一种相变纤维的制备方法与流程
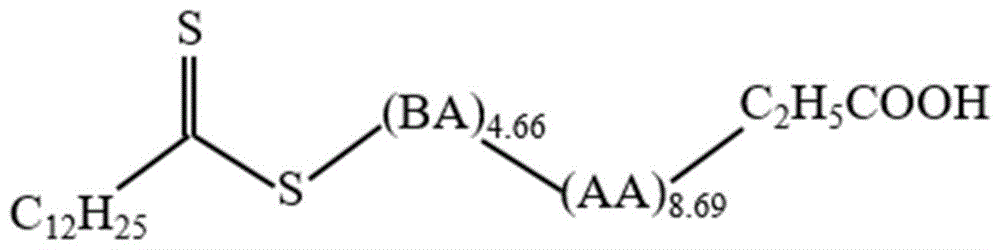
本发明涉及智能纤维领域,尤其涉及一种相变纤维的制备方法。
背景技术:
相变纤维是利用相变材料在相变过程中通过吸收或释放热量使温度保持不变的特性而研发出的一种储热调温功能纤维。该纤维织物根据环境温度自动调节织物内温度,在环境温度低时,自动调高织物内温度;在环境温度高时,自动调低织物内温度,使织物内温度处于较舒适的范围。
微/纳米胶囊是一种具有独特纳米结构的复合粒子,由一至数十纳米厚的壳层壁材和芯材组成。相变胶囊是芯材为相变材料的一种微/纳米胶囊类型,相变胶囊可以解决或缓解相变材料在使用过程中出现的易泄露、导热效率低等问题。利用微胶囊制备相变调温织物的方法主要有微胶囊涂层法、微胶囊填充织物法、微胶囊纺丝法等。
raft细乳液界面聚合是一种将raft活性自由基聚合和细乳液聚合结合制备胶囊的新方法。通过将双亲大分子raft试剂自组装在细乳化形成的油滴/水相界面上,形成的初级自由基会迅速向油滴/水相界面上的大分子raft试剂发生链转移反应,生成新的大分子自由基由于仍有双亲性会继续锚定在油滴界面上。这个自由基会与邻近的大分子raft试剂发生链转移反应或与油滴内单体发生链增长反应,反应重复发生,使得自由基始终处在油滴/水相界面上,从而限定聚合反应只在界面上发生。随着聚合反应进行,聚合物链由外向内增长,原地形成聚合物壁,而芯材留在核层。该方法解决了一般细乳液无法制备厚度均一、高交联度聚合物壳层的缺陷。
目前,利用微胶囊法制备得到的纤维织物,尽管包覆量超过50wt%,具有一定的调热性能,但限于聚合方法,制备的胶囊尺寸均在微米级甚至毫米级,使得纤维直径偏大,热响应速度慢,柔软度和舒适度较差;而在细乳液聚合和已有的raft细乳液界面聚合体系中,包覆量多低于50wt%。考虑到相变纤维的实际应用,我们希望胶囊包覆量超过50wt%,以提供更多的相变潜热;并且相变胶囊粒径小于微米级,提供更大的比表面积,快的热响应速度,并保证进一步制备得到的调热纤维具有较小的直径,从而具备较好的柔软度和舒适度。
技术实现要素:
针对现有技术的不足,本发明提供一种相变纤维的制备方法,所制备得到的相变纤维具有较好的调热性能和较小的直径,在舒适度、可编织性和力学强度等方面均有较好的表现,在智能纤维领域的应用具有巨大的应用前景。具体技术方案如下:
一种相变纤维的制备方法,方法的操作步骤如下:
(1)将1质量份的分散质置于2~9质量份分散介质中,升高温度至60~95℃,搅拌
0.1~4小时,得到纺丝原液。
所述亚微米相变胶囊水分散液由以下步骤制得:
(2)将0.01~0.5质量份的双亲性大分子raft乳化剂溶解于40~400质量份的去离子水中得到水相。
(3)将0.01~0.5质量份的油溶性引发剂、5~200质量份的芯材与1~100质量份的单体混合均匀得到油相。
(4)将上述步骤得到的水相和油相混合,搅拌预乳化5~30min,形成粗乳液。用剪切设备处理5~30min,得到细乳液。
(5)补加0.01~0.2质量份的双亲性大分子raft乳化剂,通入惰性气体5~30min,升温至60~95℃,反应6~12h,得到亚微米相变胶囊水分散液。
(6)将0.1~5质量份步骤(5)的亚微米相变胶囊水分散液与4~8质量份步骤(1)的纺丝原液混合,形成混合纺丝液;
(7)将步骤(6)的混合纺丝液经喷头压入凝固浴,经湿热拉伸、干热拉伸工艺制得相变纤维。
