掺杂硼的碳材料、导电性组合物、导电膜和蓄电元件的制作方法
1.本发明涉及一种掺杂硼的碳材料、导电性组合物、导电膜和蓄电元件。
背景技术:
2.近年来,电子学(electronics)的发展显著,对于在各种电子设备中使用的导电性材料,也逐渐要求产品的小型轻量化、低成本化、在各种使用环境下的高寿命化。例如,在制造电子设备的基础配线或连接电子设备的配线的情况下,需要导电性良好的导电性组合物,但一般是使用银或铜等金属填料的导电性组合物,但至今尚未解决成本方面的主要问题。另一方面,对使用不利用金属的导电性碳的导电性组合物也进行了各种研究,但导电性不充分,因此限于在防静电用途等半导电性用途中使用。
3.另外,关于导电性的碳材料,迄今为止也研究了各种石墨(graphite)或碳纳米管等体积电阻率低的导电材料,但与体积电阻率小于10-4
ω
·
cm的表现出非常高的导电性的金属填料相比,体积电阻率大幅提高,导电性的改善为主要问题。
4.因此,在专利文献1、专利文献2中,以改善电特性为目的,报告了通过对碳纳米管与硼化合物进行高温加热处理,而在碳纳米管中掺杂硼的研究。通过对多壁碳纳米管添加3质量%以上的硼化合物而进行最大3000℃的高温加热处理,获得了体积电阻率为10-2
ω
·
cm水平的导电性,但仍然需要改善导电性。另外,硼以哪种方式掺杂至碳纳米管的分析不充分,且关于硼的掺杂状态与导电性的关系不明确也是课题。另一方面,也有报告称,对于直径10nm以下的碳纳米管,若通过1600℃以上的高温加热处理掺杂硼,则难以维持碳纳米管的形状,且在单壁碳纳米管或双壁碳纳米管中会产生分解,因此使硼掺杂的碳材料的硼的掺杂状态与导电性的关系变得更复杂。
5.进而,专利文献3中报告了对100nm以下的厚度非常小的石墨烯纳米片添加3质量%左右的硼源来掺杂硼的研究。在所述报告中,虽然获得了体积电阻率小于10-3
ω
·
cm的优异的导电性,但与金属填料相比,仍需要改善导电性。另外,虽然公开了硼掺杂的碳材料中的硼含量,但未明示硼是以哪种方式掺杂至碳材料。
6.因此,认为通过分析硼掺杂的碳材料的硼的掺杂状态,进一步明确掺杂状态与导电性的关系,能够进一步改善硼掺杂的碳材料的导电性。
7.现有技术文献
8.专利文献
9.专利文献1:日本专利特开2000-2813238号公报
10.专利文献2:日本专利特开2009-256118号公报
11.专利文献3:国际公开第2014/185496号
技术实现要素:
12.发明所要解决的问题
13.本发明的目的是提供一种导电性优异的碳材料。
14.解决问题的技术手段
15.本发明涉及一种碳材料,以碳六角网面为基本骨架、以置换碳元素的方式掺杂有硼元素,且所述碳材料的特征在于,碳材料中的硼元素的含量为0.005摩尔%~15摩尔%,当将以置换碳材料表面的碳元素的方式掺杂的硼元素的含量设为x(摩尔%),将碳材料中的硼元素的含量设为y(摩尔%)时,x/y<0.8。
16.另外,本发明涉及碳材料包含选自由石墨、石墨烯纳米片、石墨烯、及碳纳米管所组成的群组中的至少一种的所述碳材料。
17.另外,本发明涉及0.01《x/y<0.4的所述碳材料。
18.另外,本发明涉及一种导电性组合物,其包含所述碳材料、及粘合剂树脂或溶媒的至少一者。
19.另外,本发明涉及一种包含碳材料不同的两种以上的碳材料的导电性组合物。
20.另外,本发明涉及一种还包含导电助剂的导电性组合物。
21.另外,本发明涉及还含有活性物质,且作为用于形成蓄电元件用的正极或负极的复合材料油墨使用。
22.另外,本发明涉及一种由所述导电性组合物形成的导电膜。
23.另外,本发明涉及一种蓄电元件,所述蓄电元件包括具有由所述导电性组合物形成的复合材料层的电极。
24.发明的效果
25.根据所述本发明,可提供一种碳材料,以含有硼元素的碳六角网面为基本骨架,且含有以置换表面的碳元素的方式掺杂的硼元素,所述碳材料通过特定的硼含量而导电性优异。
附图说明
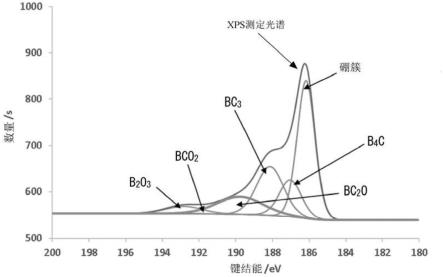
26.[图1]图1是表示x射线光电子光谱法(x-ray photoelectron spectroscopy,xps)中的b1s光谱的峰、及峰分离的例子的图。
[0027]
[图2]图2是表示激光拉曼光谱(ramam spectrum)中的碳材料的g波段与d波段的峰的图。
具体实施方式
[0028]
以下详细说明本发明。再者,本说明书中,有时将“以碳六角网面为基本骨架,以置换碳元素的方式掺杂有硼元素的碳材料”简称为“硼掺杂的碳材料”。
[0029]
《硼掺杂的碳材料》
[0030]
对本发明的硼掺杂的碳材料进行说明。硼掺杂的碳材料是以形成有碳原子共价键结为六角网状的网平面的碳六角网面为基本骨架,在这些构成单元之间具有物理性、化学性相互作用(键结),且含有硼元素的材料,并且是至少含有以置换碳元素的方式掺杂的硼元素的碳材料。再者,“以置换碳元素的方式掺杂的硼元素”表示与位于碳六角网面中的碳元素进行置换的硼元素、与位于碳材料表面的碳六角网面边缘的碳元素进行置换的硼元素、与以空位的形式存在于碳材料表面的碳六角网面中的缺陷或缺损部等的碳元素键结的硼元素、以将碳材料表面附近的碳六角网面的面之间架桥的方式与碳元素键结的硼元素,
这些硼元素能够通过后述的x射线光电子光谱法(xps)进行测定。
[0031]
本发明的硼掺杂的碳材料中的硼的含量(碳材料整体中的硼的含量)为0.005摩尔%~15摩尔%,优选为0.01摩尔%~10摩尔%,更优选为0.08摩尔%~5摩尔%,进而优选为0.1摩尔%~2摩尔%。硼的含量小于0.005摩尔%时,无法获得硼的掺杂效果,导电性得不到改善。硼的含量为15摩尔%以上时,过剩的硼阻碍电子的移动,引起导电性的降低。因此,若处于0.005摩尔%~15摩尔%的范围,则可表现出良好的导电性。
[0032]
另外,通过碳材料含有硼,提高了对溶媒或粘合剂树脂的润湿性,分散液的分散性提升。另外,硼含量多时,发现碳材料的硬度增加的倾向,对冲击等外力的耐久性提升,因此分散稳定性或膜强度改善。另一方面,硼含量多时,处于难以分散的倾向,因此就碳材料的强度及分散性的观点而言,硼含量优选为2摩尔%以下,更优选为1.1摩尔%以下。
[0033]
硼掺杂的碳材料中的硼的含量可通过感应偶合等离子体(inductively coupled plasma,icp)发光光谱分析、icp质量分析等方法求出。作为一例,可列举基于日本工业标准(japanese industrial standards,jis)-r7223的测定方法。另外,硼掺杂的碳材料中的硼的含量表示碳材料的表面或内部含有的硼单质、以置换碳材料中的碳元素的方式掺杂的硼、硼化合物的全部硼量。
[0034]
接着,对以置换本发明的硼掺杂的碳材料的表面的碳元素的方式掺杂的硼进行说明。以与硼掺杂的碳材料的表面的碳元素进行置换的方式掺杂的硼的含量可通过x射线光电子光谱法(xps)等方法求出。
[0035]
已知通过xps测定而获得的硼的b1s光谱出现在硼元素的b1s电子的键结能范围(185ev~197ev附近),大致包括4个成分。图1是表示xps中的b1s光谱的峰及峰分离的例子的图。如图1所示,各成分的键结能的值(峰顶)出现在:硼簇为186ev~187ev,碳化硼(b4c)为187ev~188ev,以与六角网面为基本骨架的碳元素进行置换的方式掺杂的硼(bc3)为188ev~189.3ev,各种氧化硼、即,bc2o为189.5ev~190.5ev,bco2为191.5ev~192ev,b2o3为192.5ev~193ev。在这些峰重叠的情况下,以高斯函数(gaussian function)的形式将峰强度、峰位置、峰半值全宽作为参数将各成分进行最优化,由此进行拟合而分离峰,从而可求出比例。因此,进行b1s峰分离,可分析硼掺杂的碳材料表面的硼的状态。
[0036]
因此,以置换本发明的硼掺杂的碳材料表面的碳元素的方式掺杂的硼的键结能的值出现在188ev~189.3ev。
[0037]
另外,将以置换硼掺杂的碳材料表面的碳元素的方式掺杂的硼元素的含量设为x、将硼掺杂的碳材料整体的硼元素的含量设为y时的比率优选为x/y<0.8,进而优选为x/y<0.6,进而优选为0.01<x/y<0.4,更优选为0.02<x/y<0.2。若x/y处于0.8以上的范围内,则由于过量的空穴的导入而使导电性降低、或者为了使硼在碳材料的表面键结和/或进行置换反应,需要局部大的能量,有时也会部分地破坏碳材料的六角网面等基本骨架。因此,若处于x/y<0.8的范围内,则可表现出良好的导电性。
[0038]
通过所述xps测定而获得的数据与碳材料表面有关,据说通常的深度分辨率为数nm左右,最大达到10nm。另一方面,通过icp测定而获得的数据与碳材料整体有关,通过xps测定而算出的硼量与通过icp测定而算出的硼量不同。
[0039]
即,关于本发明的碳材料,是以碳六角网面为基本骨架,以置换碳元素的方式掺杂有硼元素的碳材料,碳材料整体中的硼的含量为规定范围量,并且,相对于碳材料整体中的
硼的含量,以置换碳材料表面的碳元素的方式掺杂的硼元素的含量小于规定值,由此发挥优异的导电性。
[0040]
另外,使用本发明的碳材料的导电性组合物不仅分散性优异,对分散时的冲击等外力的耐久性也提高,因此可减轻对碳材料的损伤,发挥非常优异的导电性。
[0041]
另外,本发明的硼掺杂的碳材料是以碳六角网面为基本骨架的碳材料,可通过拉曼光谱测定及x射线衍射测定进行确认。
[0042]
图2是表示激光拉曼光谱中碳材料的g波段及d波段的峰的图。在拉曼光谱测定中,例如通过基于激发激光波长532nm的g波段(1560cm-1
~1620cm-1
)的确认,可确认是以碳六角网面为基本骨架的碳材料。另外,与表示碳材料缺陷的d波段(1330cm-1
~1370cm-1
)的强度比(g/d比)高时,硼掺杂的碳材料的结晶度高,导电性也高。g/d比优选为1以上,更优选为1.7以上,进而优选为2.0以上。
[0043]
另外,在将激发激光波长532nm的激光拉曼光谱中的g波段的拉曼位移设为p,将d波段的拉曼位移设为q时,若拉曼位移的差[p-q]为226cm-1
以下,则预估通过硼掺杂而关系到导电性提升的载流子增加,因此优选。拉曼位移的差[p-q]优选为218cm-1
~226cm-1
,更优选为222cm-1
~226cm-1
。
[0044]
在x射线衍射(x-ray diffraction,xrd)测定中,在将cuka射线作为x射线源而获得的硼掺杂的碳材料的xrd图中,根据衍射角(2θ)出现在24.0
°
~27.0
°
附近的(002)面衍射峰的确认,可确认是以碳六角网面为基本骨架的碳材料。另外,通过所获得的峰算出的平均面间距离d002优选为0.338nm以下,进而优选为0.336nm以下。通常的碳材料的平均面间距离d002越小,有导电性越优异的倾向,但本发明的硼掺杂的碳材料的平均面间距离d002在0.335nm~0.336nm的情况下,导电性优异。
