一种超小亚纳米孔多孔石墨烯的制备方法与流程
本发明属于多孔石墨烯材料制备技术领域,涉及一种超小亚纳米孔多孔石墨烯的制备方法。
背景技术:
石墨烯是一种由碳原子以sp2杂化轨道组成六角型呈蜂巢晶格的平面薄膜,只有一个碳原子厚度的二维材料。2004年,英国曼彻斯特大学物理学家安德烈·海姆和康斯坦丁·诺沃肖洛夫,成功地在实验中从石墨中分离出石墨烯。石墨烯是可以做到单层碳原子二维排列,石墨烯蜂巢晶格常牢固坚硬,室温下,传递电子的速度比已知导体都快。石墨烯的原子尺寸结构非常特殊,由于石墨烯实质上是一种透明、良好的导体,也适合用来制造透明触控屏幕、光板、太阳能电池,在半导体物理、催化等方面有重要应用价值。石墨烯材料还是一种优良的改性剂,在新能源领域如超级电容器、锂离子电池方面,由于其高传导性、高比表面积,可适用于作为电极材料。由于石墨烯具有特高的表面面积对质量比例,石墨烯可以用于超级电容器的导电电极。科学家认为这种超级电容器的储存能量密度会大于现有的电容器。但是石墨烯是一种片状的二维材料,在使用石墨烯的过程中,往往涉及到物质的传递和扩散,例如电解质的扩散、载流子的传递、气体的扩散。因为二维片状材料的堆叠,使用过程中不利于物质传递,严重影响石墨烯性能的发挥。此外石墨烯中的电子在层间传递速度快,但是电子从石墨烯注入到电解质或其他吸附的物质上时比较困难。
原始石墨烯或化学修饰的石墨烯片易于通过π-π堆积相互作用和范德华力彼此重叠形成不可逆的团聚体,导致其性质降低,包括严重降低的比表面积和低得多的质量扩散速率。此外,石墨烯片制造最终产品时,需要额外的处理或者添加剂,这可能进一步降低石墨烯的整体性能。具有高比表面积和设计孔隙率的可加工石墨烯及其大块材料的可扩展制备对于许多实际应用而言至关重要。多孔石墨烯,是石墨烯的结构衍生物,是指在石墨烯的二维平面内具有纳米级孔洞的材料。多孔石墨烯片层内独特的纳米孔结构,使其具有更高的比表面积;另外,纳米孔的存在有利于物质在多孔石墨烯各个方向上传输,促进碳层在液相体系中与其他物质充分接触。多孔石墨烯优异的特性使其在能源储存与转换材料(锂离子电池、超级电容器等)、生物传感、水污染处理及水污染检测等领域具有巨大的应用潜力。发展简单、经济、可控的多孔石墨烯宏量制备方法对其应用非常关键。目前多孔石墨烯的制备方法主要有模板法、离子束轰击法和化学溶液刻蚀,但是用上述方法制备多孔石墨烯时,通常难以控制孔径,也很难实现批量生产。以化学溶液刻蚀为例,是通过一种方便的温和缺陷-刻蚀反应来制备表面具有丰富平面纳米孔的多孔石墨烯。一般是在搅拌条件下加热氧化石墨烯和刻蚀氧化剂制备。但是,刻蚀材料与氧化石墨烯反应,刻蚀过程不易控制,容易产生很多碎片,制备的多孔石墨烯的表面孔的大小、数目不能控制。因此,迫切需要一种更加有效、纯净、量产且能够高精度控制孔径的制备多孔石墨烯的方法。
技术实现要素:
本发明的目的在于提供一种更加有效、纯净、量产的超小亚纳米孔多孔石墨烯的制备方法,且采用该方法制备多孔石墨烯时,能够高精度控制孔径。
为实现上述目的,本发明所采用的技术方案是:一种超小亚纳米孔多孔石墨烯的制备方法,具体为:
1)按质量比1︰0.5~5,分别取氧化石墨烯粉末和固体羰基金属,氧化石墨烯粉末加入氮-氮二甲基甲酰胺中,超声分散0.5~5小时,配成质量体积浓度为0.5~5g/l的氧化石墨烯悬浮液;再按1毫克氧化石墨烯粉末需配1.25微升辛胺的比例,取辛胺;将固体羰基金属和辛胺加入氮-氮二甲基甲酰胺中,超声分散0.5~5小时,配成总体积为氧化石墨烯悬浮液体积20%~50%的第一分散溶液,将第一分散溶液与氧化石墨烯悬浮液混合均匀,在100℃~200℃的温度下溶剂热反应0.5~5小时,制得金属氧化物纳米点负载石墨烯材料;