进一步地,上述步骤(1)中,分散质为聚乙烯醇、(苯乙烯-丙烯酸正丁酯-苯乙烯)嵌段共聚物、(苯乙烯-丁二烯-苯乙烯)嵌段共聚物或聚丙烯腈,分散介质为水、四氢呋喃、乙醇、甲苯、甲醇、乙醚、二甲基亚砜、n,n-二甲基甲酰胺或n,n-二甲基乙酰胺。
进一步地,上述步骤(3)中,所述凝固浴为硫酸钠水溶液、硫氰酸钠水溶液、氯化钠水溶液、硫酸镁水溶液、乙醇-水混合物、丙酮-水混合物、n,n-二甲基甲酰胺或n,n-二甲基乙酰胺。
进一步地,所述步骤(2)中,双亲性大分子raft乳化剂的结构式为:r-(mn1-co-nn2)-x或r-(mn1-b-nn2)-x;
其中,r为异丙酸基、乙酸基、2-氰基乙酸基或2-胺基乙酸基;mn1中,m为甲基丙烯酸单体或丙烯酸单体单元,n1为m的平均聚合度,n1=5~100;nn2中,n为苯乙烯单体、丙烯酸正丁酯单体、丙烯酸甲酯、丙烯酸异辛酯或甲基丙烯酸甲酯单体单元,n2为n的平均聚合度,n2=5~100;x基团为烷基二硫代酯基团或烷基三硫代酯基团。
进一步地,所述步骤(3)中,油溶性引发剂为偶氮类引发剂、过氧化物类引发剂;芯材为c5~c28的烷烃或其混合物;单体为含乙烯基单体。
进一步地,含乙烯基单体优选由丙烯酸甲酯、苯乙烯、丙烯酸正丁酯、甲基丙烯酸甲酯、甲基丙烯酸丁酯、丙烯酸乙酯、丙烯酸羟基乙酯、二乙烯基苯、二甲基丙烯酸乙二醇酯、二丙烯酸乙二醇酯或乙烯基吡咯烷酮、二甲基丙烯酸三甘醇中的一种或几种单体按任意比例组成。
本发明的有益效果是,本发明利用raft细乳液界面聚合,可以制备粒径小于1微米的亚微米级相变胶囊;通过改变芯材和单体的质量比,可以调节亚微米相变胶囊的包覆量;通过简单调节双亲性大分子乳化剂用量,可以在100-1000nm范围内调节相变胶囊的尺寸;胶囊的尺寸固定时,芯材质量越大,单体质量越小,壳层更容易塌陷,难以实现较高的包覆量;当尺寸增大时,壳层壁材的界面张力降低,壳层不容易塌陷,可以制备高包覆量的亚微米相变胶囊。将得到的高包覆量亚微米相变胶囊与纺丝原液直接共混,利用湿法纺丝,经喷丝头将混合纺丝液压入凝固浴,再经湿热拉伸、干热拉伸的工艺,将纤维收集到辊轮。
具有以下几个特点:
1、方法采用raft细乳液界面聚合技术,可同时实现高包覆量(>50wt%)和小粒径(<1μm)的胶囊制备,且聚合过程中壳层聚合物分子量可控,所得相变胶囊壳层厚度均一。
2、利用微胶囊纺丝法,避免了微胶囊涂层法和微胶囊填充织物法所造成的使用中胶囊易脱落、织物整体透气性差、柔软度差等问题;而利用raft细乳液界面聚合制备得到的高包覆量亚微米胶囊,则解决了其他聚合方法所制备微胶囊粒径大的问题,使得后续制备的相变纤维具备更好的柔软度、舒适度和更快的热响应速度。
3、将亚微米相变胶囊水分散液与纺丝原液直接共混,大大提高了相变胶囊在纤维中的分散程度,制备得到的相变调热纤维调热性能稳定。
附图说明
图1是本发明实施例1所用双亲性大分子raft乳化剂分子结构示意图;
图2是本发明实施例1所用双亲性大分子raft乳化剂核磁共振氢谱图;
图3是本发明实施例1-4所合成不同包覆量亚微米相变胶囊的形貌图;
图4是本发明实施例1-4所合成不同包覆量亚微米相变胶囊的粒径分布图;
图5是本发明实施例1-4所合成不同包覆量亚微米相变胶囊的结晶热和熔融热曲线;
图6是本发明实施例1(a)、实施例5(b)、实施例6(c)所制备相变调热纤维的形貌图;
图7是本发明实施例1、实施例5、实施例6所制备相变调热纤维的结晶热和熔融热曲线图;