[0045]
另外,本发明的硼掺杂的碳材料的结构(形状)只要是满足所述条件的碳材料,则并无特别限定,例如可分类为碳黑(乙炔黑、科琴黑、炉黑、中热碳黑、石墨化碳黑)、石墨、石墨烯纳米片、石墨烯、碳纤维、碳纳米管、碳纳米纤维、碳纳米角、碳纳米刷、活性碳、多孔质碳、纳米多孔碳等,优选为石墨化碳黑、石墨、石墨烯纳米片、石墨烯、碳纤维、碳纳米管、碳纳米纤维,进而优选为石墨、石墨烯纳米片、石墨烯、碳纳米管。
[0046]
本发明的硼掺杂的碳材料的平均粒径并无特别限定,为0.15μm~500μm,优选为0.5μm~100μm,进而优选为1μm~50μm。另外,本发明中所说的平均粒径是指在体积粒度分布中,自粒径细的粒子累计所述粒子的体积比例时,为50%的粒径(d50),通过一般的粒度分布计、例如动态光散射方式的粒度分布计(日机装公司制造的“麦奇克(microtrac)upa”)等进行测定。
[0047]
本发明的硼掺杂的碳材料的平均厚度并无特别限定。
[0048]
碳源为石墨时,所述平均厚度优选为1nm~500μm,更优选为150nm~50μm,进而优选为500nm~10μm,特别优选为500nm~5μm。
[0049]
另外,本发明中所说的平均厚度可根据利用光学显微镜或电子显微镜等随机提取的厚度的平均值算出,也可使用图像分析等算出。为了抑制由粒子引起的偏差,优选为由100个以上的粒子算出平均值。
[0050]
另外,在碳源为碳纳米管的情况下,其平均外径优选为2nm~500nm,更优选为3nm~250nm,进而优选为5nm~50nm,特别优选为5nm~25nm。
micron)公司制造的粒子复合化装置“纳米固化(nanocure)”、“诺必塔(nobilta)”、“机械融合机(mechanofusion)”;奈良机械制作所公司制造的粉体表面改质装置“混合型研磨系统(hybridization system)”、“微型机械(mechano micros)”“米拉罗(miralo)”等。
[0076]
另外,在使用干式混合装置时,可在作为母体的原料粉体中以粉体的状态直接添加其他原料,但为了制作更均匀的混合物,也可使用预先将其他原料溶解或分散在少量的溶媒中,一边分解作为母体的原料粉体的凝聚粒子一边添加的方法,也可进行加温以进一步提高处理效率。
[0077]
作为湿式混合装置,例如可列举:分散机、均质混合机、或行星式混合机等混合机类;爱慕科技(m-technique)公司制造的“库莱尔密克斯(clearmix)”、或谱莱密克斯(primix)公司制造的“菲鲁密克斯(filmix)”等均化器类;红魔鬼(reddevil)公司制造的涂料搅拌机、球磨机、新丸企业(shinmaru enterprises)公司制造的“戴诺磨机(dyno-mill)”等砂磨机类;磨碎机、或共球磨机等介质型分散机;吉纳苏(jenius)公司制造的“吉纳苏(jenius)py”、杉野机械(sugino machine)公司制造的“斯塔帕斯特(starburst)”、纳诺米泽(nanomizer)公司制造的“纳诺米泽(nanomizer)”等湿式喷射磨机类;爱慕科技(m-technique)公司制造的“库莱尔(clear)ss-5”、或奈良机械制作所公司制造的“麦克洛斯(micros)”等无介质分散机类;或其他辊磨机、捏合机、超声波分散机等,但并不限定于这些。另外,作为湿式混合装置,有时优选使用实施了防止来自装置的金属混入处理的装置。
[0078]
例如,当使用介质型分散机时,优选为使用搅拌器及容器为陶瓷制或树脂制的分散机的方法,或者使用对金属制搅拌器及容器表面施以碳化钨喷镀或树脂涂布等处理的分散机。而且,作为介质,优选使用玻璃珠、或氧化锆珠或氧化铝珠等陶瓷珠。另外,对于使用辊磨机的情况,也优选使用陶瓷制辊。分散装置既可仅使用一种,也可将多种装置组合起来使用。
[0079]
另外,在原料无法均匀溶解或分散的情况下,为了提高各原料对溶媒的润湿性、分散性,可根据需要并用多种溶媒,也可添加分散剂进行分散、混合。
[0080]
特别是对于已碳化(石墨化)的碳源,硼源难以均匀混合,因此硼掺杂反应发生时的碳源与硼源的混合(接触)状态很重要,优选为将碳源与硼源均匀处理。
[0081]
对所述碳源与硼源的混合物进行加热处理的条件根据作为原料的碳源或硼源的种类、量而不同,并无特别限定,加热温度为1000℃~3200℃,优选为1500℃~3000℃,进而优选为1800℃~2500℃。另外,加热时间并无特别限定,为10分钟~72小时,优选为30分钟~10小时。
[0082]
为了防止原料氧化等副反应,加热处理步骤中的环境优选为氮气、氩气等惰性气体、或真空下的环境。
[0083]
另外,加热处理步骤不仅是对固定的环境、温度及时间以一个阶段进行的处理步骤,也可为针对环境、温度、温度,以多个阶段进行的处理步骤。
[0084]
《导电性组合物》
[0085]
本发明的导电性组合物含有所述的碳材料、及粘合剂树脂或溶媒的至少一者。导电性组合物可并用不同的两种以上的碳材料,也可根据需要含有导电助剂。
[0086]
《粘合剂树脂》
[0087]
作为粘合剂树脂,并无特别限制,例如可包含选自由聚氨基甲酸酯系树脂、聚酰胺
系树脂、丙烯腈系树脂、丙烯酸系树脂、丁二烯系树脂、聚乙烯系树脂、聚乙烯丁醛系树脂、聚烯烃系树脂、聚酯系树脂、聚苯乙烯系树脂、乙烯-乙酸乙烯酯共聚物(ethylene vinyl acetate copolyme,eva)系树脂、聚偏二氟乙烯系树脂、聚四氟乙烯系树脂、硅酮系树脂、聚醚系树脂、羧甲基纤维素等纤维素系树脂等所组成的群组中的至少一种。
[0088]
所述粘合剂树脂可单独使用一种,也可并用两种以上。
[0089]
就体积电阻率、对基材的密接性及耐久性的观点而言,粘合剂树脂优选为包含选自由聚氨基甲酸酯系、聚酰胺系树脂及聚酯系树脂所组成的群组中的至少一种,更优选为包含聚氨基甲酸酯系树脂。粘合剂树脂优选为在基材上印刷或涂布导电性组合物后进行压制或热压(以下记载为(热)压制)时,适度软化或流动。通过使用此种树脂,导电性组合物在维持涂膜的平面图案形状的同时沿厚度方向流动,膜中的空隙减少,碳材料彼此的接触点增加,因此可获得体积电阻率低的导电膜。
[0090]
[聚氨基甲酸酯树脂]
[0091]
聚氨基甲酸酯树脂的合成方法并无特别限定,例如可列举:使多元醇化合物(a)与二异氰酸酯(b)反应的方法;使多元醇化合物(a)、二异氰酸酯(b)、及具有羧基的二醇化合物(c)反应,获得具有异氰酸酯基的氨基甲酸酯预聚物(d)的方法;使所述氨基甲酸酯预聚物(d)进一步与聚氨基化合物(e)反应的方法;或者在所述三种方法中,根据需要使反应停止剂反应的方法。
[0092]
作为多元醇化合物(a),可使用作为构成聚氨基甲酸酯树脂的多元醇成分而公知的各种聚醚多元醇类、聚酯多元醇类、聚碳酸酯多元醇类、聚丁二烯二醇类、或这些的混合物等。
[0093]
作为聚醚多元醇类,例如可列举氧化乙烯、氧化丙烯、四氢呋喃等聚合物或共聚物。
[0094]
作为聚酯多元醇类,可列举:乙二醇、1,2-丙二醇、1,3-丙二醇、1,3-丁烷二醇、1,4-丁烷二醇、新戊二醇、戊二醇、3-甲基-1,5-戊二醇、己二醇、辛二醇、1,4-丁二醇、二乙二醇、三乙二醇、二丙二醇、二聚物二醇等饱和及不饱和的低分子二醇类;以及正丁基缩水甘油醚、2-乙基己基缩水甘油醚类的烷基缩水甘油醚类;新癸酸缩水甘油酯等单羧酸缩水甘油酯类;己二酸、邻苯二甲酸、间苯二甲酸、对苯二甲酸、马来酸、富马酸、琥珀酸、草酸、丙二酸、戊二酸、庚二酸、辛二酸、壬二酸、癸二酸等二羧酸类;或将这些酸酐类脱水缩合而获得的聚酯多元醇类;或将环状酯化合物开环聚合而获得的聚酯多元醇类。
[0095]
作为聚碳酸酯多元醇类,可使用(1)二醇或双酚与碳酸酯的反应产物、及(2)二醇或双酚在碱的存在下与光气的反应产物。作为碳酸酯,可列举:碳酸二甲酯、碳酸二乙酯、碳酸二苯酯、碳酸亚乙酯、碳酸亚丙酯等。作为二醇,可列举:乙二醇、丙二醇、二丙二醇、二乙二醇、三乙二醇、丁二醇、3-甲基-1,5-戊二醇、2-甲基-1,8-辛二醇、3,3
′‑
二羟甲基庚烷、聚氧乙二醇、聚氧丙二醇、丙二醇、1,3-丁烷二醇、1,4-丁烷二醇、1,5-戊二醇、1,6-己二醇、1,9-壬二醇、新戊二醇、辛二醇、丁基乙基戊二醇、2-乙基-1,3-己二醇、环己烷二醇、3,9-双(1,1-二甲基-2-羟乙基)、2,2,8,10-四氧代螺〔5.5〕十一烷等。另外,作为双酚,可列举:双酚a、双酚f、将环氧乙烷、环氧丙烷等环氧烷加成到双酚类中而得的双酚类等。
[0096]
所述多元醇化合物的数量平均分子量(mn)考虑制造导电性组合物时的聚氨基甲酸酯树脂的溶解性、要形成的导电膜的耐久性、或对基材的接着强度等来适当决定,但通常
优选为580~8,000的范围,更优选为1,000~5,000的范围。
[0097]
所述多元醇化合物可单独使用,也可并用两种以上。进而,在不丧失聚氨基甲酸酯树脂性能的范围内,也可将所述多元醇化合物的一部分替换为低分子二醇类,例如所述多元醇化合物的制造中使用的各种低分子二醇。
[0098]
作为二异氰酸酯化合物(b),可使用芳香族二异氰酸酯、脂肪族二异氰酸酯、脂环族二异氰酸酯、或这些的混合物。优选为脂环族二异氰酸酯,更优选为异佛尔酮二异氰酸酯。作为芳香族二异氰酸酯,可列举:1,5-亚萘基二异氰酸酯、4,4
′‑
二苯基甲烷二异氰酸酯、4,4
′‑
二苯基二甲基甲烷二异氰酸酯、4,4
′‑
苄基异氰酸酯、二烷基二苯基甲烷二异氰酸酯、四烷基二苯基甲烷二异氰酸酯、1,3-亚苯基二异氰酸酯、1,4-亚苯基二异氰酸酯、甲苯二异氰酸酯、亚二甲苯基二异氰酸酯等。
[0099]
作为脂肪族二异氰酸酯,例如,可列举:丁烷-1,4-二异氰酸酯、六亚甲基二异氰酸酯、2,2,4-三甲基六亚甲基二异氰酸酯、赖氨酸二异氰酸酯。
[0100]
作为脂环族二异氰酸酯,例如,可列举:环己烷-1,4-二异氰酸酯、异佛尔酮二异氰酸酯、降冰片烷二异氰酸甲酯、双(4-异氰酸酯环己基)甲烷、1,3-双(异氰酸酯甲基)环己烷、甲基环己烷二异氰酸酯。
[0101]
作为具有羧基的二醇化合物(c),例如可列举:二羟甲基乙酸、二羟甲基丙酸、二羟甲基丁酸、二羟甲基戊酸等二羟甲基烷酸、二羟基琥珀酸、二羟基苯甲酸。特别是就反应性、溶解性方面而言,优选为二羟甲基丙酸、二羟甲基丁酸。
[0102]
使多元醇化合物(a)、二异氰酸酯(b)及具有羧基的二醇化合物(c)反应,获得具有异氰酸酯基的氨基甲酸酯预聚物(d)时的条件除了过量存在异氰酸酯基以外,并无特别限制,但是异氰酸酯基/羟基的当量比优选为1.05/1~3/1的范围内。更优选为1.2/1~2/1。另外,反应通常在常温~150℃的范围内进行,进而,就制造时间、副反应的控制的方面而言,优选为在60℃~120℃的范围内进行。
[0103]
聚氨基化合物(e)用作扩链剂,例如可使用:乙二胺、丙二胺、六亚甲基二胺、二乙三胺、三乙四胺、异佛尔酮二胺、二环己基甲烷-4,4
