一种棉花锌指蛋白(Czf6)及其编码基因与应用的制作方法与工艺
一种棉花锌指蛋白(Czf6)及其编码基因与应用技术领域本发明涉及植物蛋白及其编码基因与应用,特别是涉及一个来源于棉花的锌指蛋白(Czf6)及其编码基因,以及其在培育抗旱性提高的转基因植物中的应用。
背景技术:
非生物胁迫,如干旱、盐渍、极端温度、化学污染和氧损伤等能够对植物的生长发育造成严重的危害,对作物产量造成极大损失。其中干旱对作物产量的影响,在诸多自然逆境中占首位,其危害相当于其它灾害之和,是许多地区是农业发展的瓶颈。据统计,世界干旱、半干旱地区占陆地面积的34%;我国干旱、半干旱地区约占国土面积的52%,年受旱面积达200-270万公顷,全国灌溉区每年缺水约30亿立方米,因缺水而少收粮食350-400亿公斤;特别是我国主要产粮区如华北、东北和西北,是我国缺水最严重的地区,春旱频繁达到十年九遇。由于植物的耐胁迫性大多属于数量性状,现有可利用的种质资源匮乏,采用常规育种技术改良植物胁迫耐性的难度相当大,培育出真正的耐胁迫品种就尤为困难。近年来,随着对植物抗逆分子机理研究的不断深入和分子生物学技术的迅猛发展,抗逆研究已经从生理水平深入到分子水平,促进了植物抗逆基因工程的发展。当植物在受到胁迫时会产生相应的应答反应,来降低或消除给植株带来的危害。植物的这种应答反应是一个涉及多基因、多信号途径、多基因产物的复杂过程。这些基因及其表达产物可以分为3类:(1)参与信号级联放大系统和转录控制的基因及产物;(2)直接对保护生物膜和蛋白质起作用的基因及其表达产物;(3)与水和离子的摄入和转运相关的蛋白质。近年来,通过转基因技术提高植物对胁迫耐受能力的研究,以及对胁迫具有耐受能力的农作物、旱生植物和盐生植物的研究都取得了显著的成果,对胁迫相关基因和信号转导系统也有了更进一步的了解(LiuQ.1998.Twotranscriptionfactors,DREB1andDREB2,withanEREBP/AP2DNAbindingdomain,separatetwocellularsignaltransductionpathwaysindrought-andlowtemperature-responsivegeneexpression,respectively,inArabidopsis.PlantCell,10:1391-1406;KANGJY.2002.Arabidopsisbasicleucinezipperproteinsthatmediatestress-responsiveabscisicacidsignaling.PlantCell,14:343-357;ABEH.2003.ArabidopsisAtMYC2(bHLH)andAtMYB2(MYB)functionastranscriptionalactivatorsinabscisicacidsignaling.PlantCell,15:63-78.)。但就目前的研究状况而言,由于其机制十分复杂,许多植物对逆境下的生物化学和生理学上的响应机制仍有待深入研究。在抗逆应答基因的功能及表达调控方面的研究将对植物抗逆相关的信号传递途径及信号传递网络系统的研究提供重要的基础。
技术实现要素:
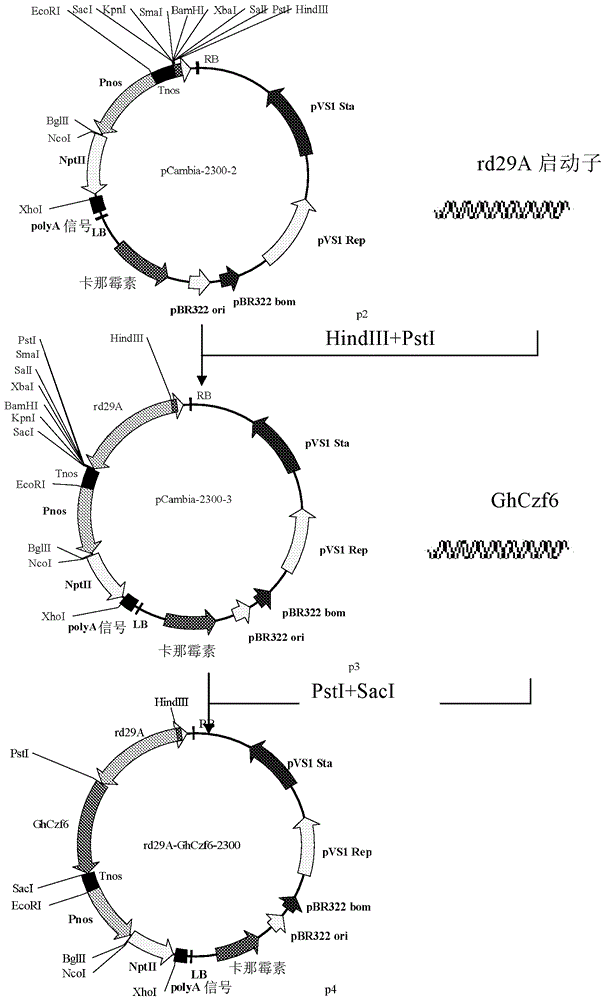
本发明人利用SSH(抑制差减杂交)与RACE(cDNA末端快速扩增)相结合的方法克隆出了棉花的一个锌指蛋白(本文命名为Czf6)的编码基因,并测定了其DNA序列。并且发现通过转基因将其导入植株后,可明显改善转基因植株的抗旱性,而且这些性状可稳定遗传。本发明第一方面提供棉花的一个锌指蛋白Czf6的编码基因(本文命名为GhCzf6),其序列为SEQIDNO:2。本发明第二方面提供一种重组表达载体,其含有本发明第一方面所述的基因并且所述基因的核苷酸序列与所述表达载体的表达控制序列可操作地连接;优选地,所述载体为附图2所示的rd29A-GhCzf6-2300载体。本发明第三方面提供一种重组细胞,其含有本发明第一方面所述的基因或者本发明第二方面所述的重组表达载体;优选地,所述重组细胞为重组农杆菌细胞。