一种具有网络结构的镍钴硒纳米材料及其制备方法和应用
1.本发明涉及新能源材料制备领域,特别涉及一种具有网络结构的镍钴硒纳米材料及其制备方法和应用。
背景技术:
2.近年来,工业的快速发展带来了严重的环境污染,对清洁能源的需求日益增加。由于清洁能源(如风能、太阳能)的不稳定性,高效储能装置已成为人们关注的主要焦点。到目前为止,锂离子电池被认为是最商业化的储能设备,尤其是在便携式设备和电动汽车中。然而,随着锂离子电池应用的日益广泛,锂资源的需求量和成本大大增加。由于钠和锂在同一主族,其物化性质及电化学反应原理类似,使得钠离子电池(sibs)在未来的储能器件市场有很好的发展前景。
3.钠离子电池的性能在很大程度上取决于负极材料。由于钠离子半径较大,锂离子电池常用的碳基负极材料难以提供大量适宜的脱嵌位点,因而发展高容量、长寿命的负极材料成为钠电领域的主要研究目标。目前,钠离子电池负极材料的研究主要分为三类:插入材料、合金化反应材料和转化材料。在转化材料中,镍钴硒化物由于其较高的理论容量和良好的导电性而备受关注。而由于硒元素的低电负性,金属硒化物容易与钠离子反应。同时,硒元素的电导率(1
×
10-3
s m-1
)高于硫元素(5
×
10-28
s m-1
)。因此,用se替换o和s是sibs应用程序的明智选择。与单一金属硒化物相比,双金属硒化物具有更高的比容量和能量密度,不同金属之间的协同作用为电化学反应提供了更丰富的氧化还原位点。特别是,二元镍钴硒化物由于其高氧化还原活性而受到研究关注。而镍钴硒化物还原过程中形成的钴和镍纳米粒子可以用作na2se可逆转化的催化剂。但是,在电池充放电的过程中,镍钴硒化物在转化反应中易于发生结构形变,巨大的电极应力使得活性物质结构变形、坍塌甚至从集流体脱落,导致电荷和离子转移受阻,容量迅速衰减,电池寿命急剧下降,破坏了其作为电极材料具有的优秀电化学性能。
技术实现要素:
4.为了解决现有技术中存在的不足,本发明的目的在于提供一种具有网络结构的镍钴硒纳米材料及其制备方法和应用,本发明(ni,co)se2纳米材料具有循环稳定性能好、速率性能优异的特点。
5.本发明采用的技术方案如下:
6.一种具有网络结构的镍钴硒纳米材料的制备方法,包括如下过程:
7.准备a液和b液:其中,所述a液的制备过程包括:将镍源和钴源溶于去离子水中,得到所述a液;将硒源与还原剂溶于碱性溶剂,直至硒源完全溶解,得到所述b液;
8.将所述a液、b液混合,使镍源和钴源与硒源充分反应,得到c液;
9.向c液中加入表面活性剂并混合均匀、得到d液;
10.将所述d液进行超声、水热反应、过滤、洗涤、干燥,获得所述具有网络结构的镍钴
硒纳米材料,所述具有网络结构的镍钴硒纳米材料为(ni,co)se2纳米材料,(ni,co)se2纳米材料中,ni、co和se的摩尔比为(0.6-1.4):(0.6-1.4):2。
11.优选的,所述镍源采用硝酸镍或乙酸镍;钴源采用硝酸钴或乙酸钴。
12.优选的,所述硒源采用硒粉或二氧化硒;所述还原剂采用水合肼或硼氢化钠。
13.优选的,所述碱性溶剂采用ph值为14~15的naoh水溶液。
14.优选的,将所述a液、b液混合时,续搅拌b液,将a液缓慢加入b液,直至a液耗尽。
15.优选的,所述表面活性剂采用ctab、pvp、sds或peg-500。
16.优选的,将超声混匀后的d液进行水热反应时,反应温度为160℃-200℃,反应时间为12-36h,得到黑色沉淀。
17.优选的,将所述d液进行超声、水热反应、过滤、洗涤后,干燥时,干燥温度为40-80℃,干燥时间为6-18h。
18.本发明还提供了一种具有网络结构的镍钴硒纳米材料,该具有网络结构的镍钴硒纳米材料通过本发明如上所述的制备方法制得。
19.本发明还提供了一种具有网络结构的镍钴硒纳米材料的应用,该具有网络结构的镍钴硒纳米材料用作钠离子电池负极材料。
20.与现有技术相比,本发明具有如下优点与技术效果:
21.本发明的制备方法制得的具有网络结构的镍钴硒纳米材料即(ni,co)se2纳米材料拥有纳米级微晶颗粒;极小的纳米颗粒堆垛形成的网络结构能够促进电极与电解液反应过程中电子与离子的转移与扩散,提高材料的导电性;本发明向c液中加入表面活性剂并混合均匀,利用表面活性剂可诱导材料中部分纳米颗粒生长成为多边形片状颗粒,实现了材料的形貌改善。独特的多边形片状颗粒能够与纳米网络结构协同作用,为na
+
提供更多的反应活性位点以及较短的扩散路径,提高材料的电导率;而规则的片状结构则有效地限制了na
+
脱嵌时材料的形变,缓解了材料的体积效应,提高了材料的循环稳定性,因此,本发明合成的(ni,co)se2纳米材料制成钠离子电池负极时,表现出较高的比容量、优异的循环稳定性以及良好的电导率;并且本发明得到的材料为一步水热热法制备,工艺简单,成本低,环境友好,产率高。
附图说明
22.图1为本发明实施例1中(ni,co)se2纳米材料的xrd图;
23.图2为本发明实施例1中(ni,co)se2纳米材料的sem图;
24.图3为本发明实施例1中(ni,co)se2纳米材料的循环性能图;
25.图4为本发明实施例1中(ni,co)se2纳米材料的eis图;
26.图5为本发明实施例2中(ni,co)se2纳米材料的xrd图
27.图6为本发明实施例2中(ni,co)se2纳米材料的sem图;
28.图7为本发明实施例2中(ni,co)se2纳米材料的eds图;
29.图8为本发明实施例2中(ni,co)se2纳米材料的循环性能图;
30.图9为本发明实施例2中(ni,co)se2纳米材料的eis图;
31.图10为本发明实施例3中(ni,co)se2纳米材料的xrd图;
32.图11为本发明实施例3中(ni,co)se2纳米材料的sem图;
33.图12为本发明实施例3中(ni,co)se2纳米材料的循环性能图;
34.图13为本发明实施例3中(ni,co)se2纳米材料的eis图;
具体实施方式
35.下面结合附图对本发明作进一步详细说明。
36.本发明提供了(ni,co)se2纳米材料及其制备方法及应用,本发明通过一步水热法合成该材料,其制备工艺简单、操作方便、产率较高,(ni,co)se2纳米材料表现出高比能和优异的电化学性能。本发明合成的(ni,co)se2纳米材料能够作为钠离子电池的负极材料。
37.本发明具有网络结构的镍钴硒纳米材料的制备方法,包括如下过程:
38.准备a液、b液:其中,所述a液的制备过程包括:将镍源、钴源溶于去离子水中,得到所述a液;将硒源与还原剂溶于碱性溶剂,直至硒源完全溶解,得到所述b液;
39.将a液、b液混合反应,反应完全后得到c液,向c液中加入表面活性剂,混合均匀,得到d液;
40.将d液进行超声、水热反应、过滤、洗涤、干燥获得所述(ni,co)se2纳米材料。
41.本发明的上述方案具体步骤包括:
42.步骤(1),取2.4-5.6mmol镍源与2.4-5.6mmol钴源溶于去离子水中,再进行充分搅拌一段时间后,使镍源与钴源完全溶解,将得到的混合液作为a液。其中,镍源采用硝酸镍或乙酸镍,钴源采用硝酸钴或乙酸钴。
43.步骤(2),取一定的naoh加入20ml去离子水中并搅拌15分钟,制成碱性水溶液,调控ph值为14~15。取5-15ml(水合肼)或16-24mmol(硼氢化钠)还原剂溶于碱性水溶液中,再搅拌5分钟后,取8mmol硒源加入,充分搅拌15-60分钟使硒源完全溶解。将得到的混合液作为b液。其中,硒源采用硒粉或二氧化硒;还原剂采用水合肼或硼氢化钠。
44.步骤(3),将a液、b液混合,搅拌30-120分钟,使镍源和钴源与硒源充分反应,得到c液,在具体操作时,持续搅拌b液,向b液中缓慢加入a液,直至a液加完;再向c液中加入表面活性剂,随后进行超声分散30-120分钟,进一步混匀,得到d液,之后将d液加入溶剂热釜中,在160℃-200℃下反应12-36h,使物料充分反应,之后过滤得到黑色沉淀。其中,表面活性剂采用ctab、pvp、sds或peg-500。
45.步骤(4),将所得黑色沉淀充分洗涤后,加入真空干燥箱在40-80℃时,干燥6-18h个小时,即可得所述(ni,co)se2纳米材料,(ni,co)se2纳米材料中,ni、co和se的摩尔比为(0.6-1.4):(0.6-1.4):2。