′‑
二胺、降冰片烷二胺,除此之外也使用2-(2-氨基乙基氨基)乙醇、2-羟基乙基乙二胺、2-羟基乙基丙二胺、二-2-羟基乙基乙二胺、二-2-羟基丙基乙二胺等具有羟基的胺类。其中,优选使用异佛尔酮二胺。
[0104]
在使具有异氰酸酯基的氨基甲酸酯预聚物(d)与聚氨基化合物(e)反应而合成聚氨基甲酸酯树脂时,可并用反应停止剂以调整所获得的聚氨基甲酸酯树脂的分子量。作为反应停止剂,可使用二-正丁基胺等二烷基胺类;二乙醇胺等二烷醇胺类;乙醇、异丙醇等醇类。
[0105]
使具有异氰酸酯基的氨基甲酸酯预聚物(d)与聚氨基化合物(e)、及视需要的反应停止剂反应时的条件并无特别限制,但是于将在氨基甲酸酯预聚物的两末端存在的游离异氰酸酯基设为1当量的情况下,聚氨基化合物(e)及反应停止剂中的氨基的合计当量优选为0.5~1.3的范围内。进而优选为0.8~0.995的范围内。
[0106]
就涂布性或处理性的观点而言,聚氨基甲酸酯树脂的重量平均分子量优选为5,000~200,000的范围。
[0107]
[聚酰胺树脂]
[0108]
聚酰胺树脂是通过二元酸与二胺的缩聚、氨基羧酸的缩聚、或内酰胺的开环聚合
等各种反应而获得的具有酰胺键的高分子的总称,以各种改性聚酰胺为首,由部分氢化的反应物制造,且可使用由其他单体部分共聚而成的聚合物、或混合了各种添加剂等其他物质的树脂。
[0109]
聚酰胺树脂并无特别限定,优选为使以二聚物酸为主要成分的二元酸与多胺类缩聚而获得的二聚物酸改性聚酰胺树脂。作为制造二聚物酸改性聚酰胺树脂时的二聚物酸,工业上广泛使用将妥尔油脂肪酸、大豆油脂肪酸等中含有的天然的一元性不饱和脂肪酸聚合而成的二聚物酸,但原理上可为饱和脂肪族、不饱和脂肪族、脂环式、或芳香族等各种二羧酸等。作为所述二聚物酸的市售品,可列举:哈利迪莫(haridimer)200、哈利迪莫(haridimer)300(哈利玛(harima)化成公司制造)、巴萨戴姆(varsadime)228、巴萨戴姆(varsadime)216、艾普罗(empol)1018、艾普罗(empol)1019、艾普罗(empol)1061、艾普罗(empol)1062(科宁(cognis)公司制造)等。进而,也可使用氢化的二聚物酸,作为氢化二聚物酸的市售品,可列举:普利普罗(pripol)1009(日本禾大(croda japan)股份有限公司制造)、艾普罗(empol)1008(科宁(cognis)公司制造)等。
[0110]
除了上述二聚物酸以外,为了制成具有适当柔软性的聚酰胺树脂,可使用各种二羧酸作为二元酸。作为二羧酸,具体而言,可使用草酸、丙二酸、琥珀酸(酐)、马来酸(酐)、戊二酸、己二酸、庚二酸、辛二酸、壬二酸、癸二酸、对苯二甲酸、间苯二甲酸、邻苯二甲酸、萘二羧酸、1,3-或1,4-环己烷二羧酸、1,18-十八烷二羧酸、1,16-十六烷二羧酸等。
[0111]
也可还使用具有酚性羟基者作为二元酸。通过使用具有酚性羟基的二元酸,可在聚酰胺树脂的侧链导入酚性羟基,可用于与硬化剂的反应。
[0112]
作为具有酚性羟基的二元酸,可列举:2-羟基间苯二甲酸、4-羟基间苯二甲酸、5-羟基间苯二甲酸等羟基间苯二甲酸、2,5-二羟基间苯二甲酸、2,4-二羟基间苯二甲酸、4,6-二羟基间苯二甲酸等二羟基间苯二甲酸;2-羟基对苯二甲酸、2,3-二羟基对苯二甲酸、2,6-二羟基对苯二甲酸等二羟基对苯二甲酸;4-羟基邻苯二甲酸、3-羟基邻苯二甲酸等羟基邻苯二甲酸;3,4-二羟基邻苯二甲酸、3,5-二羟基邻苯二甲酸、4,5-二羟基邻苯二甲酸、3,6-二羟基邻苯二甲酸等二羟基邻苯二甲酸等。
[0113]
进而,也可列举这些化合物的酸酐或例如多元酸甲酯那样的酯衍生物等。
[0114]
其中,就共聚性、获得容易性等方面而言,优选为5-羟基间苯二甲酸。
[0115]
进而,为了制成在加热时具有适当的流动性的聚酰胺树脂,视需要使用各种单羧酸。作为单羧酸,具体而言使用丙酸、乙酸、辛酸(caprylic acid)、硬脂酸、油酸等。
[0116]
作为制造所述二聚物酸改性聚酰胺树脂时的反应物的多胺类,例如为脂肪族、脂环式、芳香族等的各种二胺、三胺、多胺等。
[0117]
作为所述二胺的具体例,可列举:乙二胺、丙二胺、丁二胺、三乙二胺、四乙二胺、六亚甲基二胺、对二甲苯二胺或间二甲苯二胺、4,4
′‑
亚甲基双(环己基胺)、2,2-双-(4-环己基胺)、聚乙二醇二胺、异佛尔酮二胺、1,2-环己烷二胺、1,3-环己烷二胺、或1,4-环己烷二胺、1,4-双-(2
′‑
氨基乙基)苯、n-乙基氨基哌嗪、哌嗪等。
[0118]
另外,三胺可列举二亚乙基三胺等,多胺可列举三亚乙基四胺、四亚乙基五胺、五亚乙基六胺等。进而,也可使用将经二聚体化的脂肪族的腈基转换并进行氢还原而得到的二聚物二胺。
[0119]
另外,作为多胺化合物,可列举将具有碳数20~48的环状或非环状的烃基的多元
酸化合物的羧基转化为氨基而得的化合物,作为市售品的例子,例如可列举:日本禾大(croda japan)股份有限公司制造的“普利敏(priamine)1071”、“普利敏(priamine)1073”、“普利敏(priamine)1074”、“普利敏(priamine)1075”或日本科宁(cognis japan)股份有限公司制造的“维萨敏(versamine)551”等。
[0120]
二胺中也可并用烷醇胺。烷醇胺可列举乙醇胺、丙醇胺、二乙醇胺、丁醇胺、2-氨基-2-甲基-1-丙醇、2-(2-氨基乙氧基)乙醇等。另外,可使用骨架中具有氧的聚醚二胺。所述聚醚可由通式:h2n-r
1-(ro)
n-r
2-nh2(式中,n为2~100,r1、r2为碳原子数为1~14个的烷基或脂环式烃基,r为碳原子数为1~10个的烷基或脂环式烃基。烷基可为直链状,也可为支链状)表示。作为所述醚二胺,可列举聚氧丙烯二胺等,作为市售品,有捷夫胺(jeffamine)类(太阳科技化学(sun techno chemical)公司制造)。另外,也可列举双-(3-氨基丙基)-聚四氢呋喃。
[0121]
所述多胺类与二聚物酸或各种二羧酸通过常规方法加热缩合,并通过伴随脱水的酰胺化步骤来制造以二聚物酸改性聚酰胺树脂为首的各种聚酰胺树脂。一般,反应温度为100℃~300℃左右,反应时间为1~8小时左右。
[0122]
[聚酯树脂]
[0123]
聚酯树脂是包含作为单量体的多元羧酸及多元醇的聚合物。聚酯树脂可采用公知者,具体而言,就确保树脂的凝聚力的方面而言,重量平均分子量优选为1,000~100,000。另外,就密接的观点而言,玻璃化转变温度优选为-10℃~200℃。
[0124]
作为多价羧酸成分,例如可列举芳香族二羧酸、脂肪族二羧酸、不饱和二羧酸、三价以上的羧酸等,可自这些中选择一种或两种以上使用。另一方面,作为多元醇成分,可列举脂肪族二醇、醚二醇类、三价以上的多元醇等,可自这些中选择一种、或一种以上使用。
[0125]
作为聚酯树脂的市售品,可列举:拜龙(vylon)(东洋纺股份有限公司制造、“拜龙(vylon)”是注册商标)、波利斯特(polyester)(日本合成化学工业股份有限公司制造、“波利斯特(polyester)”是注册商标)、泰斯莱克(teslac)(日立化成聚合物股份有限公司制造、“泰斯莱克(teslac)”是注册商标)等。
[0126]
作为粘合剂树脂,就加热时的流动性、体积电阻率、对基材的密接性及耐久性的观点而言,也优选为包含具有能够与异氰酸酯基反应的官能基的、侧链具有选自由聚醚、聚酯、聚碳酸酯及聚丁二烯所组成的群组中的至少一种结构的乙烯基系聚合物。侧链的导入方法并无特别限定,可通过各种合成方法而获得。
[0127]
作为能够与异氰酸酯基反应的官能基,可列举羟基、氨基、羧基、环氧基、n-羟甲基、n-烷氧基甲基等,但就反应性的方面而言,羟基优选。
[0128]
能够与异氰酸酯基反应的官能基可导入乙烯基系聚合物的侧链或主链,导入方法并无特别限定,可通过各种合成方法导入。用于需要高韧性、耐久性的用途时,理想的是在乙烯基系聚合物的主链中直接导入能够与异氰酸酯反应的官能基,由此可提高树脂的交联密度。
[0129]
乙烯基系聚合物的聚苯乙烯换算的重量平均分子量优选为5,000~500,000,更优选为10,000~100,000。重量平均分子量为500,000以下时,在溶媒中的溶解性提高,为5,000以上时,(热)压制后可获得充分的涂膜强度。
[0130]
粘合剂树脂也可使用在将粘合剂树脂应用于基材后,经过硬化(交联)反应的硬化
性树脂。
[0131]
作为在硬化性树脂中使用的交联剂,并无特别限定,可列举具有两个以上异氰酸酯基的聚异氰酸酯化合物等。作为聚异氰酸酯化合物并无特别限定,在室外使用时,为了防止涂膜随时间劣化,优选为仅使用脂环族或脂肪族的化合物。
[0132]
作为脂环族聚异氰酸酯化合物,例如可列举:异佛尔酮二异氰酸酯、氢化甲亚苯基二异氰酸酯、氢化4,4
′‑
二苯基甲烷二异氰酸酯等。
[0133]
作为脂肪族聚异氰酸酯化合物,例如可列举:三甲基六亚甲基二异氰酸酯、4,4
′‑
二苯基甲烷二异氰酸酯、六亚甲基二异氰酸酯、赖氨酸二异氰酸酯等。
[0134]
作为芳香族聚异氰酸酯化合物,例如可列举:二苯基甲烷二异氰酸酯、甲苯二异氰酸酯、萘-1,5-二异氰酸酯、邻二甲苯二异氰酸酯、间二甲苯二异氰酸酯、对二甲苯二异氰酸酯、三苯基甲烷三异氰酸酯、聚亚甲基聚苯基异氰酸酯等。
[0135]
作为聚异氰酸酯化合物,可使用所述化合物与二醇类或二胺类的两末端异氰酸酯加合物、缩二脲改性体、异氰脲酸酯改性体。
[0136]
特别是在聚异氰酸酯化合物包含异氰脲酸酯改性体、特别是含有异氰脲酸酯环的三异氰酸酯的情况下,在热压制后可获得充分的涂膜强度,因此优选。作为含有异氰脲酸酯环的三异氰酸酯,具体而言,可列举:异氰脲酸酯改性异佛尔酮二异氰酸酯(例如住友拜耳(bayer)氨基甲酸酯股份有限公司制造的德士模都(desmodur)z4470)、异氰脲酸酯改性六亚甲基二异氰酸酯(例如,住友拜耳(bayer)氨基甲酸酯股份有限公司制造的斯米都(sumidur)n3300)、异氰脲酸酯改性甲苯二异氰酸酯(例如住友拜耳氨基甲酸酯股份有限公司制造的斯米都(sumidur)fl-2、斯米都fl-3、斯米都fl-4、斯米都hlba)。
[0137]
聚异氰酸酯化合物根据所要求的性能,可以异氰酸酯基的总数相对于粘合剂树脂官能基的总数优选为0.1倍~5.0倍,更优选为0.5倍~3.0倍,特别优选为0.8倍~2.0倍的比率,使用一种或混合两种以上使用。
[0138]
粘合剂树脂可以是溶解于溶媒中的溶解性树脂、及不溶解于溶媒中而以微粒的状态存在的分散型树脂微粒(乳液)的任意形态。
[0139]
