液放入80ml的带有聚四氟乙烯内衬的高压反应釜中,再加入20ml去离子水以确保将纤维浸入液体中,随后,将高压反应釜密封并在烘箱中在175℃下反应7h,待反应釜冷却后,取出样品,抽滤,用去离子水洗涤8次直至滤液无荧光反应,将获得的纤维在50℃的真空干燥箱内干燥12h,最终得到长度为6mm的荧光纤维。
52.实施例4
53.s1、将4g淀粉和3g三聚氰胺在30ml去离子水中混合以形成均匀溶液,然后将溶液转移到以聚四氟乙烯为内衬的100ml高压反应釜中,将高压反应釜封紧后放入烘箱中,反应温度设置为165℃后,反应时间为5h,反应结束后,关闭烘箱,待反应釜冷却至室温后,得到黄棕色液体;
54.s2、将产物转移至超纯水中搅拌均匀并以9000rpm离心30min以除去较大的颗粒杂质,将滤液放置冰箱中冷藏12h,再用2um滤膜过滤除去不溶性物质,使用透析膜(分子截留量为mwco:1000)对黄色上清液透析24h,每隔12h更换一次超纯水,以除去未反应的原料,最终得到平均粒径为5.5nm的碳点水溶液;
55.s3、将2g长度为20mm的尼龙纤维放入无水乙醇中超声2h,之后用去离子水冲洗,将冲洗后的纤维基材浸入到浓度为2.4mol/l的2-甲基-1,5-二氨基戊烷溶液中,在80℃水浴锅中反应20h进行氨基化改性处理,取出氨基化改性的纤维,用去离子水冲洗后将其放入10ml30%(体积分数)的戊二醛溶液中,在25℃摇床中摇晃反应5h后用去离子水冲洗,之后置于温度约为5℃的冰箱中待用;
56.s4、将表面改性后的尼龙纤维与26ml碳点溶液放入80ml的带有聚四氟乙烯内衬的高压反应釜中,再加入14ml去离子水以确保将纤维浸入液体中,随后,将高压反应釜密封并在烘箱中在175℃下反应8h,待反应釜冷却后,取出样品,抽滤,用去离子水洗涤8次直至滤液无荧光反应,将获得的纤维在50℃的真空干燥箱内干燥12h,最终得到长度为12mm的荧光纤维。
57.实施例5
58.s1、将5g聚氨酯和2g氨水在30ml去离子水中混合以形成均匀溶液,然后将溶液转移到以聚四氟乙烯为内衬的100ml高压反应釜中,将高压反应釜封紧后放入烘箱中,反应温度设置为170℃后,反应时间为9h。反应结束后,关闭烘箱,待反应釜冷却至室温后,得到黄棕色液体;
59.s2、将产物转移至超纯水中搅拌均匀并以10000rpm离心25min以除去较大的颗粒杂质,将滤液放置冰箱中冷藏24h,再用1um滤膜过滤除去不溶性物质,使用透析膜(分子截留量为mwco:1000)对黄色上清液透析24h。每隔12h更换一次超纯水,以除去未反应的原料,最终得到平均粒径为8.6nm的碳点水溶液;
60.s3、将2g长度为8mm的芳纶放入无水乙醇中超声4h,之后用去离子水冲洗,将冲洗后的纤维基材浸入到浓度为3mol/l的2-甲基-1,5-二氨基戊烷溶液中,在80℃水浴锅中反
应25h进行氨基化改性处理。取出氨基化改性的纤维,用去离子水冲洗后将其放入20ml30%(体积分数)的戊二醛溶液中,在25℃摇床中摇晃反应5h后用去离子水冲洗,之后置于温度约为5℃的冰箱中待用;