或者,按质量比1︰0.5~5,分别取氧化石墨烯粉末和液体羰基金属,将氧化石墨烯粉末加入氮-氮二甲基甲酰胺中,超声分散0.5~5小时,配成质量体积浓度为0.5~5g/l的氧化石墨烯悬浮液;将液体羰基金属加入氮-氮二甲基甲酰胺中,超声分散0.5~5小时,配成质量体积浓度为0.5~5g/l的第二分散溶液,第二分散溶液和氧化石墨烯悬浮液混合均匀,在100~200℃的温度下溶剂热反应0.5~5小时,制得金属氧化物纳米点负载石墨烯;
2)金属氧化物纳米点负载石墨烯置于通入高纯氩气保护气的环境中,以5℃/分钟的升温速率升温至300~1000℃并保温1~5h,自然冷却至室温,盐酸洗涤,再用蒸馏水洗涤,烘干,制得超小亚纳米孔多孔石墨烯。
本发明制备方法以羰基金属为前驱体,在石墨烯表面负载均匀分布的金属氧化物纳米颗粒,并以此为模板,通过碳热反应实现了亚纳米孔石墨烯的制备,并通过对羰基金属的选择、加入的羰基金属的比例、退火温度和时间进行控制,得到控制在纳米级别的不同孔径的多孔石墨烯纳米材料,从而精准调控孔径大小,提高了利用效率,适合工业化生产。孔的引入可以有效阻止石墨烯片重新堆叠,使制得的并具有良好的导电性,在能源储存与转换材料(锂离子电池、超级电容器等)、生物传感、水污染处理及水污染检测等领域具有巨大的应用潜力。
附图说明
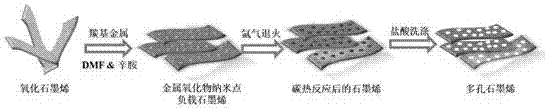
图1为采用本发明制备方法合成多孔石墨烯的工艺流程示意图。
图2为本发明制备方法中采用的氧化石墨烯纳米材料的表征图,其中a,b为扫面电子显微镜图;c,d为透射电子显微镜图。
图3为实施例1制得的多孔石墨烯纳米材料的表征图,其中:a为xrd表征;b为拉曼光谱表征。
图4为实施例1制得的多孔石墨烯纳米材料的电化学性能图,其中:a为循环伏安法测定的结果;b为恒电流充放电发测定的结果;c为无孔石墨烯与多孔石墨烯在相同扫描速率下的循环伏安法的对比结果;d为无孔石墨烯与多孔石墨烯电化学阻抗的对比结果。
图5为实施例1中羰基钴与氧化石墨烯溶剂热后产物的透射电子显微镜图与颗粒粒径分布统计结果图,其中:a,b为氧化钴负载石墨烯的透射电子显微镜图,c为颗粒粒径分布统计结果图。
图6为实施例1羰基钴与氧化石墨烯材料650℃刻孔后的透射电子显微镜图与孔径径分布统计结果,其中,a、b为亚纳米孔多孔石墨烯的透射电子显微镜表征图,c为孔径分布统计结果。
图7为实施例2中羰基镍与氧化石墨烯溶剂热后产物的透射电子显微镜表征图与颗粒粒径分布统计结果图,其中a、b为氧化镍负载石墨烯的透射电子显微镜图,c为颗粒粒径分布统计结果图。
图8为实施例2中羰基铁前驱体负载氧化石墨烯材料并700℃刻孔后的透射电子显微镜图与孔径分布统计结果图,其中a、b为多孔石墨烯的透射电子显微镜图,c为孔径分布统计结果图。
图9为实施例2和实施例3中羰基铁与氧化石墨烯溶剂热后产物的透射电子显微镜图与颗粒粒径分布统计结果图。
图10为实施例4中羰基铁前驱体负载氧化石墨烯在800℃高温刻孔后的透射电子显微镜图与孔径分布统计结果图,其中a,b为多孔石墨烯的透射电子显微镜图,c为孔径分布统计结果图。
图11为实施例4中羰基铁前驱体负载氧化石墨烯在900℃高温刻孔后的透射电子显微镜图。
具体实施方式