分散型树脂微粒的粒子结构也可设为多层结构,即,所谓的核壳粒子。例如,通过使主要由具有官能基的单量体聚合而成的树脂局部存在于核部或壳部,或根据核和壳设置tg、组成的差异,可提高硬化性、干燥性、成膜性、粘合剂的机械强度。就粘接性或粒子的稳定性的观点而言,树脂微粒的平均粒径优选为10nm~1000nm,优选为10nm~300nm。另外,若含有大量超过1μm的粗大粒子,则粒子的稳定性受损,因此超过1μm的粗大粒子优选为最多5%以下。
[0140]
再者,所述的平均粒径表示体积平均粒径,可通过动态光散射法测定。基于动态光散射法的平均粒径的测定能够以如下方式进行。根据树脂微粒的固体成分,用与分散媒相同的分散液预先稀释至200~1000倍。将所述稀释分散液约5ml注入测定装置(日机装公司制造的纳米莱克(nanotrac))的池中,输入与样品对应的分散媒及树脂的折射率条件后,进行测定。可通过此时获得的体积粒径分布数据(柱状图)的峰进行测定。
[0141]
作为分散型树脂微粒,优选为包含交联型树脂微粒。交联型树脂微粒表示具有内部交联结构(三维交联结构)的树脂微粒,重要的是在粒子内部交联。另外,通过交联型树脂微粒含有特定的官能基,可有助于与基材的密接性。进而通过调整交联结构或官能基的量,
可获得优异的耐久性的涂膜。
[0142]
就环境负荷等观点而言,优选为水系溶剂,优选为可在水中使用的水溶性树脂及水性树脂微粒。另外,就导电性组合物的浆料稳定性或涂布性等观点而言,进而优选为并用水溶性树脂及水性树脂微粒。
[0143]
[水溶性树脂]
[0144]
水溶性树脂是指,在25℃的水99g中加入树脂1g并搅拌,在25℃下放置24小时后,不分离析出而完全能够溶解于水中的树脂。水溶性树脂具有提高碳材料的分散性的效果,因此可利用少的树脂量获得稳定的组合物。
[0145]
水溶性树脂大致分为阴离子性树脂、阳离子性树脂、同时具有阴离子性及阳离子性性质的两性树脂、以及除此之外的非离子性树脂,而且所述树脂也可包含多个单量体。另外,水溶性树脂可单独使用一种,也可并用两种以上。
[0146]
作为阴离子性树脂,例如可列举含有羧基、磺基、磷酸基及将这些的一部分或全部中和了的骨架的树脂。若进行例示,则可列举:(甲基)丙烯酸、衣康酸、富马酸、马来酸、甲基丙烯酸2-磺乙酯、酸式磷酸2-(甲基)丙烯酰氧基乙酯等聚合性单量体的均聚物、或与其他聚合性单量体的共聚物、羧基甲基纤维素、及这些的碱中和物等。
[0147]
作为阳离子性树脂,例如可列举:含有包含环状的氨基及中和了氨基的一部分或者全部的骨架、或四级铵盐的树脂等。若进行例示,则可列举:(甲基)丙烯酸n,n-二甲基氨基乙酯、(甲基)丙烯酸n,n-二乙基、乙烯基吡啶等聚合性单量体的均聚物、或与其他聚合性单量体的共聚物及这些的酸中和物。
[0148]
作为两性树脂,例如可列举:同时含有所述阴离子性骨架及所述阳离子性骨架的树脂。若进行例示,则可列举:苯乙烯-马来酸-n,n-二甲基氨基乙基(甲基)丙烯酸酯的共聚物等。
[0149]
非离子性树脂是所述阴离子性、阳离子性及两性树脂以外的树脂。若进行例示,则可列举:聚乙烯吡咯烷酮、聚乙烯醇、聚乙烯丁醛、聚丙烯酰胺、聚-n-乙烯基乙酰胺、聚烷二醇等。
[0150]
水溶性树脂的分子量并无特别限定,但优选为质量平均分子量为5,000~2,500,000。质量平均分子量(mw)表示凝胶渗透色谱术(gel permeation chromatography,gpc)中的聚环氧乙烷换算分子量。
[0151]
[水性树脂微粒]
[0152]
水性树脂微粒(水性乳液)是树脂在水中不溶解而以微粒的状态存在的分散型树脂微粒,例如可列举:(甲基)丙烯酸系乳液、腈系乳液、氨基甲酸酯系乳液、聚烯烃系乳液、氟系乳液(聚偏二氟乙烯(polyvinylidene fluoride,pvdf)、聚四氟乙烯(polytetrafluoroethylene,ptfe)等)、二烯系乳液(苯乙烯-丁二烯橡胶(styrene-butadienerubber,sbr)等)。再者,(甲基)丙烯酸是指甲基丙烯酸或丙烯酸。
[0153]
包含水性树脂微粒的导电性组合物在形成涂膜的情况下,可提供粒子间及与基材的密接性优异、强度高的涂膜。进而,由于密接性优异,因此所需水性树脂微粒少量即可,结果导电性组合物的导电性提高。为了获得所述效果,作为水性树脂微粒,优选为粒子间的粘接性及柔软性(膜的可挠性)优异的(甲基)丙烯酸系乳液或氨基甲酸酯系乳液。
[0154]
(甲基)丙烯酸系乳液是指含有10质量份以上具有(甲基)丙烯酰基的单量体的乳
化聚合物,优选为可含有20质量份以上,进而优选为可含有30质量份以上。具有丙烯酰基的单量体的反应性优异,因此可比较容易地制作树脂微粒。因此,作为水性树脂微粒,(甲基)丙烯酸系乳液特别优选。
[0155]
《溶媒》
[0156]
本发明的导电性组合物在使碳材料分散时、或使碳材料与粘合剂树脂均匀混合时,可适当使用溶媒。作为此种溶媒,只要是可溶解树脂的溶媒、或可稳定地分散树脂微粒乳液的溶媒,就并无特别限定,可列举水或有机溶剂。
[0157]
有机溶剂可自甲醇、乙醇、丙醇、丁醇、乙二醇甲醚、二乙二醇甲醚等醇类,丙酮、甲乙酮、甲基异丁基酮、环己酮等酮类,四氢呋喃、二噁烷、乙二醇二甲醚、二乙二醇二甲醚等醚类,己烷、庚烷、辛烷等烃类,苯、甲苯、二甲苯、枯烯等芳香族类,乙酸乙酯、乙酸丁酯等酯类等中,根据导电性组合物的组成而使用适当者。
[0158]
另外,溶媒可将水与有机溶剂组合使用,也可将两种以上的有机溶剂组合使用。
[0159]
使用水溶性树脂或水性树脂微粒时,就溶解性或分散性的观点而言,优选使用水作为溶媒,根据需要,也可添加与水相容的液状介质。作为与水相容的液状介质,优选为碳数为4以下的醇系溶剂。
[0160]
另外,本发明的树脂组合物中,可视需要,在不妨碍本发明的效果的范围内添加紫外线吸收剂、紫外线稳定剂、自由基补充剂、填充剂、触变性赋予剂、抗老化剂、抗氧化剂、抗静电剂、阻燃剂、导热性改良剂、塑化剂、防松弛剂、防污剂、防腐剂、杀菌剂、消泡剂、流平剂、抗粘连剂、硬化剂、增粘剂、分散剂、硅烷偶合剂等各种添加剂。
[0161]
《不同的两种以上的碳材料》
[0162]
如上所述,本发明的导电性组合物可含有本发明碳材料中不同的两种以上的碳材料。
[0163]
所述不同的两种以上的碳材料的组合并无特别限定,优选为比表面积不同的两种以上的碳材料的并用;石墨与碳黑、石墨与碳纳米管、石墨与石墨烯(石墨烯纳米片)那样的碳种类不同的两种以上的碳材料的并用;或者是这些的组合。
[0164]
并用比表面积不同的两种以上的碳材料时,比表面积最大的碳材料的比表面积优选为5m2/g~1500m2/g,更优选为20m2/g~1300m2/g,进而优选为110m2/g~900m2/g。
[0165]
由包含本发明的不同的两种以上的碳材料的导电性组合物形成的导电膜是均匀且碳材料之间的封装性高、密度高的导电膜,因此推测膜中的碳材料之间的导电性网络及导电膜的耐久性提高。进而,推测通过并用两种以上的碳材料,润湿性提高,与粘合剂树脂的相互作用发生变化,粘合剂树脂不易被覆碳材料表面,从而在碳材料之间的接点上不易存在粘合剂树脂,碳材料的接触电阻减少。另外,若并用两种以上的碳材料,由于分散性的提高,粘度有降低的倾向,可看到分散体的处理变得容易的效果,而且涂布性也提高。
[0166]
通过这些作用,推测通过使用不同的两种以上的碳材料,可形成导电性极其优异的导电膜。
[0167]
《导电助剂》
[0168]
本发明的导电性组合物根据需要可还含有本发明的碳材料以外的导电助剂。导电助剂只要并不相当于本发明特定的硼掺杂的碳材料即可,例如,可列举:碳黑、活性碳、石墨、导电性碳纤维(碳纳米管、碳纳米纤维等)、碳纳米角、石墨烯、石墨烯纳米片、纳米多孔
5”、或奈良机械公司制造的“麦克洛斯(micros)”等无介质分散机;或其他辊磨机、捏合机、超声波分散机等,但并不限于这些。
[0181]
例如,使用介质型分散机时,优选使用搅拌器及容器为陶瓷制或树脂制的分散机的方法,或者使用对金属制搅拌器及容器表面施以碳化钨喷镀或树脂涂布等处理的分散机。而且,作为介质,优选使用玻璃珠、或者氧化锆珠或氧化铝珠等陶瓷珠。分散装置可仅使用一种,也可将多种装置组合起来使用。
[0182]
《导电膜》
[0183]
本发明的导电膜是由导电性组合物形成的膜,可通过在基材上涂布导电性组合物,根据需要进行干燥来形成。
[0184]
(基材)
[0185]
导电膜成形中使用的基材的形状并无特别限定,可适当选择符合用途的形状。基材优选为片状,更优选为绝缘性的树脂膜。
[0186]
基材的材质并无特别限定,例如可列举:pet(聚对苯二甲酸乙二酯)、pen(聚萘二甲酸乙二酯)、聚酰亚胺、聚氯乙烯、聚酰胺、尼龙、opp(拉伸聚丙烯)、cpp(未拉伸聚丙烯)。
[0187]
另外,作为形状,一般使用平板状的膜,但也可使用表面经粗糙面化者、经底漆处理者、开孔状者、以及网状的基材。
[0188]
作为在基材上涂布导电性组合物的方法,并无特别限制,可使用公知的方法。作为此种涂布方法,例如可列举模涂法、浸涂法、辊涂法、刮涂法、刀片涂法、喷涂法、凹版涂布法、网版印刷法、静电涂装法。作为干燥方法,例如可列举放置干燥、送风干燥、暖风干燥、红外线加热、远红外线加热,但并不特别限定于这些。
[0189]
另外,也可在涂布后通过平版压机或砑光辊等进行轧制处理,为了使导电膜软化而容易压制,轧制处理可一边加热一边进行。
[0190]
导电膜的厚度一般为0.1μm以上且1mm以下,优选为1μm以上且200μm以下。
[0191]
(导电膜的体积电阻率)
[0192]
本发明的导电膜的体积电阻率优选为小于5
×
10-3
ω
·
cm,更优选为小于2
×
10-3
ω
·
cm,进而优选为小于1
×
10-3
ω
·
cm。通过体积电阻率小于5
×
10-3
ω
·
cm,作为导电性非常高的组合物,可用于电池的电极、集电体、或电池、电子设备的配线等。
[0193]
《蓄电元件》
[0194]
本发明的导电性组合物可通过根据需要调配活性物质等,制成用于形成蓄电元件用的正极或负极的组合物。然后,可使用由用于形成所述正极或负极的组合物形成的正极或负极的至少一者来获得蓄电元件。
[0195]
作为此种蓄电元件,可列举二次电池或电容器。作为二次电池,例如可列举:锂离子二次电池、钠离子二次电池、镁二次电池、碱性二次电池、铅蓄电池、钠硫二次电池、锂空气二次电池等。作为电容器,例如可列举:双电层电容器、锂离子电容器。
[0196]
所述二次电池及电容器的结构并无特别限制,通常包括:正极及负极、根据需要设置的隔板,可根据使用目的自纸型、圆筒型、按钮型、层叠型等各种形状中选择。
[0197]
[复合材料油墨]
[0198]
接着,对作为用于形成所述蓄电元件用正极或负极的组合物的优选方式之一的、以活性物质为必要成分的复合材料油墨进行说明。
[0199]
复合材料油墨有正极复合材料油墨或负极复合材料油墨,导电性组合物中含有正极或负极用的活性物质,根据需要,可还含有粘合剂树脂、溶媒、其他添加剂。
[0200]
(活性物质)
[0201]