图8是本发明实施例1、实施例5、实施例6所制备相变调热纤维的力学拉伸曲线图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚明白,以下结合附图和具体实施例,对本发明进一步详细说明。
采用的亚微米相变胶囊水分散液通过raft细乳液界面聚合制备得到,操作步骤如下:
(a)将0.01~0.5质量份的双亲性大分子raft乳化剂溶于40~400质量份的去离子水中,搅拌溶解;
(b)将0.01~0.5质量份的油溶性引发剂、5~200质量份的芯材,1~100质量份的单体混合,搅拌溶解;
(c)将水相和油相混合,搅拌预乳化5~30min,形成粗乳液。用剪切设备处理5~30min,得到细乳液;剪切设备可以采用细胞粉碎机、均质机、高速剪切乳化机,但不限于此。
(d)补加0.01~0.2质量份的双亲性大分子raft乳化剂,通入惰性气体5~30min,升温至60~95℃,反应6~12h,得到亚微米相变胶囊水分散液。
通过改变芯材和单体的质量比,可以调节亚微米相变胶囊的包覆量;通过简单调节双亲性大分子raft乳化剂用量,可以在100-1000nm范围内调节相变胶囊的尺寸。胶囊的尺寸固定时,芯材质量越大,单体质量越小,包覆量越高,壳层越容易塌陷;当减少双亲性大分子raft乳化剂用量时,尺寸增大,壳层壁材的界面张力降低,壳层不容易塌陷,可以制备高包覆量的亚微米相变胶囊。
上述步骤(a)中采用的双亲性大分子raft乳化剂是通过将小分子raft、m、n单体分散到二氧六环、甲醇、乙醇、水或其混合物中聚合一步制备得到,但不限于此。其中,小分子raft具有以下结构特征:r-x;双亲性大分子raft乳化剂具有以下结构特征:r-(mn1-co-nn2)-x或r-(mn1-b-nn2)-x,
其中,r为异丙酸基、乙酸基、2-氰基乙酸基或2-胺基乙酸基;mn1中,m为甲基丙烯酸单体或丙烯酸单体单元,n1为m的平均聚合度,n1=5~100;nn2中,n为苯乙烯单体、丙烯酸正丁酯单体、丙烯酸甲酯、丙烯酸异辛酯或甲基丙烯酸甲酯单体单元,n2为n的平均聚合度,n2=5~100;x基团为烷基二硫代酯基团或烷基三硫代酯基团;
上述步骤(b)油溶性引发剂为偶氮类引发剂、过氧化物类引发剂;芯材为c5~c28的烷烃或其混合物;单体为含乙烯基单体,具体指:丙烯酸甲酯、苯乙烯、丙烯酸正丁酯、甲基丙烯酸甲酯、甲基丙烯酸丁酯、丙烯酸乙酯、丙烯酸羟基乙酯、二乙烯基苯、二甲基丙烯酸乙二醇酯、二丙烯酸乙二醇酯或乙烯基吡咯烷酮、二甲基丙烯酸三甘醇或以上单体的混合物。
采用亚微米相变胶囊水分散液制备相变纤维的操作步骤如下:
(1)将1质量份的分散质置于2~9质量份分散介质中,升高温度至60~95℃,搅拌0.1~4小时,得到纺丝原液;
(2)将0.1~5质量份的亚微米相变胶囊水分散液与4~8质量份纺丝原液混合,形成混合纺丝液;
(3)将混合纺丝液经喷头压入凝固浴,经湿热拉伸、干热拉伸工艺,用辊轮收集纤维。
上述步骤(1)所采用的纺丝原液,分散质为聚乙烯醇、(苯乙烯-丙烯酸正丁酯-苯乙烯)嵌段共聚物、(苯乙烯-丁二烯-苯乙烯)嵌段共聚物或聚丙烯腈,分散介质为水、四氢呋喃、乙醇、甲苯、甲醇、乙醚、二甲基亚砜、n,n-二甲基甲酰胺或n,n-二甲基乙酰胺。
上述步骤(3)所采用的凝固浴为硫酸钠水溶液、硫氰酸钠水溶液、氯化钠水溶液、硫酸镁水溶液、乙醇-水混合物、丙酮-水混合物、n,n-二甲基甲酰胺或n,n-二甲基乙酰胺。
双亲性大分子raft乳化剂的结构通过核磁共振氢谱确定,仪器型号为brukeravancedmx500,核磁试剂为二甲基亚砜(dmso)。