本发明第四方面提供一种改善植物抗旱性的方法,包括:将本发明第一方面所述基因或者本发明第二方面所述的重组表达载体导入植物或植物组织并使所述基因表达;优选地,所述植物是烟草。本发明第五方面提供一种制备转基因植物的方法,包括:在有效产生植物的条件下培养含有本发明第一方面所述基因或者本发明第二方面所述的重组表达载体的植物或植物组织;优选地,所述植物是烟草。本发明第六方面提供本发明第一方面所述的基因、本发明第二方面所述的重组表达载体或者本发明第三方面所述的重组细胞用于改善植物抗旱性以及用于植物育种的用途;优选地,所述植物是烟草。本发明第七方面提供本发明第一方面所述的基因编码的蛋白质,其氨基酸序列如SEQIDNO:1所示。附图说明图1是GhCzf6的植物表达载体(rd29A-GhCzf6-2300)的构建流程。图2是GhCzf6的植物表达载体(rd29A-GhCzf6-2300)的质粒图。图3是对照烟草和转基因烟草的抗旱性生长情况;CK(左):对照烟草;T1F9(中)和T1F12(右):转基因烟草株系。图4是耐干旱和不耐干旱T1代转基因烟草植株在转录水平上的验证结果。M为Marker,1-8为耐干旱的T1代转基因烟草植株,9-12为不耐干旱的T1代转基因烟草植株。具体实施方式提供以下实施例,以方便本领域技术人员更好地理解本发明。所述实施例仅出于示例性目的,并非意在限制本发明的范围。实施例1干旱胁迫下棉花SSH文库构建:具体方法为:利用Clontech公司的PCR-selectTMcDNASubtractionKit所示的方法通过抑制差减杂交方法构建差减文库。在实验过程中以干旱处理的棉花幼苗的叶片中提取的mRNA作为样本(tester),以未处理的棉花幼苗的叶片中提取的mRNA作为对照(driver)。具体步骤简述如下:(1)供试材料:非洲棉(国家棉花中期库,获取单位中国棉花研究所,统一编号:ZM-06838)播种到灭过菌的蛭石上,在25℃、光周期16h/8h(光强2000-3000Lx)条件下培养,每周浇1/2MS培养基(含有9.39mMKNO3,0.625mMKH2PO4,10.3mMNH4NO3,0.75mMMgSO4,1.5mMCaCl2,50μMKI,100μMH3BO3,100μMMnSO4,30μMZnSO4,1μMNa2MoO4,0.1μMCoCl2,100μMNa2EDTA,100μMFeSO4)一次。当苗株高达25-30cm时用于实验。(2)材料处理:将供试幼苗分为2组,每组4盆,每盆1株。第一组为对照组,在25℃、光周期16h/8h(光强2000-3000Lx)条件下培养,正常浇灌。第二组为干旱处理组,25℃、光周期16h/8h(光强2000-3000Lx)条件下培养,停止浇灌,处理10天,处理完毕后及时剪取两组幼苗顶端1/3的叶片,用液氮迅速冷冻后,于-70℃冰箱中保存。(3)总RNA提取:分别取对照组和干旱处理组的棉花叶片0.5g,用植物RNA提取试剂盒(购自invitrogen)提取棉花叶片的总RNA。用HITACHI公司的紫外分光光度计U-2001测定总RNA在260nm和280nm的吸光度值,OD260/OD280比值为1.8-2.0,表明总RNA纯度较高,用1.0%的琼脂糖凝胶电泳检测总RNA的完整性,28S条带的亮度约为18S条带的2倍,表明RNA的完整性良好。使用Qiagen公司的OligotexmRNA纯化试剂盒(purificationofpolyA+RNAfromtotalRNA)分离mRNA。(4)抑制差减杂交:按Clontech公司的PCR-selectTMcDNASubtractionKit试剂盒所示的方法进行抑制差减杂交。应用Clontech的PCR-SelectcDNASubtractionKits差减杂交试剂盒,先将DrivermRNA和TestermRNA分别反转录,得到双链cDNA,再以2μgTestercDNA和2μgDrivercDNA作为起始材料进行差减杂交。在37℃水浴下分别将TestercDNA和DrivercDNA用RsaI酶切1.5h,然后将酶切后的TestercDNA分成两等份,连接上不同的接头,而DrivercDNA不连接头。两种连有不同接头的TestercDNA分别与过量的Driver混合,进行第一次正向差减杂交。