作为锂离子二次电池用的正极活性物质,并无特别限定,可使用能够掺杂或嵌入锂离子的金属氧化物、金属硫化物等金属化合物、以及导电性高分子等。
[0202]
例如可列举:fe、co、ni、mn等过渡金属的氧化物、与锂的复合氧化物、过渡金属硫化物等无机化合物等。具体而言,可列举mno、v2o5、v6o
13
、tio2等过渡金属氧化物粉末、层状结构的镍酸锂、钴酸锂、锰酸锂、镍、钴及锰三成分与锂的复合氧化物即三元系活性物质、尖晶石结构的锰酸锂等的锂与过渡金属的复合氧化物粉末、作为橄榄石结构的磷酸化合物的磷酸铁锂系材料、tis2、fes等过渡金属硫化物粉末等。
[0203]
另外,也可使用聚苯胺、聚乙炔、聚吡咯、聚噻吩等导电性高分子。另外,也可混合使用所述的无机化合物或有机化合物。
[0204]
作为锂离子二次电池用的负极活性物质,只要能够掺杂或嵌入锂离子,则并无特别限定。例如,可列举:金属li;作为其合金的锡合金、硅合金、铅合金等合金系;lixfe2o3、lixfe3o4、li
x
wo2、钛酸锂、钒酸锂、硅酸锂等金属氧化物系;聚乙炔、聚对亚苯等导电性高分子系;软碳或硬碳等非晶系碳质材料、或高石墨化碳材料等人造石墨、或者天然石墨等碳质粉末、碳黑、中间相(mesophase)碳黑、树脂烧成碳材料、气层成长碳纤维、碳纤维等碳系材料。这些负极活性物质也可使用一种或组合多种使用。
[0205]
另外,作为碱性二次电池用的正极活性物质及负极活性物质,可适当选择以往公知的活性物质。
[0206]
作为双电层电容器用的电极活性物质,并无特别限定,可列举:活性碳、多并苯、碳晶须及石墨等,可使用这些的粉末或纤维等。双电层电容器用的优选的电极活性物质为活性碳,具体而言可列举将苯酚系、椰子壳系、人造丝系、丙烯酸系、煤炭/石油系沥青焦炭、中间相碳微球(mesocarbon microbeads,mcmb)等活化的活性碳。
[0207]
作为锂离子电容器用的正极活性物质,只要是能够可逆地掺杂、脱掺杂锂离子及阴离子的材料,则并无特别限定,例如可列举活性碳粉末。
[0208]
作为锂离子电容器用的负极活性物质,只要是能够可逆地掺杂、脱掺杂锂离子的材料,则并无特别限定,例如可列举人造石墨、天然石墨等石墨系材料。
[0209]
(粘合剂树脂)
[0210]
作为可含有复合材料油墨的粘合剂树脂,可引用所述的《粘合剂树脂》项的记载。另外,作为所述粘合剂树脂,例如可列举:丙烯酸树脂、聚氨基甲酸酯树脂、聚酯树脂、酚树脂、环氧树脂、苯氧基树脂、尿素树脂、三聚氰胺树脂、醇酸树脂、甲醛树脂、硅树脂、氟树脂、羧甲基纤维素等纤维素树脂;苯乙烯-丁二烯橡胶或氟橡胶等合成橡胶;聚苯胺或聚乙炔等导电性树脂;聚偏二氟乙烯、聚氟乙烯、及四氟乙烯等含有氟原子的高分子化合物,也可为这些树脂的改性物、混合物或共聚物。这些粘合剂也可使用一种或组合多种使用。
[0211]
而且,可根据需要在复合材料油墨中调配分散剂、成膜助剂、消泡剂、流平剂、防腐剂、ph调整剂、粘性调整剂等。
[0212]
复合材料油墨的粘度可根据涂布方法适当调整,但通常在固体成分30质量%~90质量%的范围内,优选设为100mpa
·
s以上且30,000mpa
·
s以下。
[0213]
在能够涂布的粘度范围内,活性物质的含量优选为尽可能多,活性物质相对于复合材料油墨的固体成分的比例优选为80质量%~99质量%。另外,本发明的碳材料相对于复合材料油墨的固体成分的比例优选为0.01质量%~15质量%。
[0214]
(分散机、混合机)
[0215]
作为获得复合材料油墨时使用的装置,可使用颜料分散等中通常使用的分散机、混合机。
[0216]
[电极]
[0217]
蓄电元件中使用的正极或负极的至少一个电极可通过将所述复合材料油墨涂布在集电体上并进行干燥、形成复合材料层而获得。
[0218]
(集电体)
[0219]
电极所使用的集电体的材质或形状并无特别限定,可适宜选择适合各种蓄电元件的材质、形状。例如,作为集电体的材质,可列举铝、铜、镍、钛或不锈钢等金属或合金。在锂离子电池的情况下,作为正极材料优选为铝,作为负极材料优选为铜。
[0220]
[电解液]
[0221]
电解液可适宜选择公知的电解液使用。例如,作为锂离子二次电池中使用的电解液,可列举将含有锂的电解质溶解在非水系的溶剂中而成的电解液。
[0222]
作为所述电解质,并无特别限制,例如可列举:libf4、liclo4、lipf6、liasf6、lisbf6、licf3so3、li(cf3so2)2n、lic4f9so3、li(cf3so2)3c、lii、libr、licl、lialcl、lihf2、liscn或libph4。
[0223]
作为所述非水系的溶剂,并无特别限定,例如可列举:碳酸亚乙酯、碳酸亚丙酯、碳酸亚丁酯、碳酸二甲酯、碳酸乙基甲基酯、碳酸二乙酯等碳酸酯类;γ-丁内酯、γ-戊内酯、γ-辛内酯等内酯类;四氢呋喃、2-甲基四氢呋喃、1,3-二氧杂环戊烷、4-甲基-1,3-二氧杂环戊烷、1,2-甲氧基乙烷、1,2-乙氧基乙烷、1,2-二丁氧基乙烷等乙二醇二甲醚类;甲酸甲酯、乙酸甲酯、丙酸甲酯等酯类;二甲基亚砜、环丁砜等亚砜类;乙腈等腈类。
[0224]
这些溶剂可分别单独使用,也可混合两种以上使用。
[0225]
进而,也可将所述电解液保持在聚合物基质中,制成凝胶状的高分子电解质。作为聚合物基质,可列举具有聚环氧烷链段的丙烯酸酯系树脂、具有聚环氧烷链段的聚磷腈系树脂、及具有聚环氧烷链段的聚硅氧烷等,但不限于这些。
[0226]
另外,只要能够进行离子传导,也可使用固体电解质代替电解液。作为此种固体电解质,并无特别限制,例如可列举氧化物系固体电解质、或硫化物系固体电解质等。
[0227]
[隔板]
[0228]
作为隔板,例如可列举聚乙烯无纺布、聚丙烯无纺布、聚酰胺无纺布以及对这些实施了亲水性处理的隔板,但并不特别限定于这些。
[0229]
实施例
[0230]
以下,通过实施例更详细地说明本发明。再者,只要并无特别说明,份、%表示质量份、摩尔%。
[0231]
《碳材料的制造》
[0232]
[实施例a1]
[0233]
使用球磨机将84份作为碳源的粒状天然石墨cgb-20(日本石墨工业公司制造)、及
16份作为硼源的硼酸(富士胶片和光纯药公司制造)进行干式混合,制作了前体(1)。
[0234]
接着,向石墨坩埚中填充前体(1),利用烧成炉在氩气环境下、在2100℃下进行1小时热处理,获得碳材料(1)。
[0235]
[实施例a2]
[0236]
将作为硼源的硼酸15份溶解在作为溶媒的水30份、乙醇270份中后,添加作为碳源的粒状天然石墨cgb-50(日本石墨工业公司制造)85份,使用行星式混合机进行搅拌,进行湿式混合。接着,在80℃下使溶媒挥发,制作前体(2)。
[0237]
接着,向石墨坩埚中填充前体(2),利用烧成炉在氩气环境下、在2100℃下进行1小时热处理,获得碳材料(2)。
[0238]
[实施例a3~实施例a7、比较例a1~比较例a3]
[0239]
使用表1所示的原料及条件,将原料进行干式混合时与实施例a1同样,将原料进行湿式混合时与实施例a2同样,制作了碳材料(3)~碳材料(10)。
[0240]
[比较例a4]
[0241]
使用离子注入装置使硼离子化而对100份作为碳源的多层石墨烯(石墨烯纳米片)xgnp-c-750(xg科技(sciences)公司制造)进行掺杂,获得碳材料(11)。
[0242]
《碳材料的评价》
[0243]
对所述的碳材料进行了以下评价。其结果如表2所示。
[0244]
(碳材料表面的硼元素的含量)
[0245]
利用xps(赛默飞世尔科技(thermo fisher scientific)公司制造、k-alpha)测定碳材料表面的硼元素的含量(%)。
[0246]
由于硼1s电子的光谱出现在键结能185ev~194ev处,因此可通过求出其峰面积来定量表面的硼元素的含量。另外,详细而言,硼簇出现在186ev~187ev,碳化硼出现在187ev~188ev,以与将六角网面作为基本骨架的碳元素置换的方式掺杂的硼元素(bc3)出现在188ev~189.3ev,各种氧化硼、即,bc2o出现在189.5ev~190.5ev,bco2出现在191.5ev~192ev,b2o3出现在192.5ev~193ev(参照图1)。
[0247]
因此,进行硼1s的峰分离,可分析碳材料表面的硼的掺杂状态。
[0248]
(碳材料中的硼元素的含量)
[0249]
利用icp发光光谱分析(斯派克(spectro)公司制造的斯派克阿克斯(spectroarcos)fhs12)测定碳材料中的硼元素的含量(%)。所获得的值表示碳材料的整体含有的硼元素的量(%)。
[0250]
(碳材料的体积电阻率)
[0251]
使用粉体电阻测定系统(三菱化学分析技术(mitsubishi chemical analytech)公司制造的mcp-pd51型)测定碳材料的体积电阻率。体积电阻率是在测定池中加入碳材料后,施加20kn的负荷时的值。
[0252]
(碳材料的碳六角网面的基本骨架)
[0253]
利用x射线衍射装置(理学(rigaku)公司制造、斯莫特莱伯(smartlab)),通过以cukα射线为x射线源的测定,在2θ=24.0
°
~27.0
°
附近确认到来自石墨骨架的(002)面的峰,而确认到碳材料(1)~碳材料(11)以及碳材料(12)~碳材料(45)具有碳六角网面的基本骨架。
[0254]
[表1]
[0255]
表1.
[0256] 碳源质量份硼源质量份溶媒质量份混合条件热处理条件碳材料实施例a1cgb-2084硼酸16无0干式混合2100℃-1小时(1)实施例a2cgb-5085硼酸15水/乙醇30/270湿式混合2100℃-1小时(2)实施例a3fb-15099.5碳化硼0.5乙醇10湿式混合2100℃-10分钟(3)实施例a4cgb-2099硼酸1乙醇20湿式混合2100℃-20分钟(4)实施例a5cgb-5060硼酸40无0干式混合2000℃-1小时(5)实施例a6cgb-5099.2硼酸0.8水16湿式混合2000℃-2小时(6)实施例a7cgb-2084碳化硼16乙醇1000湿式混合2200℃-10小时(7)比较例a1pag-599.9硼酸0.1乙醇5湿式混合2000℃-5分钟(8)比较例a2pag-550碳化硼50无0干式混合2000℃-5小时(9)比较例a3xgnp-m-596硼酸4无0干式混合2900℃-2小时(10)
[0257]
cgb-20:粒状天然石墨、粒径20μm、日本石墨工业公司制造
[0258]
cgb-50:粒状天然石墨、粒径50μm、日本石墨工业公司制造
[0259]
fb-150:薄片状天然石墨、粒径50μm、日本石墨工业公司制造
[0260]
pag-5:粒状人造石墨、粒径50μm、日本石墨工业公司制造
[0261]
xgnp-m-5:多层石墨烯(石墨烯纳米片)、粒径5μm、xg科技(sciences)公司制造
[0262]
[表2]
[0263]
表2.