46.实施例1:
47.选择使用5ml的peg-500作为表面活性剂制备具有网络结构的镍钴硒纳米材料,其制备过程包括如下步骤:
48.步骤(1)量取15ml去离子水作为溶剂,分别称取2.4mmol的四水合乙酸镍和2.4mmol四水合乙酸钴并溶于去离子水中,搅拌15分钟,作为a液。
49.步骤(2)量取20ml去离子水作为溶剂,称取0.02mol的naoh并溶于去离子水中,搅拌15分钟至naoh完全溶解。量取10ml的水合肼作为还原剂并将其缓慢加入氢氧化钠溶液中,搅拌5分钟使溶液均匀。称取8mmol硒粉加入上述含有水合肼的naoh溶液,搅拌30分钟,得到b液。
50.步骤(3)持续搅拌b液,用塑料滴管将a液缓慢滴入b液直至a液耗尽,搅拌30分钟,得到c液。量取5ml的peg-500加入c液,随后搅拌30分钟,再超声分散60分钟,得到d液。将d液倒入聚四氟乙烯的反应釜中,在160℃下反应12小时,过滤后得到黑色沉淀。
51.步骤(4)将所得黑色沉淀加入真空干燥箱,在40℃下干燥6个小时即可得(ni,co)se2纳米材料。
52.步骤(5)将super-p、镍钴硒材料与和聚偏氟乙烯按质量比为2:7:1称量,先在研钵中研磨均匀,然后加入无水nmp作分散剂,制备出粘稠浆料,再采用刮涂法涂于铜箔上。将涂好的极片真空干燥箱,在60℃下真空干燥12小时。然后冲出12mm直径大小的电极极片。制备钠离子扣式电池时,使用玻璃纤维(whatmangf/d)作为隔膜,1mol l-1
的nacf3so3溶解在二乙二醇二甲醚(degdme)的溶液作为电解液,金属钠圆片作为对电极,在充满氩气的手套箱内组装成2032型纽扣电池。
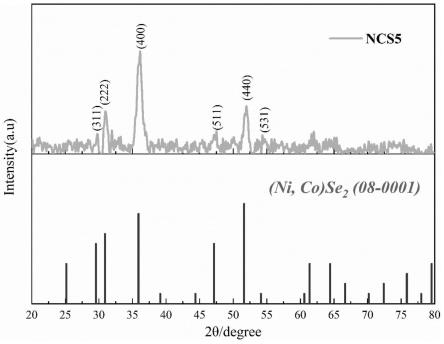
53.由实例1所制的(ni,co)se2纳米材料进行分析后发现,通过xrd分析,并且由图1可知,在5ml的peg-500条件下制备的(ni,co)se2纳米材料,存在(ni,co)se2物相的峰,其在2θ=29.6
°
、30.916
°
、35.891
°
、47.149
°
、51.626
°
、54.127
°
处出现衍射峰,分别对应(311)、(222)、(400)、(511)、(440)、(531)晶面。由此可知经过一步水热法成功制备出(ni,co)se2,由图2可知,通过sem分析可以发现(ni,co)se2纳米材料整体呈纳米颗粒状,小颗粒堆积重叠,并且纳米颗粒的普遍直径较小,直径范围大约在50nm-300nm之间。材料的电池循环性能如图3所示,材料首圈充电比容量与放电比容量分别为776.8mah
·
g-1
与501.3mah
·
g-1
,首圈库伦效率为64.54%,经过100圈的循环后,充放电比容量达到226.7mah
·
g-1
与236.2mah
·
g-1
。材料的阻抗测试结果如图4所示,通过图中数据计算可知,其电荷转移阻抗为146ω。
54.实施例2:
55.选择使用10ml的peg-500作为表面活性剂制备具有网络结构的镍钴硒纳米材料,其制备过程包括如下步骤:
56.步骤(1)量取15ml去离子水作为溶剂,分别称取4mmol的四水合乙酸镍和四水合乙酸钴并溶于去离子水中,搅拌15分钟,作为a液。
57.步骤(2)量取20ml去离子水作为溶剂,称取0.1mol的naoh并溶于去离子水中,搅拌15分钟至naoh完全溶解。量取10ml的水合肼作为还原剂并将其缓慢加入氢氧化钠溶液中,搅拌5分钟使溶液均匀。称取8mmol硒粉加入上述含有水合肼的naoh溶液,搅拌30分钟,得到b液。
58.步骤(3)持续搅拌b液,用塑料滴管将a液缓慢滴入b液直至a液耗尽,搅拌30分钟,得到c液。量取10ml的peg-500加入c液,随后搅拌30分钟,再超声分散60分钟,得到d液。将d液倒入聚四氟乙烯的反应釜中,在180℃下反应24小时,过滤后得到黑色沉淀。