下面对本发明实施例中的技术方案进行清楚、完整的描述。
本发明提供了一种超小亚纳米孔及孔径可调控的多孔石墨烯的制备方法,其工艺流程如图1所示,该制备方法具体为:
1)按质量比1︰0.5~5,分别取氧化石墨烯粉末和固体羰基金属,将氧化石墨烯粉末加入氮-氮二甲基甲酰胺中(dmf),超声分散0.5~5小时,配成质量体积浓度为0.5~5g/l的氧化石墨烯悬浮液;再按1毫克氧化石墨烯粉末需要1.25微升辛胺的比例,取辛胺;将固体羰基金属和辛胺加入氮-氮二甲基甲酰胺中,超声分散0.5~5小时,配成总体积为氧化石墨烯悬浮液体积20%~50%的第一分散溶液,将第一分散溶液与氧化石墨烯悬浮液混合均匀,装入50毫升反应釜中,将该反应釜置于烘箱中,在100~200℃的温度下溶剂热反应0.5~5小时,制得金属氧化物纳米点负载石墨烯材料;
或者,按质量比1︰0.5~5,分别取氧化石墨烯粉末和液体羰基金属,将氧化石墨烯粉末加入氮-氮二甲基甲酰胺中(dmf),超声分散0.5~5小时,配成质量体积浓度为0.5~5g/l的氧化石墨烯悬浮液;将液体羰基金属加入氮-氮二甲基甲酰胺中,超声分散0.5~5小时,配成质量体积浓度为0.5~5g/l的第二分散溶液,第二分散溶液和氧化石墨烯悬浮液混合均匀,在100~200℃的温度下溶剂热反应0.5~5小时,制得金属氧化物纳米点负载石墨烯;
固体羰基金属采用羰基钴、羰基锰、羰基钨、羰基钼中的一种、两种、三种或者四种,当采用一种以上时,各组分以任意质量比混合。
液体羰基金属采用羰基铁、羰基镍中的一种或者两种;当采用两种时,羰基铁和羰基镍以任意质量比混合。
2)金属氧化物纳米点负载石墨烯放入瓷舟中,将该瓷舟置于管式炉内,通过高纯氩气保护气,以5℃/分钟的升温速率升温至300~1000℃并保温1~5h,自然冷却至室温,用盐酸洗涤所得粉末,以去除表面的金属氧化物,再用蒸馏水洗涤,以去除盐酸,烘干,制得超小亚纳米孔(<1nm)多孔石墨烯。
本发明制备方法的机理:石墨烯的氮-氮二甲基甲酰胺分散液与羰基金属前驱体混合,并视前驱体的种类选择是否加入辛胺,然后进行溶剂热反应,反应过程中,羰基金属脱羰基化反应,并在原子级别层面与石墨烯进行复合,根据控制前驱体和表面活性剂的加入量以及反应温度、时间等参数,可以精确控制金属氧化物纳米点的成核均匀程度以及大小;退火时,金属氧化物由于与石墨烯表面直接通过金属-氧-碳键相结合,随着温度的升高,会发生碳热反应,石墨烯上的碳原子会形成一氧化碳或二氧化碳而被吹扫掉,形成的孔洞被金属氧化物纳米点占据;退火完成后利用盐酸将金属氧化物去除后,成功获得多孔石墨烯。
由于要控制制得的多孔石墨烯表面孔径小于1纳米,如果选择的金属氧化物前驱体不合适,在溶剂热过程中难以形成小于1纳米以下的金属氧化物纳米点,因此需要选择的金属氧化物负载石墨烯前驱体是由羰基钴、羰基铁或羰基镍中的一种或多种得到的。
采用本发明制备方法制得的多孔石墨烯中含有氮基和氧基官能团。
本发明制备方法步骤1)中采用通过改进的hummers方法制备的大片少层氧化石墨烯,该大片少层氧化石墨烯的扫面电子显微镜图(sem),如图2的a图和b图所示,该大片少层氧化石墨烯的透射电子显微镜图(tem),如图2的c图和d图所示。从图2可以看出,该氧化石墨烯为大片石墨烯,氧化石墨烯的片层数为1~2层,这种大片厚度可控的氧化石墨烯为后续石墨烯表面刻孔提供了基础。并且从高分辨透射电子显微镜图片上观察,氧化石墨烯表面没有任何杂质。
实施例1