[0264][0265]
[实施例a8]
[0266]
通过日本专利特开2019-108256的段落[0147]、段落[0148]中记载的方法,制作了碳纳米管(carbon nanotube,cnt)合成用催化剂(1)。然后,在能够加压、能够利用外部加热器加热的、内容积10l的卧式反应管的中央部,设置了散布有1g所述cnt合成用催化剂的石英玻璃制耐热皿。一边注入氮气一边进行排气,用氮气置换反应管内的空气,加热至卧式反应管中的环境温度达到700℃。达到700℃后,以每分钟2l的流速将乙烯气体作为烃导入反应管内,使其接触反应15分钟。反应结束后,用氮气置换反应管内的气体,将反应管的温度冷却至100℃以下并取出,从而获得cnt(1)。
[0267]
接着,使用超声波均质器(先进数字索尼法伊厄(advanced digital sonifer)(注
册商标)、模式(model)450da、必能信(branson)公司制造)将96份作为碳源的cnt(1)、4份作为硼源的硼酸(富士胶片和光纯药公司制造)、作为溶媒的9500份n-甲基吡咯烷酮(n-methyl pyrrolidone,nmp)、400份乙醇均匀分散后,进行干燥来制作前体(12)。再者,在表3及表7中,对与前体(12)同样的混合方法,标记为处理(1)。
[0268]
接着,向石墨坩埚中填充前体(12),利用烧成炉在氩气环境下、1750℃下进行1小时热处理,从而获得碳材料(12)。
[0269]
[实施例a9、实施例a10]
[0270]
除了使用表3所示的原料、条件以外,与实施例a8同样地制作碳材料(13)、碳材料(14)。
[0271]
[实施例a11]
[0272]
使用超声波均质器将95份作为碳源的cnt(2)(多层碳纳米管100p、锦湖石化(kumho petrochemical)公司制造)、5份作为硼源的硼酸(富士胶片和光纯药公司制造)、作为溶媒的9500份nmp、400份乙醇、及0.9份作为分散剂的pvp(polyvinylpyrrolidone)(1)(聚乙烯吡咯烷酮、k-15、西格玛奥德里奇(sigma-aldrich)公司制造)均匀分散后,使其干燥,制作前体(15)。再者,在表3及表7中,对与前体(15)同样的混合方法,标记为处理(2)。
[0273]
接着,向石墨坩埚中填充前体(15),利用烧成炉在氩气环境下、1700℃下进行1小时热处理,从而获得碳材料(15)。
[0274]
[实施例a12]
[0275]
使用超声波均质器将96.2份作为碳源的cnt(3)(多层碳纳米管jenotube 8s、杰奥(jeio)公司制造)、3.8份作为硼源的硼酸(富士胶片和光纯药公司制造)、作为溶媒的9500份nmp、400份乙醇、0.8份作为分散剂的pvp(1)均匀分散,使溶媒干燥后,使用粒子复合化装置机械融合机(细川密克朗(hosokawa micron)公司制造)进行复合化,制作了前体(16)。再者,在表3及表7中,对与前体(16)同样的混合方法,标记为处理(3)。
[0276]
接着,向石墨坩埚中填充前体(16),利用烧成炉在氩气环境下、1700℃下进行1小时热处理,从而获得碳材料(16)。
[0277]
[实施例a13]
[0278]
除了使用表3所示的原料、条件以外,与实施例a12同样地制作碳材料(17)。
[0279]
[比较例a5]
[0280]
将99.5份作为碳源的cnt(4)(碳纳米管tuball、奥克西尔(ocsial)公司制造)、及0.5份作为硼源的碳化硼(富士胶片和光纯药公司制造)用玛瑙乳钵混合后,填充到石墨坩埚中,利用烧成炉在氩气环境下,2800℃下,进行20分钟热处理,从而获得碳材料(18)。
[0281]
[比较例a6]
[0282]
除了使用表3所示的原料、条件以外,与比较例a5同样地制作碳材料(19)。
[0283]
[比较例a7]
[0284]
在能够加压、能够利用外部加热器加热的、内容积10l的卧式反应管的中央部,设置了散布有1g cnt合成用催化剂(1)的石英玻璃制耐热皿。一边注入氮气一边进行排气,用氮气置换反应管内的空气,加热至卧式反应管中的环境温度达到700℃。达到700℃后,以每分钟2l的流速将乙烯气体、硼酸三甲酯气体的混合气体导入反应管内,使其接触反应15分钟。反应结束后,用氮气置换反应管内的气体,将反应管的温度冷却至100℃以下,从而获得
碳材料(20)。
[0285]
[比较例a8]
[0286]
使用离子注入装置使硼离子化,对作为碳源的碳黑(cb(1))(科琴黑ec-600jd、狮王特殊化学(lion specialty chemicals)公司制造)进行掺杂,从而获得碳材料(21)。
[0287]
对所述碳材料进行了所述《碳材料的评价》。评价结果如表4所示。
[0288]
[表3]
[0289]
[0290]
[表4]
[0291]
表4.
[0292][0293]
[实施例a14]
[0294]
将99份作为碳源的up-20(天然石墨、日本石墨工业公司制造)、1份作为硼源的硼酸(富士胶片和光纯药公司制造)、1800份作为溶媒的甲苯、200份乙醇放入混合机中混合,进一步放入砂磨机进行分散。
[0295]
对分散液进行取样,持续分散至前体的碳的平均厚度达到90nm后,使溶媒干燥而制作前体(22)。再者,在表5中,对与前体(22)同样的混合方法,标记为处理(4)。
[0296]
接着,向石墨坩埚中填充前体(22),利用烧成炉在氩气环境下、2100℃下进行1小时热处理,从而获得碳材料(22)。
[0297]
[实施例a15、实施例a16]
[0298]
除了以前体的碳的平均厚度分别成为355nm、825nm的方式分散以外,与实施例a14同样地制作碳材料(23)、碳材料(24)。
[0299]
[实施例a17]
[0300]
将98.8份作为碳源的f#1(天然石墨、日本石墨工业公司制造)、1.2份作为硼源的碳化硼(富士胶片和光纯药公司制造)、1000份作为溶媒的甲苯、0.1份作为分散剂的pvp(1)放入混合机进行混合,进一步放入砂磨机进行分散。
[0301]
对分散液进行取样,持续分散至前体的碳平均厚度达到3.5μm后,将溶媒干燥来制作前体(25)。再者,在表5中,对与前体(25)同样的混合方法,标记为处理(5)。
[0302]
接着,向石墨坩埚中填充前体(25),利用烧成炉在氩气环境下、2000℃下进行1小时热处理,从而获得碳材料(25)。
[0303]
[实施例a18]
[0304]
除了以前体的碳的平均厚度为11.6μm的方式进行分散以外,与实施例a17同样地制作碳材料(26)。
[0305]
[比较例a9]
[0306]
将92份作为碳源的石油焦炭、8份作为硼源的氧化硼(富士胶片和光纯药公司制造)用玛瑙乳钵混合后,填充到石墨坩埚中,利用烧成炉在氩气环境下、2800℃下进行1小时热处理,从而获得碳材料(27)。
[0307]
对所述碳材料进行了所述《碳材料的评价》及以下的评价。结果如表6所示。
[0308]
(g/d比)
[0309]
使用激光拉曼分光光度计(日本分光公司制造、nrs-3100)评价g/d比。在激发激光波长532nm的条件下进行测定,根据获得的各样品的拉曼光谱(参照图2)的d波段(1330cm-1
~1370cm-1
)及g波段(1560cm-1
~1620cm-1
)的峰强度之比(ig/id),算出g/d。
[0310]
(拉曼位移的差[p-q])
[0311]
使用激光拉曼分光光度计(日本分光公司制造、nrs-3100)评价拉曼位移的差(p-q)。在激发激光波长532nm的条件下进行测定,根据获得的各样品的拉曼光谱的d波段(1330cm-1
~1370cm-1
)的峰顶的拉曼位移与g波段(1560cm-1
~1620cm-1
)的峰顶的拉曼位移之差,算出(p-q)。
[0312]
[表5]
[0313]
[0314]
[表6]
[0315]
表6.
[0316][0317]
[实施例a19]
[0318]
将1.1份作为硼源的碳化硼(富士胶片和光纯药公司制造)、及1000份作为溶媒的甲苯放入混合机中混合,进而放入砂磨机进行分散后,加入98.9份作为碳源的f#1(天然石墨、日本石墨工业公司制造),用混合机进行混合。将溶媒干燥后,利用粒子复合化装置机械融合机(细川密克朗(hosokawa micron)公司制造)进行复合化,制作出前体(28)。再者,在表7中,对与前体(28)同样的混合方法,标记为处理(6)。
[0319]
接着,向石墨坩埚中填充前体(28),利用烧成炉在氩气环境下、2000℃下进行1小时热处理,从而获得碳材料(28)。
[0320]
[实施例a20、实施例a34]
[0321]
除了使用表7所示的原料、条件以外,与实施例a19同样地制作碳材料(29)、碳材料(43)。
[0322]
[实施例a21、实施例a22、实施例a25~实施例a30、实施例a33、实施例a35、实施例a36]
[0323]
除了使用表7所示的原料、条件以外,与实施例a8同样地制作碳材料(30)、碳材料(31)、碳材料(34)~碳材料(39)、碳材料(42)、碳材料(44)、碳材料(45)。
[0324]
[实施例a23]
[0325]
利用高剪切混合机(high shear mixer)(l5m-a、西仑弗尔(silverson)公司制造)将97.5份作为碳源的f#1(天然石墨、日本石墨工业公司制造)、0.5份作为硼源的碳化硼(富士胶片和光纯药公司制造)、2份硼酸(富士胶片和光纯药公司制造)、作为溶媒的900份甲苯、100份乙醇、及0.1份作为分散剂的聚乙烯醇缩丁醛(polyvinyl butyral,pvb)(s-lec bl-10、积水化学工业公司制造)进行分散。将溶媒干燥后,利用粒子复合化装置机械融合机(细川密克朗(hosokawa micron)公司制造)进行复合化,制作出前体(32)。再者,在表7中,对与前体(32)同样的混合方法,标记为处理(7)。
[0326]
接着,向石墨坩埚中填充前体(32),利用烧成炉在氩气环境下、2000℃下进行1小时热处理,从而获得碳材料(32)。
[0327]
[实施例a24]
[0328]
除了使用表7所示的原料、条件以外,与实施例a23同样地制作碳材料(33)。
[0329]
[实施例a31]
[0330]
将作为硼源的碳化硼(富士胶片和光纯药公司制造)0.2份、及作为溶媒的甲苯1000份放入混合机中混合,进而放入砂磨机进行分散后,加入99.8份作为碳源的fb-150(天
然石墨、日本石墨工业公司制造),利用超声波均质器进行分散后,将溶媒干燥,而制作前体(40)。再者,在表7中,对与前体(40)同样的混合方法标记为处理(8)。
[0331]
接着,向石墨坩埚中填充前体(40),利用烧成炉在氩气环境下、2400℃下进行1小时热处理,从而获得碳材料(40)。
[0332]
[实施例a32]
[0333]
除了使用表7所示的原料、条件以外,与实施例a31同样地制作碳材料(41)。
[0334]
对所述的碳材料进行了所述的《碳材料的评价》及(g/d比)、(拉曼位移的差〔p-q〕)的评价。结果如表8所示。
[0335]
[表7]
[0336]
japan)股份有限公司制造)、5.5份5-羟基间苯二甲酸、146.4份作为多胺化合物的普利敏(priamine)1074(日本禾大(croda japan)股份有限公司制造)、100份离子交换水后,进行搅拌直至产生热量的温度变得恒定。温度稳定后升温至110℃,确认水流出后,在30分钟后将温度升温至120℃,然后一边每隔30分钟升温10℃,一边继续脱水反应。温度达到230℃后,在所述温度下持续反应3小时,在约2kpa的真空下保持1小时,使温度降低。最后,添加抗氧化剂,获得重量平均分子量24,000、酸值13.2mgkoh/g、羟基值5.5mgkoh/g、玻璃化转变温度-32℃的聚酰胺树脂。
[0348]
将获得的聚酰胺树脂用甲苯/2-丙醇(质量比:2/1)稀释,获得固体成分20质量%的聚酰胺树脂溶液。