61.s4、将表面改性后的芳纶与15ml碳点溶液放入入80ml的带有聚四氟乙烯内衬的高压反应釜中,再加入25ml去离子水以确保将纤维浸入液体中。随后,将高压反应釜密封并在烘箱中在165℃下反应10h。冷却后,抽滤,洗涤直至呈中性,将获得的纤维在80℃条件下干燥12h,最终得到长度为6mm的荧光纤维。
62.本发明所列举的各原料,以及本发明各原料的上下限、区间取值,以及工艺参数(如温度、时间等)的上下限、区间取值都能实现本发明,在此不一一列举实施例。
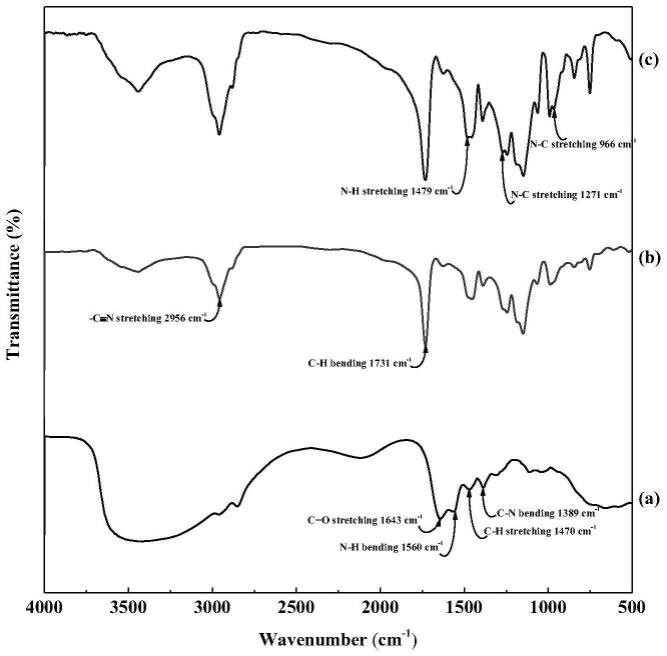
63.参照图1,图1为本发明实施例一制备的碳点的红外谱图,图1中(a)、(b)和(c)分别是本发明实施例一聚丙烯腈纤维改性前后的红外谱图。利用傅里叶红外光谱仪对cds、panf和panf-g-cds进行分析,发现cds红外光谱图中在1643cm-1、1560cm-1、1470cm-1、1389cm-1处有明显的特征峰。根据相关资料表明,1643cm-1处的峰归属于c=o伸缩振动峰,1560cm-1处的特征峰归属于n-h弯曲振动峰,1470cm-1处归属于c=h伸缩振动峰,1389cm-1处归属于c-n伸缩振动峰。panf红外光谱图中在2956cm-1、1731cm-1处有明显的特征峰。根据相关资料表明,2956cm-1处的峰归属于-c≡n伸缩振动峰、1731cm-1处的峰归属于c-h弯曲振动峰。pan-g-cds与pan相比,pan-g-cds在1479cm-1和1271cm-1处显示两个新的特征峰,分别对应于仲酰胺上n-h和n-c的伸缩振动,这说明cds成功在pan上进行了接枝反应。
64.参照图2,图2为本发明实施例一制备的碳点的粒径分布图,通过动态光散射仪测定的粒径分布测试结果显示碳点颗粒分布均匀,表明碳点在水溶液中具有良好的分散性,没有发生团聚。碳点的粒径分布较窄,颗粒大小均匀,平均粒径为5.5nm。
65.参照图3,图3中(a)为本发明实施例一制备的碳点的平均粒径,(b)为本发明实施例二制备的碳点的平均粒径,(c)为本发明实施例三制备的碳点的平均粒径,(d)为本发明实施例四制备的碳点的平均粒径。通过动态光散射仪测定的粒径分布测试结果显示,本发明制备的碳点的粒径平均粒径大小在5.5nm左右,符合常见cds的粒径大小(小于10nm)。
66.参照图4,图4中(a)为本发明实施例一制备的碳点的荧光发射光谱图,(b)为本发明实施例一制备的panf-g-cds的荧光发射光谱图。为了对比cds和panf-g-cds的荧光性能用波长为365nm的激发光对样品进行了荧光发射光谱测试,结果如图4所示,可以观察到cds在440nm处有明显的发射峰,panf-g-cds的发射峰出现在454nm处,证明了两者具有荧光特性,具有防伪用途。相较于cds,panf-g-cds略微红移这可能是由于panf-g-cds周围环境产生变化对其上负载的cds产生了影响。
67.以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1