取20毫克氧化石墨烯粉末、10毫克羰基钴和25微升辛胺,将氧化石墨烯粉末加入氮-氮二甲基甲酰胺中,超声分散5小时,配成质量体积浓度为1g/l的氧化石墨烯悬浮液;将羰基钴和辛胺加入氮-氮二甲基甲酰胺中,超声分散3小时,配成总体积为氧化石墨烯悬浮液体积20%~50%的第一分散溶液,第一分散溶液与氧化石墨烯悬浮液混合均匀,装入50毫升反应釜中,将该反应釜置于烘箱中,在170℃的温度下溶剂热反应0.5~5小时,制得氧化钴纳米点负载石墨烯;将氧化钴纳米点负载石墨烯放入瓷舟中,将该瓷舟置于管式炉内,通过高纯氩气保护气,以5℃/分钟的升温速率升温至650℃并保温2h,自然冷却至室温,盐酸洗涤所得粉末,再用蒸馏水洗涤,烘干,制得超小亚纳米孔及孔径可调控的多孔石墨烯。
在溶剂热反应过程中,羰基钴伴随着温度的升高发生脱羰基化反应,从而形成原子级别的钴原子,在表面活性剂以及氧化石墨烯表面官能团的帮助下,在石墨烯表面成核并均匀分布,形成氧化钴纳米点,同时dmf分解形成二甲胺,对石墨烯进行氮掺杂反应,氮原子的引入可以提升材料的导电性,使得该多孔石墨烯具有良好的电化学性能。
对实施例1制得的多孔石墨烯进行表征,其xrd图,如图3中的a图,图中显示,该多孔石墨烯在2θ为24.5°处出现了衍射峰,与文献(k.h.liao,a.mittal,s.bose,c.leighton,k.a.mkhoyanandc.w.macosko,acsnano,2011,5,1253.)中石墨的衍射峰位置一致,说明采用本发明制备方法制得的最终产物为石墨烯;但是衍射峰有所宽化,这是因为石墨片层的无序性增大且晶体结晶度的完整性下降所致。该多孔石墨烯的拉曼光谱图,如图3中的b图,可以看到,该多孔石墨烯具有2个明显的拉曼峰(g峰和d峰),分别代表sp2碳原子和sp3碳原子,d峰与g峰的强度比值较大,表明多孔石墨烯表面有着明显的缺陷。
实施例1制得的多孔石墨烯的电化学性能图,如图4所示。图4中的a图为循环伏安法测定结果曲线图;图4中的b图为恒电流充放电测定结果曲线图;图4中的c图为无孔石墨烯与多孔石墨烯的循环伏安测定结果对比图;图4中的d图为无孔石墨烯与多孔石墨烯的电化学阻抗对比结果图。在进行电化学性能测试时,所用电解液为摩尔体积浓度为1摩尔/升的硫酸钠溶液,电压窗口选择为-1~0v,测得多孔石墨烯材料的最高比电容是170f/g,表明该种材料具有优异的电容特性。循环伏安曲线从小扫速到大扫速下的形状均接近矩形,表明该种材料具有优异的电容特性和倍率性能。通过将无孔石墨烯与多孔石墨烯在相同扫描速率下的循环伏安曲线对比可知,多孔石墨烯具有更高的电化学比容量,这是由于石墨烯表面具有丰富的孔结构后,有利于电极表面与电解液的接触,能够利用双电层电容行为存储更多的电荷,从而具有更高的比电容,另外,通过二者的电化学阻抗谱分析可知,虽然无孔石墨烯与多孔石墨烯的等效串联阻抗相差不大,但是在低频区,多孔石墨烯比无孔石墨烯表现出更小的电荷转移阻抗,这也表明采用本发明制备方法制得的多孔石墨烯具有优异的电化学性能。可以被用于超级电容器电极材料、电池电极导电添加剂、污水处理和生物传感材料等等。
实施例1制得的氧化钴纳米点负载石墨烯的透射电子显微镜图,如图5中的a图和b图所示,从图中可以看出石墨烯表面没有大颗粒的团聚,氧化钴纳米点分布非常均匀,该氧化钴纳米点负载石墨烯表面的颗粒平均直径在0.86纳米左右,如图5中的c图。
实施例1制得亚纳米孔多孔石墨烯的透射电子显微镜图,如图6所示,图6中的a图和b图显示石墨烯表面的纳米点去除,并且在表面留有非常均匀的孔洞,该多孔石墨烯表面孔径的大小约在0.9纳米左右,见图7c。