[0349]
[制造例3]聚酯树脂溶液
[0350]
用甲苯/甲乙酮(质量比:1/1)稀释拜龙(vylon)200(东洋纺公司制造、聚酯树脂),获得固体成分20质量%的聚酯树脂溶液。
[0351]
再者,树脂的评价如下进行。
[0352]
(重量平均分子量(mw))
[0353]
重量平均分子量的测定使用了东曹(tosoh)股份有限公司制造的凝胶渗透色谱仪(gel permeation chromatograph,gpc)“hpc-8020”。gpc是通过其分子大小的差来对溶解于溶剂(thf:四氢呋喃)中的物质进行分离定量的液相色谱仪。本发明的测定是串联连接2根“lf-604”(昭和电工股份有限公司制造:快速分析用gpc管柱:6mmid
×
150mm尺寸)而用作管柱,并在流量0.6ml/min、管柱温度40℃的条件下进行,重量平均分子量的决定是通过聚苯乙烯换算来进行。
[0354]
(酸值(av))
[0355]
在带磨口塞的锥形烧瓶中精密地量取试样约1g,添加甲苯/乙醇(体积比:甲苯/乙醇=2/1)混合液100ml而进行溶解。向其中添加酚酞试液作为指示剂,并保持30秒。其后,利用0.1n醇性氢氧化钾溶液进行滴定直至溶液呈现出淡红色为止,通过下式求出酸值。
[0356]
酸值(mgkoh/g)=(5.611
×a×
f)/s
[0357]
其中,
[0358]
s:试样的采集量(g)
[0359]
a:0.1n醇性氢氧化钾溶液的消耗量(ml)
[0360]
f:0.1n醇性氢氧化钾溶液的滴定度
[0361]
(羟基值(oxhydryl value,ohv))
[0362]
羟基值是通过用于中和使羟基乙酰化时与羟基键结的乙酸所需的氢氧化钾的量(mg)表示1g含有羟基的树脂中所含的羟基的量。羟基值可依据日本工业标准(japanese industrial standard,jis)k0070而测定,在本发明中,如下述所示地考虑酸值而求出。
[0363]
在带磨口塞的锥形烧瓶中精密地量取试样约1g,添加甲苯/乙醇(体积比:甲苯/乙醇=2/1)混合液100ml而进行溶解。进而准确地添加5ml的乙酰化剂(将25g的乙酸酐以吡啶加以溶解而成为体积100ml的溶液),搅拌约1小时。在其中加入酚酞试液作为指示剂,持续30秒。其后,利用0.1n醇性氢氧化钾溶液进行滴定直至溶液呈现出淡红色为止,通过下式求出羟基值。
[0364]
羟基值(mgkoh/g)=[{(b-a)
×f×
28.05}/s]+d
[0365]
其中,
[0366]
s:试样的采集量(g)
[0367]
a:0.1n醇性氢氧化钾溶液的消耗量(ml)
[0368]
b:空白实验的0.1n醇性氢氧化钾溶液的消耗量(ml)
[0369]
f:0.1n醇性氢氧化钾溶液的滴定度
[0370]
d:酸值(mgkoh/g)
[0371]
(玻璃化转变温度(tg))
[0372]
树脂的玻璃化转变温度使用将溶剂干燥除去的树脂,使用梅特勒
·
托利多(mettler toledo)(股)制造的“dsc-1”,以2℃/分钟升温至-80℃~150℃,进行测定。
[0373]
《导电性组合物、导电膜的制造》
[0374]
[实施例b1]
[0375]
将88份碳材料(2)、60份(树脂固体成分12份)作为粘合剂树脂的聚氨基甲酸酯树脂溶液、152份作为溶剂的甲苯/甲乙酮/2-丙醇(质量比:1/1/1)放入混合机中进行混合,进而放入砂磨机中进行分散,获得导电性组合物(1)。
[0376]
接着,使用刮刀将所述导电性组合物(1)涂布在厚度100μm的pet膜基材上后,在烘箱中使其干燥。然后,在线压300kg/cm的条件下进行辊压制,获得导电膜(1)。
[0377]
[实施例b2~实施例b5、比较例b1、比较例b2]
[0378]
除了变更为表9-1所示的配方组成以外,通过与实施例b1同样的方法获得导电性组合物(2)~导电性组合物(7)、导电膜(2)~导电膜(7)。
[0379]
[实施例b6]
[0380]
将5份碳材料(12)、1份作为粘合剂树脂的pvp(2)(聚乙烯吡咯烷酮、富士胶片和光纯药公司制造、k-30)、94份作为溶剂的nmp放入混合机进行混合,进而放入砂磨机进行分散,获得导电性组合物(8)。
[0381]
接着,使用刮刀将所述导电性组合物(8)涂布在厚度100μm的pet膜基材上后,在烘箱中使其干燥,获得导电膜(8)。
[0382]
[实施例b7~实施例b10、比较例b3~比较例b5]
[0383]
以表9-2所示的配方组成比,通过与实施例b6同样的方法,获得导电性组合物(9)~导电性组合物(12)、导电性组合物(17)~导电性组合物(19)、导电膜(9)~导电膜(12)、导电膜(17)~导电膜(19)。
[0384]
[实施例b11]
[0385]
将3份碳材料(13)、0.6份作为粘合剂树脂的聚乙烯醇(pva)(可乐丽(kuraray)公司制造、可乐丽波瓦尔(kuraraypoval)sd1000)、96.4份作为溶剂的离子交换水加入混合机进行混合,进而放入砂磨机进行分散,获得导电性组合物(13)。
[0386]
接着,使用刮刀将所述导电性组合物(13)涂布在厚度100μm的pet膜基材上后,在烘箱中使其干燥,获得导电膜(13)。
[0387]
[实施例b12~实施例b14、比较例b6~比较例b8]
[0388]
以表9-2所示的配方组成比,通过与实施例b11同样的方法,获得导电性组合物(14)~导电性组合物(16)、导电性组合物(20)~导电性组合物(22)、导电膜(14)~导电膜(16)、导电膜(20)~导电膜(22)。
[0389]
[实施例b15]
[0390]
将57份碳材料(29)、13份作为导电助剂的cb(1)、150份(树脂固体成分30份)作为粘合剂树脂的聚氨基甲酸酯树脂溶液、作为溶剂的甲苯/甲乙酮/2-丙醇(质量比:1/1/1)以组合物中的固体成分成为30质量%的方式放入混合机中,进行混合,进而放入砂磨机中进行分散,获得导电性组合物(23)。
[0391]
接着,使用刮刀将所述导电性组合物(23)涂布在厚度100μm的pet膜基材上后,在烘箱中使其干燥,获得导电膜(23)。
[0392]
[实施例b16~实施例b37、实施例b39、比较例b9~比较例b13]
[0393]
以表9-3、表9-4所示的配方组成比,通过与实施例b15同样的方法,获得导电性组合物(24)~导电性组合物(45)、导电性组合物(47)~导电性组合物(52)、导电膜(24)~导电膜(45)、导电膜(47)~导电膜(52)。
[0394]
[实施例b38]
[0395]
将56份碳材料(41)、18份作为导电助剂的cb(3)(乙炔黑hs-100、登卡(denka)公司制造)、300份(树脂固体成分6份)溶解了2质量%的作为粘合剂树脂的水溶性树脂cmc(大赛璐米拉斯(daicelmiraizu)公司制造、羧甲基纤维素#1240)的水溶液放入混合机中进行混合,进而放入砂磨机进行分散。
[0396]
接着,追加40份(树脂固体成分20份)作为粘合剂树脂的水性树脂微粒(聚丙烯酸乳液w-168、东洋化工(toyo chem)公司制造、固体成分50质量),用混合机混合,获得导电性组合物(46)。
[0397]
接着,使用刮刀将所述导电性组合物(46)涂布在厚度100μm的pet膜基材上后,在烘箱中使其干燥,获得导电膜(46)。
[0398]
《导电性组合物、导电膜的评价》
[0399]
利用以下方法对获得的导电性组合物及导电膜进行评价。评价结果示于表9-1~表9-4。
[0400]
(导电性组合物的分散稳定性评价)
[0401]
根据将导电性组合物在25℃下静置7天保存后的液体性状的变化评价分散稳定性。根据用抹刀搅拌时的搅拌难易度判断液体性状的变化。
[0402]
判定基准
[0403]
○
:液体性状无变化(良好)
[0404]
δ:粘度上升,但没有凝胶化(可)
[0405]
×
:凝胶化(不良)
[0406]
(涂布性评价)
[0407]
利用视频显微镜(video microscopic)vhx-900(基恩士(keyence)公司制造)以500倍观察所获得的导电膜,按照下述基准判定涂布不均及针孔。根据膜表面的浓淡判断涂布不均。针孔根据膜中有无未涂布的缺陷进行判断。
[0408]
《涂布不均》
[0409]
○
:未确认到膜表面的浓淡(良好)
[0410]
δ:膜表面的浓淡有2~3处,但为极其微小的区域(可)
[0411]
×
:确认到大量膜表面的浓淡,或者确认到一个以上浓淡条纹的长度为5mm以上的
膜表面的浓淡(不良)
[0412]
《针孔》
[0413]
○
:未确认到针孔(良好)
[0414]
δ:有2~3个针孔,但极其微小(可)
[0415]
×
:确认到有多个针孔,或者确认到1个以上直径1mm以上的针孔(极其不良)
[0416]
(膜的体积电阻率)
[0417]
导电膜的体积电阻率依据jis-k7194,使用罗莱斯塔(loresta)gp(日东精工分析科技(nittoseiko analytech)公司制造)通过四端子法进行测定。
[0418]
(膜的耐久性)
[0419]
导电膜的耐久性依据jis k5600-5-4:1999,使用硬度为hb的铅笔通过划痕硬度(铅笔法)进行了评价。
[0420]
○
:不发生塑性变形及凝聚破坏(良好)
[0421]
δ:部分发生塑性变形或凝聚破坏(可)
[0422]
×
:发生塑性变形及凝聚破坏(极其不良)
[0423]
[表9-1]
[0424]
[0425]
[0426]
[0427][0428]
根据所述实施例b的结果,使用材料中的硼在本发明的范围内的硼掺杂的碳材料的导电性组合物具有优异的分散稳定性及涂布性,由所述导电性组合物形成的导电膜兼顾
了高导电性及耐久性。
[0429]
另外,通过比较实施例b1、实施例b3~实施例b5、或实施例b7、实施例b8,判明碳材料的硼的含有状态的不同对导电性组合物、导电膜的特性有很大的影响。根据硼含量的不同,有分散性良好的倾向,因此分散稳定性、涂布性良好。进而,根据实施例b20、实施例b22、实施例b27或实施例b35、实施例b36的比较,关于石墨与碳黑或碳纳米管的并用,通过并用两种以上处于本发明范围内的不同的硼掺杂的碳材料,获得了涂布性良好的导电性组合物,因此可获得无不均的、均匀的涂膜。
[0430]
因此,这些结果暗示,起因于碳材料的导电性以外的因素作出贡献。虽然现阶段不清楚详细情况,但推测本发明的导电性组合物不仅硼掺杂的碳材料的导电性优异,而且分散性及分散稳定性良好,进而涂布性也良好,因此碳材料在膜中有效率地形成导电网络,从而表现出优异的导电性。另外,推测碳材料的均匀的网络提高碳材料之间的封装性,使致密的膜形成成为可能,也关系到导电膜的耐久性改善。
[0431]
《正极用复合材料油墨的制造》
[0432]
[实施例c1]
[0433]
以导电性组合物中的碳材料、pvdf、正极活性物质的组成(质量比)成为0.25/1.5/98.25的方式,搅拌表10所示的导电性组合物(8)、及溶解了8质量%的pvdf(索尔(solef)#5130、苏威(solvey)股份有限公司制造)的nmp后,添加正极活性物质ncm523(日本化学工业股份有限公司制造、组成:lini0.5co0.2mn0.3o2)并搅拌。进而,以正极用复合材料油墨的固体成分为75质量%的方式添加nmp并搅拌,获得正极用复合材料油墨(1)。所述搅拌均使用自转公转混合机。
[0434]
[实施例c2~实施例c5、比较例c1~比较例c3]
[0435]
除了变更为表10所示的导电性组合物以外,与实施例c1同样地获得正极用复合材料油墨(2)~正极用复合材料油墨(8)。
[0436]
《正极用复合材料油墨的评价》
[0437]
(正极复合材料层的体积电阻率)
[0438]