将两种第一次差减杂交的产物混合,再与新鲜变性的DrivercDNA进行第二次正向差减杂交,通过两次抑制性PCR扩增差异表达的片段,使其得到富集。(5)cDNA差减文库的构建与初步筛选、克隆、鉴定依照pGEM-TEasy试剂盒的程序,将所述正向差减杂交cDNA片段的第二次PCR产物(使用QIAquickPCRPurificationKit纯化,购自Qiagen)与pGEM-TEasy(购自Promega试剂盒)载体连接,其具体步骤如下:用200μlPCR管依次加入下列成分:纯化的正向差减杂交cDNA片段的第二次PCR产物3μl,2×T4连接酶缓冲液5μl,pGEM-TEasy载体1μl,T4DNA连接酶1μl,于4℃连接过夜。取10μl连接反应产物,加入到100μl感受态大肠杆菌JM109(购自TAKARA)中,冰浴30min、热休克60秒、冰浴2min,另加250μlLB培养液(含有1%Tryptone(购自OXOID),0.5%YeastExtract(购自OXOID),1%NaCl(购自国药))置37℃水浴中,以225rpm振荡培养30min,取200μl菌液接种于含50μg/ml氨苄青霉素的LB(同上)/X-gal/IPTG(X-gal/IPTG购自TAKARA)培养板上,37℃培育18h。计数培养板中直径>1mm的清晰白色及蓝色菌落数,随机挑取540个白色菌落(编号:Gh-D2-001至Gh-D2-540)。将所有白色克隆接种于含有50μg/ml氨苄青霉素的LB液体培养基的96孔细胞培养板(CORNING)中,37℃培养过夜后加甘油至终浓度20%,于-80℃保存备用。以巢式PCR引物Primer1和Primer2R(Clontech公司的PCR-selectTMcDNASubtractionKit试剂盒自带)进行菌液PCR扩增,得到452个阳性克隆,然后将所有阳性克隆送英潍捷基(上海)贸易有限公司测序。(6)差异克隆的cDNA测序分析:将DNA测序结果去除载体和不明确序列及冗余的cDNA后,共得到405个有效EST(unigene)。实施例2棉花锌指蛋白基因GhCzf6的克隆克隆子Gh-D2-153去掉冗余DNA后,序列为SEQIDNo:3,序列分析表明该序列编码的蛋白属于锌指蛋白,本文将SEQIDNo:3序列对应的全长编码基因命名为GhCzf6,对应的蛋白命名为Czf6。SEQIDNo:3GhCzf6全长编码基因的克隆根据已经获得的SEQIDNo:3序列,设计如下三条特异性引物,作为5’RACE的3’端特异性引物。GhCzf6GSP3:SEQIDNO:4:TCACGATCTTCTCTGCTTTGACGhCzf6GSP4:SEQIDNO:5:CACGGGATTGGCACGTGCAATCGhCzf6GSP5:SEQIDNO:6:CCCAATCTTCTTGAAATCGAAC实验步骤按试剂盒说明书操作(5’RACESystemforRapidAmplificationofcDNAEnds试剂盒购自invitrogen公司)。用SEQIDNO:5与5’通用引物AAP(试剂盒自带),以mRNA逆转录的cDNA(反转录引物SEQIDNO:4)为模板进行第一轮PCR扩增,具体步骤如下:50μlPCR反应体系:5μl10×ExBuffer、3μl2.5mM的dNTP、2.0μlmRNA反转录的cDNA、1.0μlExTaq(购自TAKARA)、10μM的引物SEQIDNO:5和AAP各2.0μl、以及35μl双蒸水。PCR反应条件:94℃预变性5min,33个循环(94℃变性30s,55℃退火30s,72℃延伸1min),72℃延伸10min。所得的PCR产物用双蒸水稀释50倍后取2.0μl作为模板,用SEQIDNO:8与3’端引物AUAP进行第二轮PCR扩增,具体步骤如下:50μlPCR反应体系:5μl10×ExBuffer、3μl2.5mM的dNTP、2.0μl稀释的第一轮PCR产物、1.0μlExTaq、10μM的引物SEQIDNO:6和AUAP各2.0μl、以及35μl双蒸水。PCR反应条件:94℃预变性5min,33个循环(94℃变性30s,58℃退火30s,72℃延伸1min),72℃延伸10min。