实施例2
取20毫克氧化石墨烯粉末,加入氮-氮二甲基甲酰胺中(dmf),超声分散0.5~5小时,配成质量体积浓度为1g/l的氧化石墨烯悬浮液;取50毫克羰基镍,分散于dmf中,配成质量体积浓度为0.5~5g/l的第二分散溶液,第二分散溶液和氧化石墨烯悬浮液混合均匀,在170℃的温度下溶剂热反应0.5~5小时,制得氧化镍纳米点负载石墨烯;金属氧化物纳米点负载石墨烯放入瓷舟中,将该瓷舟置于管式炉内,通过高纯氩气保护气,以5℃/分钟的升温速率升温至650℃并保温2h,自然冷却至室温,用盐酸洗涤所得粉末,以去除表面的金属氧化物,再用蒸馏水洗涤,以去除盐酸,烘干,制得超小亚纳米孔多孔石墨烯。
实施例2中制得的氧化镍纳米点负载石墨烯的射电子显微镜图,如图7所示,图7中的a图和b图,石墨烯表面没有明显的大颗粒团聚,氧化镍纳米点分布非常均一,其表面孔径的大小约在0.9纳米左右,见图7中的c图。
从图5和图7可以看出,由于金属氧化物纳米点大小的区别,退火后得到的多孔石墨烯的孔径大小也能够得以调控。
使用羰基镍作为前驱体制备多孔石墨烯材料的过程类似。值得注意的是,羰基镍为浅绿色液体,本身沸点低,容易挥发,在高温下极易在石墨烯表面成核生长,因此不需要添加表面活性剂,由于镍原子在石墨烯表面的吸附能与钴原子不同,金属氧化物成核所需要的形成能也有所差别,因此溶剂热后所形成的金属氧化物纳米点的大小有所差别。
实施例3
取20毫克氧化石墨烯粉末,加入氮-氮二甲基甲酰胺中(dmf),超声分散0.5~5小时,配成质量体积浓度为1g/l的氧化石墨烯悬浮液;取50毫克羰基铁,分散于dmf中,配成质量体积浓度为0.5~5g/l的羰基铁分散溶液,将羰基铁分散溶液和氧化石墨烯悬浮液混合均匀,在170℃的温度下溶剂热反应0.5~5小时,制得氧化铁纳米点负载石墨烯;氧化铁纳米点负载石墨烯放入瓷舟中,将该瓷舟置于管式炉内,通过高纯氩气保护气,以5℃/分钟的升温速率升温至650℃并保温2h,自然冷却至室温,用盐酸洗涤所得粉末,以去除表面的金属氧化物,再用蒸馏水洗涤,以去除盐酸,烘干,制得超小亚纳米孔多孔石墨烯。
实施例3制得的氧化铁纳米点负载石墨烯的透射电子显微镜图,如图8所示。图8中的图a和图b,石墨烯表面的纳米点去除,并且在表面留有非常均匀的较大的孔洞,表面孔径的大小约在7.11纳米左右,见图8中的图c。
从图6中的图c和图8中的图c可以看出,制得的氧化物纳米点负载石墨烯表面的孔径大小不同,这一差异表明不同羰基金属前驱体在溶剂热反应负载石墨烯时形成的金属氧化物纳米颗粒不同,在退火过程中,形成的纳米孔会有所差别。通过选择不同羰基金属,可以间接控制多孔石墨烯孔径的大小,方便工业化大规模制备。同样,使用羰基锰、钼、钨为前驱体也可以得到类似尺寸的金属氧化物纳米点,退火即可得到多孔石墨烯。
实施例4
取氧化石墨烯,加入dmf中,超声分散2小时,配成质量体积浓度为1g/l的氧化石墨烯悬浮液20毫升。取50毫克羰基铁分散于dmf中配成5g/l羰基铁分散液,取2毫升羰基铁分散液与氧化石墨烯悬浮液混合均匀,转移至反应釜中,170℃溶剂热2小时,反应完成后将产物利用无水乙醇和蒸馏水离心洗涤,然后冷冻干燥;制得氧化铁纳米点负载石墨烯,该氧化铁纳米点负载石墨烯的透射电子显微镜图,如图9a,b,表面的颗粒平均直径在3.16纳米左右(图9c)。