使用涂敷器将正极用复合材料油墨以电极的单位面积重量为20mg/cm2的方式涂敷于pet基材上后,在电烘箱中在120℃下干燥30分钟,获得正极。然后,使用日东精工分析科技(analytech)公司制造的罗莱斯塔(loresta)gp、mcp-t610,测定干燥后的膜的表面电阻率(ω/γ)。测定后,乘以在pet基材上形成的复合材料层的厚度,作为正极用的电极膜的体积电阻率(ω.cm)。使用膜厚计,由测定膜中的5点的平均值,减去pet基材的厚度,求出复合材料层的厚度。
[0439]
判定基准
[0440]
◎
:正极复合材料层的体积电阻率(ω
·
cm)小于8(优良)
[0441]
○
:正极复合材料层的体积电阻率(ω
·
cm)为8以上且小于12(良)
[0442]
δ:正极复合材料层的体积电阻率(ω
·
cm)为12以上且小于16(可)
[0443]
×
:正极复合材料层的体积电阻率(ω
·
cm)为16以上(不良)
[0444]
(正极复合材料层的剥离强度)
[0445]
使用涂敷器将正极用复合材料油墨以电极的单位面积重量为20mg/cm2的方式涂布在铝箔上后,在烘箱中在120℃下干燥30分钟。然后,以涂布方向为长轴切成90mm
×
20mm
的长方形2条。剥离强度的测定使用拉伸试验机,通过180度剥离试验法进行评价。具体而言,将100mm
×
30mm尺寸的双面胶带粘贴在不锈钢板上,使制作的复合材料层与双面胶带的另一个面密接,以固定速度(50mm/分钟)自下方向上方拉伸的同时剥离,将此时的应力的平均值作为剥离强度。
[0446]
判定基准
[0447]
◎
:正极复合材料层的剥离强度(n/cm)为0.3以上(优良)
[0448]
○
:正极复合材料层的剥离强度(n/cm)为0.25以上且小于0.3(良)
[0449]
δ:正极复合材料层的剥离强度(n/cm)为0.2以上且小于0.25(可)
[0450]
×
:正极复合材料层的剥离强度(n/cm)小于0.2(不良)
[0451]
《负极用复合材料油墨的制造》
[0452]
[实施例c6]
[0453]
以导电性组合物中的碳材料、cmc、sbr、负极活性物质(石墨/一氧化硅)的组成(质量比)为0.5/1/88/9/1.5的方式,将表10所示的导电组合物(13)及溶解了2质量%的cmc(大赛璐精细化学(daicel finechem)股份有限公司制造、#1190)的水溶液搅拌后,添加一氧化硅(大阪钛科技股份有限公司制造,一氧化硅(silicon monooxide)、sio1.3c 5μm)并搅拌。进一步添加石墨(日本石墨工业股份有限公司制造、cgb-20)并搅拌。然后,添加sbr(jsr股份有限公司制造、trd2001,固体成分48质量%分散液)并搅拌。最后,以使负极用复合材料油墨的固体成分为50质量%的方式添加离子交换水,进行搅拌,获得负极用复合材料油墨(1)。所述搅拌均使用自转公转混合机。
[0454]
[实施例c7~实施例c9]、比较例c4~比较例c6]
[0455]
除了变更为表10所示的导电性组合物以外,与实施例c6同样地获得负极用复合材料油墨(2)~负极用复合材料油墨(7)。
[0456]
《负极用复合材料油墨的评价》
[0457]
(负极复合材料层的体积电阻率)
[0458]
除了使用负极用复合材料油墨,将电极的单位面积重量设为8mg/cm2以外,与正极复合材料层同样地测定负极复合材料层的体积电阻率(ω
·
cm)。
[0459]
判定基准
[0460]
◎
:负极复合材料层的体积电阻率(ω
·
cm)小于0.12(优良)
[0461]
○
:负极复合材料层的体积电阻率(ω
·
cm)为0.12以上且小于0.15(良)
[0462]
δ:负极复合材料层的体积电阻率(ω
·
cm)为0.15以上且小于0.25(可)
[0463]
×
:负极复合材料层的体积电阻率(ω
·
cm)为0.25以上(不良)
[0464]
(负极复合材料层的剥离强度)
[0465]
除了使用涂敷器在铜箔上以使电极的单位面积重量为8mg/cm2的方式涂布负极用复合材料油墨以外,与正极复合材料层的剥离强度同样地测定负极复合材料层的剥离强度。
[0466]
判定基准
[0467]
◎
:负极复合材料层的剥离强度(n/cm)为0.5以上(优良)
[0468]
○
:负极复合材料层的剥离强度(n/cm)为0.4以上且小于0.5(良)
[0469]
δ:负极复合材料层的剥离强度(n/cm)为0.3以上且小于0.4(可)
[0470]
×
:负极复合材料层的剥离强度(n/em)小于0.3(不良)
[0471]
《非水电解质二次电池的制造》
[0472]
评价用的标准负极(a)及标准正极(c)通过以下方法制作。
[0473]
[标准负极(a)的制作]
[0474]
在塑料容器中加入乙炔黑(登卡股份有限公司制造、登卡黑(注册商标)hs-100)、cmc(大赛璐精细化学(daicel finechem)股份有限公司制造、羧甲基纤维素#1190)、水之后,使用自转及公转混合机进行搅拌。再添加石墨(日本石墨工业股份有限公司制造、cgb-20)作为负极活性物质,使用自转及公转混合机进行搅拌。继而,加入sbr(jsr股份有限公司制造、trd2001、固体成分48质量%分散液),使用自转及公转混合机进行搅拌,获得标准负极用复合材料油墨。标准负极用复合材料油墨的固体成分设为48质量%。标准负极用复合材料油墨中的负极活性物质:导电材:cmc:sbr的固体成分比率设为97∶0.5∶1∶1.5。
[0475]
接着,使用涂敷器将标准负极用复合材料油墨涂布在作为集电体的厚度20μm的铜箔上后,在80℃烘箱中干燥30分钟,调整为电极的每单位面积的单位面积重量为10mg/cm2。再进行利用辊压制的轧制处理,制作了负极复合材料层的密度为1.6g/cm3的标准负极(a)。
[0476]
[标准正极(c)的制作]
[0477]
在塑料容器中加入正极活性物质(巴斯夫(basf)户田电池材料(basf battery materials)联合公司制造、hed(注册商标)ncm-111 1100)93份、乙炔黑(登卡股份有限公司制造、登卡黑(注册商标)hs100)4份、pvdf(日本库雷哈电池材料(kureha battery materials japan)股份有限公司制造,库雷哈(kureha)kf聚合物w#1300)3份后,使用抹刀混合至粉末变得均匀。然后,添加20.5份nmp,使用自转/公转混合机进行搅拌。然后,使用抹刀将塑料容器内的混合物混合至均匀,使用所述自转/公转混合机进行搅拌。然后,添加14.6份nmp,使用所述自转/公转混合机进行搅拌。最后,使用分散器进行搅拌,获得标准正极用复合材料油墨。然后,使用涂敷器将标准正极用复合材料油墨涂布在作为集电体的厚度20μm的铝箔上后,在120℃烘箱中干燥30分钟,调整成电极的每单位面积的单位面积重量为20mg/cm2。进而通过辊压制进行轧制处理,制作了复合材料层的密度为3.1g/cm3的标准正极(c)。
[0478]
[实施例d1~实施例d9、比较例d1~比较例d6]
[0479]
评价用的正极及负极通过以下方法制作。
[0480]
使用涂敷器将表11所示的正极用复合材料油墨涂布在作为集电体的厚度20μm的铝箔上后,用烘箱在120℃下干燥30分钟,调整为电极的单位面积重量为20mg/cm2。再进行利用辊压制的轧制处理,制作出正极复合材料层的密度为3.1g/cm3的正极。
[0481]
另一方面,使用涂敷器将表11所示的负极用复合材料油墨涂布在作为集电体的厚度20μm的铜箔上后,利用烘箱在80℃下干燥30分钟,调整为电极的单位面积重量为10mg/cm2。再进行利用辊压制的轧制处理,制作出负极复合材料层的密度为1.6g/cm3的负极。
[0482]
将表11所示的正极及标准负极(a)、或者表11所示的负极及标准正极(c)分别冲裁成50mm
×
45mm、45mm
×
40mm。然后,将正极、隔板(多孔质聚丙烯膜)、及标准负极(a)重叠,另外,将标准正极(c)、隔板(多孔质聚丙烯膜)、及负极重叠,分别插入铝制层压袋,在电烘箱中,在70℃下干燥1小时。继而,在充满氩气的手套箱内,注入2ml电解液,封口铝制层压袋,制作非水电解质二次电池(1)~非水电解质二次电池(15)。电解液是在将碳酸亚乙酯、碳酸
二甲酯、及碳酸二乙酯以1∶1∶1(体积比)的比例混合而成的混合溶媒中,进一步加入相对于电解液100份为1份的作为添加剂的vc(碳酸亚乙烯酯)后,以1m的浓度溶解lipf6而成的非水电解液。
[0483]
《非水电解质二次电池的评价》
[0484]
对获得的非水电解质二次电池进行了以下评价。评价结果如表11所示。
[0485]
(速率特性)
[0486]
将非水电解质二次电池设置在25℃的恒温室内,使用充放电装置(北斗电工股份有限公司制造、sm-8)进行充放电测定。以充电电流10ma(0.2c)、充电终止电压4.3v进行恒电流恒电压充电(截止电流1ma(0.02c))后,以放电电流10ma(0.2c)、放电终止电压3v进行恒电流放电。重复所述操作3次后,以充电电流10ma(0.2c)、充电终止电压4.3v进行恒电流恒电压充电(截止电流(1ma(0.02c)),以放电电流0.2c及3c进行恒电流放电直到达到放电终止电压成为3.0v,分别求出放电容量。速率特性可使用0.2c放电容量及3c放电容量之比,由以下的式1表示。
[0487]
(式1)
[0488]
速率特性=3c放电容量/第三次的0.2c放电容量
×
100(%)
[0489]
判定基准
[0490]
◎
:速率特性为80%以上(极其优良)
[0491]
○
:速率特性为70%以上且小于80%(优良)
[0492]
○
δ:速率特性为60%以上且小于70%(良)
[0493]
δ:速率特性为50%以上且小于60%(可)
[0494]
×
:速率特性小于50%(不良)
[0495]
(循环特性)
[0496]
将非水电解质二次电池设置在40℃的恒温室内,使用充放电装置(北斗电工股份有限公司制造,sm-8)进行充放电测定。以25ma(0.5c)的充电电流、4.3v的充电终止电压进行恒电流恒电压充电(截止电流2.5ma(0.05c))后,以25ma(0.5c)的放电电流、3v的放电终止电压进行恒电流放电。重复所述操作200次。循环特性可使用25℃下的第3次的0.5c放电容量与第200次的0.5c放电容量之比,利用以下的式2表示。
[0497]
(式2)
[0498]
循环特性=第3次的0.5c放电容量/第200次的0.5c放电容量
×
100(%)
[0499]
判定基准
[0500]
◎
:循环特性为80%以上(极其优良)
[0501]
○
:循环特性为70%以上且小于80%(优良)
[0502]
○
δ:循环特性为60%以上且小于70%(良)
[0503]
δ:循环特性为60%以上且小于70%(可)
[0504]
×
:循环特性小于60%(不良)
[0505]
[表10]
[0506]
表10.
[0507][0508]
[表11]
[0509]
表11.
[0510][0511]
根据所述实施例c、实施例d的结果,通过使用含有本发明的碳材料的导电性组合物,获得了速率特性及循环特性优异的锂离子二次电池。
[0512]
本发明的导电性组合物的分散性及分散稳定性良好,碳纳米管可在电极中有效率地形成导电网络,因此推测循环特性良好。另外,比较例d3、比较例d6即使可形成电极中的导电网络,循环特性也差。据研究,这是因为硼在本发明范围内的碳纳米管更硬,相对于电池充放电中活性物质的反复膨胀收缩,碳纳米管的导电网络维持不变,循环特性得到了改
善。另一方面,若比较实施例d1~实施例d3、或者实施例d6~实施例d8,则显示碳纳米管的硼含量及x/y的值的不同会影响电池特性(速率特性、循环特性)。
[0513]
本技术主张以2020年4月3日提出申请的日本技术特愿2020-067315为基础的优先权,将其公开的全部内容并入本文中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1