亚微米相变胶囊的单体转化率由称重法测得。
亚微米相变胶囊的形貌采用jeoljemacro-1230透射电子显微镜表征,测试电压为80千伏。
亚微米相变胶囊和相变纤维的形貌采用su-8010扫描电子显微镜表征,测试前将样品通过导电胶粘于样品台,真空氛围下对其表面进行喷金90s处理。
亚微米相变胶囊的粒径分布由nanomeasure软件取100个样本点作图得到。
亚微米相变胶囊和相变纤维的相变焓值通过差示扫描量热仪(dsc)表征,仪器型号为dsc-200,测试时升温范围为-20~70℃,升温速率为10℃/min。
相变调热纤维的力学拉伸性能由zwick/rollz020型万能材料试验机进行测试,测试温度为20℃,拉伸速率为10mm/min,每个样品至少重复三次。
实施例1
第一步:将0.05质量份的(aa8.69-co-nba4.66)无规共聚物溶于70质量份的去离子水中,搅拌溶解;
第二步:将0.1质量份的aibn、13.4质量份的正十八烷,5.03质量份的苯乙烯,2.09质量份的二乙烯基苯混合,搅拌溶解;
第三步:将水相和油相混合,搅拌预乳化30min,形成粗乳液。用剪切设备处理30min,得到细乳液;
第四步:补加0.07质量份的双亲性大分子raft乳化剂,通入惰性气体30min,升温至70℃,反应10h,得到亚微米相变胶囊水分散液;
第五步:将1质量份的聚乙烯醇置于5.67质量份分散介质中,升高温度至90℃,搅拌2小时,得到纺丝原液;
第六步:将1.87质量份的亚微米相变胶囊水分散液与6.67质量份纺丝原液混合,形成混合纺丝液;
第七步:将混合纺丝液经喷头压入凝固浴,经湿热拉伸、干热拉伸工艺,用辊轮收集纤维。
所采用的双亲性大分子raft乳化剂分子示意图如图1所示,在其结构示意图中,r-mn1-co-nn2-x,r为异丙酸基,m单体选用丙烯酸,丙烯酸单体单元聚合度为8.69,即n1=8.69;n单体选用丙烯酸正丁酯,丙烯酸正丁酯单体单元聚合度为4.66,即n2=4.66,x基团为十二烷基三硫酯。双亲性大分子乳化剂的核磁数据如图2所示。制备得到亚微米相变胶囊的形貌如图3所示,表明形成了完整的核-壳结构;其粒径分布如图4所示,可以看出相变胶囊的尺寸均小于1μm;该条件下制备的亚微米相变胶囊的熔融热、结晶热如图5所示,表明该亚微米相变胶囊具有较好的调热性能,最高结晶热或熔融热可超过110j/g;
实施例2
第一步:将0.5质量份的(aa100-co-nba100)无规共聚物溶于400质量份的去离子水中,搅拌溶解;
第二步:将0.5质量份的aibn、200质量份的正十八烷,80质量份的苯乙烯,20质量份的二乙烯基苯混合,搅拌溶解;
第三步:将水相和油相混合,搅拌预乳化30min,形成粗乳液。用剪切设备处理30min,得到细乳液;
第四步:补加0.2质量份的双亲性大分子raft乳化剂,通入惰性气体5min,升温至60℃,反应12h,得到亚微米相变胶囊水分散液。
实施例3
第一步:将0.01质量份的(aa5-co-nba5)无规共聚物溶于40质量份的去离子水中,搅拌溶解;
第二步:将0.01质量份的aibn、5质量份的正十八烷,1质量份的苯乙烯混合,搅拌溶解;
第三步:将水相和油相混合,搅拌预乳化5min,形成粗乳液。用剪切设备处理5min,得到细乳液;
第四步:补加0.01质量份的双亲性大分子raft乳化剂,通入惰性气体30min,升温至95℃,反应6h,得到亚微米相变胶囊水分散液。
实施例4