59.步骤(4)将所得黑色沉淀加入真空干燥箱,在60℃下干燥12个小时即可得(ni,co)se2纳米材料。
60.步骤(5)将super-p、镍钴硒材料与和聚偏氟乙烯按质量比为2:7:1称量,先在研钵中研磨均匀,然后加入无水nmp作分散剂,制备出粘稠浆料,再采用刮涂法涂于铜箔上。将涂好的极片真空干燥箱,在60℃下真空干燥12小时。然后冲出12mm直径大小的电极极片。制备钠离子扣式电池时,使用玻璃纤维(whatmangf/d)作为隔膜,1mol l-1
的nacf3so3溶解在二
乙二醇二甲醚(degdme)的溶液作为电解液,金属钠圆片作为对电极,在充满氩气的手套箱内组装成2032型纽扣电池。
61.由实例2所制的(ni,co)se2纳米材料进行分析后发现,通过xrd分析,并且由图5可知,在10ml的peg-500条件下制备的(ni,co)se2纳米材料,存在(ni,co)se2物相的峰,其在2θ=29.6
°
、30.916
°
、35.891
°
、47.149
°
、51.626
°
、54.127
°
处出现衍射峰,分别对应(311)、(222)、(400)、(511)、(440)、(531)晶面。由此可知经过一步水热法成功制备出(ni,co)se2,由图6可知,通过sem分析可以发现(ni,co)se2纳米材料整体仍呈纳米颗粒状,同时出现了部分多边形的片状颗粒。小颗粒堆积重叠形成三维网络,而片状颗粒穿插其中。其中片状颗粒的边长范围大约在150nm-400nm之间,而厚度约为50nm-100nm。通过对比后发现,当peg-500的用量后提高至10ml时,部分纳米颗粒长成了表面光滑,形貌均匀的片状。规则的形貌有利于限制材料的结构变化,提高其循环稳定性。eds结果如图7所示,镍、钴、硒三种元素均匀的分布在材料的表面,证明成果的合成了(ni,co)se2纳米材料。电池循环性能如图8所示,材料首圈充电比容量与放电比容量分别为768mah
·
g-1
与557.4mah
·
g-1
,首圈库伦效率为72.58%,经过100圈的循环后,充放电比容量达到357.7mah
·
g-1
与355.7mah
·
g-1
。与材料的阻抗测试结果如图9所示,在循环前,其电荷转移阻抗为102.8ω。通过与实例1制备的材料相比,发现材料的首圈库伦效率与容量保持率都有所提高。与此同时,材料的电荷转移阻抗降低。这些结果表明实例2制备材料具有更好的电化学性能。其原因为:在实例2条件下,10ml的表面活性剂有利于诱导材料生长。特殊的片状颗粒形貌为材料提供了更多的反应活性位点,而规则的形貌有效地抑制了材料的结构变化,改善了材料循环稳定性。
62.实施例3:
63.选择使用15ml的peg-500作为表面活性剂制备具有网络结构的镍钴硒纳米材料,其制备过程包括如下步骤:
64.本实施例(ni,co)se2纳米材料的制备过程包括如下步骤:
65.步骤(1)量取15ml去离子水作为溶剂,分别称取5.6mmol的四水合乙酸镍和四水合乙酸钴并溶于去离子水中,搅拌15分钟,作为a液。
66.步骤(2)量取20ml去离子水作为溶剂,称取0.2mol的naoh并溶于去离子水中,搅拌15分钟至naoh完全溶解。量取10ml的水合肼作为还原剂并将其缓慢加入氢氧化钠溶液中,搅拌5分钟使溶液均匀。称取8mmol硒粉加入上述含有水合肼的naoh溶液,搅拌30分钟,得到b液。
67.步骤(3)持续搅拌b液,用塑料滴管将a液缓慢滴入b液直至a液耗尽,搅拌30分钟,得到c液。量取15ml的peg-500加入c液,随后搅拌30分钟,再超声分散60分钟,得到d液。将d液倒入聚四氟乙烯的反应釜中,在200℃下反应36小时,过滤后得到黑色沉淀。
68.步骤(4)将所得黑色沉淀加入真空干燥箱,在80℃下干燥18个小时即可得(ni,co)se2纳米材料。
69.步骤(5)将super-p、镍钴硒材料与和聚偏氟乙烯按质量比为2:7:1称量,先在研钵中研磨均匀,然后加入无水nmp作分散剂,制备出粘稠浆料,再采用刮涂法涂于铜箔上。将涂好的极片真空干燥箱,在60℃下真空干燥12小时。然后冲出12mm直径大小的电极极片。制备钠离子扣式电池时,使用玻璃纤维(whatmangf/d)作为隔膜,1mol l-1