取上述制得的氧化铁纳米点负载石墨烯分成三份,每一份分别放入一个瓷舟中,将一个瓷舟转移至一个管式炉中,三个管式炉中均通入高纯氩气保护气,
第一个管式炉以5℃每分钟的升温速率升温至500℃,并保温2h,然后自然冷却至室温,退火后材料的透射电子显微镜图,见图9中的d图和e图,该退火后材料表面的颗粒平均直径在5.52纳米左右,见图9f。
第二个管式炉以5℃每分钟的升温速率升温至600℃,并保温2h,然后自然冷却至室温,退火后材料的透射电子显微镜图,见图9中的g图和h图,该退火后材料表面的颗粒平均直径在7.4纳米左右,见图9i。
第三个管式炉以5℃每分钟的升温速率升温至700℃,并保温2h,然后自然冷却至室温,退火后材料的透射电子显微镜图,见图9中的j图和k图,该退火后材料表面的颗粒平均直径在16.56纳米左右,见图9l。
退火后所得黑色粉末通过盐酸洗涤去除表面的氧化铁就得到了具有超小亚纳米孔的石墨烯材料。
如果将氧化铁纳米点负载石墨烯在800℃退火,制得的多孔石墨烯的透射电子显微镜图,如图10a和10b,表面孔径的大小约在14.76纳米左右,如图10c。
氧化铁纳米点负载石墨烯在900℃退火,制得的多孔石墨烯的透射电子显微镜图,如图11,表面孔径大小约在88纳米左右。
上述结果表明,在退火过程中,金属氧化物纳米颗粒在石墨烯表面由于滑移效应,相互进行团聚,越高温度下,金属氧化物的团聚现象越明显,最终形成的纳米孔会有所差别。通过不同退火的选择,直接控制了多孔石墨烯孔径的大小。同样,这一方法同样适用于使用羰基锰、钼、钨为前驱体得到类似孔径尺寸的多孔石墨烯纳米材料。
以上描述的实施例是本发明一部分实施例,而不是全部的实施例。本发明的实施例的详细描述并非旨在限制要求保护的本发明范围,而是仅仅表示本发明的选定实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种氧化石墨烯表面原位生长二氧化硅的纳米杂化填料及其制备方法与流程
- 一种基于纳米纤维素晶须MoS2/石墨烯复合对电极材料及其制备方法和应用与流程
- 一种改性石墨烯与纳米纤维素复合温敏材料的制备方法与制造工艺
- 一种石墨烯生产设备的制造方法与工艺
- 石墨烯掺杂改性纳米钙钛矿型La1‑xSrxMnO3复合材料及其制备方法和应用与制造工艺
- 一种氧化石墨烯/银纳米颗粒/金字塔形硅三维拉曼增强基底及制备方法和应用与制造工艺
- 纳米羧基化石墨烯复合涤纶导电织物的制备方法与制造工艺
- 硅烷纳米石墨烯陶化膜及其制备方法与制造工艺
- 纳米氧化石墨烯在促进小鼠胚胎干细胞培养和自我更新中的应用的制造方法与工艺
- 一种纳米石墨烯改性蓝藻基复合生物塑料及其制备方法与制造工艺
- 过渡金属与石墨烯的复合纳米线及其制备方法与流程
- 一种氧化石墨烯表面原位生长二氧化硅的纳米杂化填料及其制备方法与流程
- GO增强CNT覆膜砂的智能混凝土无线传感器的制造方法与工艺
- 一种基于纳米纤维素晶须MoS2/石墨烯复合对电极材料及其制备方法和应用与流程
- 聚醚砜树脂/石墨烯纳米片多孔纳米复合材料、制备方法及其用途与流程
- 一种改性石墨烯与纳米纤维素复合温敏材料的制备方法与制造工艺
- 石墨烯掺杂改性纳米钙钛矿型La1‑xSrxMnO3复合材料及其制备方法和应用与制造工艺
- 一种在二氧化锰电极表面对苯胺进行原位氧化的方法与制造工艺
- 一种氧化石墨烯/银纳米颗粒/金字塔形硅三维拉曼增强基底及制备方法和应用与制造工艺
- 纳米羧基化石墨烯复合涤纶导电织物的制备方法与制造工艺