第一步:将0.05质量份的(aa10-co-nba5)无规共聚物溶于70质量份的去离子水中,搅拌溶解;
第二步:将0.1质量份的aibn、10质量份的正十八烷,5质量份的苯乙烯混合,搅拌溶解;
第三步:将水相和油相混合,搅拌预乳化30min,形成粗乳液。用剪切设备处理30min,得到细乳液;
第四步:补加0.07质量份的双亲性大分子raft乳化剂,通入惰性气体5~30min,升温至70℃,反应10h,得到亚微米相变胶囊水分散液。
实施例1-4中不同包覆量的亚微米相变胶囊实验数据如表1所示,结果表明本发明方法可同时实现高包覆量(>50wt%)和小粒径(<1μm)的胶囊制备,且聚合过程中壳层聚合物分子量可控,所得相变胶囊壳层厚度均一。
表1
实施例5
第一步:将0.05质量份的(aa8.69-co-nba4.66)无规共聚物溶于70质量份的去离子水中,搅拌溶解;
第二步:将0.1质量份的aibn、13.4质量份的正十八烷,5.03质量份的苯乙烯,2.09质量份的二乙烯基苯混合,搅拌溶解;
第三步:将水相和油相混合,搅拌预乳化30min,形成粗乳液。用剪切设备处理30min,得到细乳液;
第四步:补加0.07质量份的双亲性大分子raft乳化剂,通入惰性气体30min,升温至70℃,反应10h,得到亚微米相变胶囊水分散液;
第五步:将1质量份的聚乙烯醇置于2质量份分散介质中,升高温度至95℃,搅拌0.1小时,得到纺丝原液;
第六步:将0.1质量份的亚微米相变胶囊水分散液与4质量份纺丝原液混合,形成混合纺丝液;
第七步:将混合纺丝液经喷头压入凝固浴,经湿热拉伸、干热拉伸工艺,用辊轮收集纤维。
实施例6
第一步:将0.05质量份的(aa8.69-co-nba4.66)无规共聚物溶于70质量份的去离子水中,搅拌溶解;
第二步:将0.1质量份的aibn、13.4质量份的正十八烷,5.03质量份的苯乙烯,2.09质量份的二乙烯基苯混合,搅拌溶解;
第三步:将水相和油相混合,搅拌预乳化30min,形成粗乳液。用剪切设备处理30min,得到细乳液;
第四步:补加0.07质量份的双亲性大分子raft乳化剂,通入惰性气体30min,升温至70℃,反应10h,得到亚微米相变胶囊水分散液;
第五步:将1质量份的聚乙烯醇置于9质量份分散介质中,升高温度至60℃,搅拌4小时,得到纺丝原液;
第六步:将5质量份的亚微米相变胶囊水分散液与8质量份纺丝原液混合,形成混合纺丝液;
第七步:将混合纺丝液经喷头压入凝固浴,经湿热拉伸、干热拉伸工艺,用辊轮收集纤维。
不同相变胶囊掺入量得到的相变调热纤维形貌如图6所示,单根纤维直径在40-50μm之间,表明本发明所制备的相变纤维尺寸小于50μm,与目前相变纤维相比具有明显的尺寸优势。不同相变胶囊掺入量制备得到的不同调热纤维的熔融热、结晶热如图7所示,表明了所制备的相变纤维均具有一定的调热性能,且调热性能与亚微米相变胶囊的掺入量成正相关,相变纤维最高结晶热或熔融热可超过20j/g;不同相变胶囊掺入量制备得到的不同调热纤维的力学拉伸性能如图8所示,表明不同调热能力的相变纤维均具有一定的拉伸强度和断裂伸长率,并且从图中可看出,相变纤维的力学拉伸性能与相变胶囊的掺入量有关,应力最高可到60mpa,对应的断裂伸长率为40%。上述实施例用来解释说明本发明,而不是对本发明进行限制,在本发明的精神和权利要求的保护范围内,对本发明作出的任何修改和改变,都落入本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!