的nacf3so3溶解在二乙二醇二甲醚(degdme)的溶液作为电解液,金属钠圆片作为对电极,在充满氩气的手套箱
内组装成2032型纽扣电池。
70.由实例3所制的(ni,co)se2纳米材料进行分析后发现,通过xrd分析,并且由图10可知,在10ml的peg-500条件下制备的(ni,co)se2纳米材料,存在(ni,co)se2物相的峰,其在2θ=29.6
°
、30.916
°
、35.891
°
、47.149
°
、51.626
°
、54.127
°
处出现衍射峰,分别对应(311)、(222)、(400)、(511)、(440)、(531)晶面。由此可知经过一步水热法成功制备出(ni,co)se2,由图11可知,(ni,co)se2纳米材料主要为颗粒状,小颗粒相互交织重叠,形成三维网络。其中颗粒直径范围大约在50nm-300nm。通过对比后发现,当peg-500的用量后提高至15ml时,纳米片结构消失。电池循环性能如图12所示,材料首圈充电比容量与放电比容量分别为573.4mah
·
g-1
与408.9mah
·
g-1
,首圈库伦效率为71.31%,经过100圈的循环后,充放电比容量达到210.1mah
·
g-1
与209.7mah
·
g-1
。与材料的阻抗测试结果如图13所示,在循环前,其电荷转移阻抗为146ω。通过与实例2制备的材料相比,发现材料的首圈库伦效率与容量保持率都有所下降。与此同时,材料的电荷转移阻抗增高。其原因为:在实例3条件下,15ml的表面活性剂并未起到增加纳米片颗粒的作用。特殊的片状颗粒形貌消失后,坍塌的纳米颗粒无法抵抗材料发生体积膨胀,不利于改善材料的循环稳定性。
71.综上,本发明利用一步水热法制备(ni,co)se2纳米材料:在密封的反应釜中,通过特有的高温高压环境诱导材料的结晶与生长,并形成较小的纳米颗粒。通过改变各种反应条件来控制产物的晶体形核与生长过程,使其形成特殊的纳米晶体结构。纳米化的(ni,co)se2微晶颗粒堆叠,形成三维的纳米网络,能够促进电极中电子与离子的转移与扩散,提高材料的导电性。而一定条件下的(ni,co)se2纳米颗粒生长出具有规则形貌与光滑表面的多边形片状结构。特殊的片状结构能够有效的限制充放电过程带来的体积膨胀,缓解材料的体积效应,保持循环过程的稳定性;光滑的表面与三维的纳米网络协同作用,为电极反应提供更多的反应活性位点,加速电解液与电极之间的接触,充分提升了材料的速率性能。本发明以10ml的peg-500为表面活性剂,诱导材料中部分纳米颗粒生长成为多边形片状颗粒,实现了材料的形貌改善。与常规的方法相比,利用适量peg-500合成的(ni,co)se2纳米材料具有更优秀的电化学性能。
72.相比于水热+固相烧结等方法,本发明工艺流程简单,操作方便,材料产率高,并且能够避免传统方法中需要高温硒化的步骤。在实验过程中,通过调节溶剂ph,制造硒离子的热力学稳定区域,同时以过量的还原剂保证硒离子不发生二次氧化。最后利用调整表面活性剂的加入量来引导(ni,co)se2纳米颗粒的生成。纳米化的(ni,co)se2颗粒能够减少颗粒发生团聚,缓解材料的体积效应。纳米颗粒构成的三维网络有益于钠离子与电荷的快速扩散,即使在高电流密度下也能提供良好的电子反应动力。在本发明的条件下生成的规则的片状结构能够与纳米网络结构协同作用,为na
+
提供更多的反应活性位点以及较短的扩散路径,提高材料的电导率;并且在放电/充电过程中抑制体积膨胀和收缩,维持结构稳定,表现出优异的循环稳定性。
73.以上详细描述了本发明的具体实施例。应当理解,本领域的普通技术无需创造性劳动就可以根据本发明的构思作出诸多修改和变化。因此,凡本技术领域中技术人员依本发明的构思在现有技术的基础上通过逻辑分析、推理或者有限的实验可以得到的技术方案,皆应在本发明的权利要求保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1