回收第二次PCR产物中片段约为500bp的条带(GelExtractionKit购自OMEGA),并将其连接于pGEM-TEasyVector,然后转化到JM109(具体方法同上),随机挑取10个白色菌落接种于含有50μg/ml氨苄青霉素的LB液体培养基中,37℃培养过夜后加甘油至终浓度20%,-80℃保存备用。用SEQIDNO:8与3’端引物AUAP进行菌液PCR扩增(反应体系及反应条件同上),得到7个阳性克隆,选取其中4个克隆送至英潍捷基(上海)贸易有限公司测序测序,获得该基因的cDNA的5’端。所得的5’RACE产物克隆测序后,将其与SEQIDNo:3序列拼接。获得GhCzf6全长cDNA序列SEQIDNo:7:根据SEQIDNO:7序列设计一对引物如下:GhCzf6:SEQIDNO:8:ATGGCGGAGGAACATAAATGCCGhCzf6:SEQIDNO:9:TCAAATCCTCACGATCTTCTCT通过SEQIDNO:8和SEQIDNO:9来克隆GhCzf6全长编码基因。采用TaKaRa的PrimeSTARHSDNA聚合酶,以棉花的cDNA为模板进行PCR反应。50μlPCR反应体系:10μl5×PSBuffer、3μl2.5mM的dNTP、2.0μlcDNA、1.0μlPrimeSTAR、10μM的引物SEQIDNO:8和SEQIDNO:9各2.0μl、以及30μl双蒸水。PCR反应条件:94℃预变性5min,33个循环(94℃变性30s,58℃退火30s,72℃延伸1min),72℃延伸10min。PCR扩增产物加A尾:PCR产物中加入2.5倍体积的无水乙醇,-20℃放置10分钟,离心,去上清,晾干,然后用21μl双蒸水溶解。然后向其中加入2.5μl10×ExBuffer、0.5μl5mM的dATP、2.5μl10×ExTaq。反应条件:70℃反应30分钟。将得到的约540bp的DNA片段回收(Omega回收试剂盒),并将其连接至pGEMT-easy载体上,然后转化JM109(方法同上),随机挑取10个白色菌落接种于含有50μg/ml氨苄青霉素的LB液体培养基中,37℃培养过夜后加甘油至终浓度20%,-80℃保存备用。用SEQIDNO:8与SEQIDNO:9进行菌液PCR扩增(反应体系及反应条件同上),得到7个阳性克隆,选取其中4个阳性克隆送至英潍捷基(上海)贸易有限公司测序,序列为SEQIDNO:2,其编码的蛋白质的氨基酸序列为SEQIDNO:1。Czf6蛋白的氨基酸序列:SEQIDNO:1GhCzf6编码基因的核苷酸序列:SEQIDNO:2实施例3GhCzf6基因植物表达载体构建选择植物双元表达载体pCAMBIA2300(购自北京鼎国昌盛生物技术有限责任公司)作为植物表达载体,用Pnos启动子替换NPTII基因含双增强子的35S启动子,以降低NPTII蛋白在植物中的表达。选择诱导型的rd29A启动子及终止子Tnos分别作为GhCzf6基因的启动子和终止子。用引物SEQIDNO:10和SEQIDNO:11以植物表达载体pBI121(购自北京华夏远洋科技有限公司)为模板扩增Pnos,采用TaKaRa的PrimeSTARHSDNA聚合酶。50μlPCR反应体系:10μl5×PSBuffer、3μl2.5mM的dNTP、1.0μlpBI121、1.0μlPrimeSTAR、10μM的引物SEQIDNO:10和SEQIDNO:11各2.0μl、以及31μl的双蒸水。PCR反应条件:94℃预变性5min,33个循环(94℃变性30s,56℃退火30s,72℃延伸30s),72℃延伸10min。通过EcoRI、BglII酶切将所得的PCR产物连接(promega,T4连接酶盒)到pCAMBIA2300获得pCAMBIA2300-1。SEQIDNO:10:GCACGAATTCATACAAATGGACGAACGGATSEQIDNO:11:ATCCAGATCTAGATCCGGTGCAGATTATTTG用引物SEQIDNO:12和SEQIDNO:13以pBI121为模板扩增Tnos,采用TaKaRa的PrimeSTARHSDNA聚合酶。50μlPCR反应体系:10μl5×PSBuffer、3μl2.5mM的dNTP、1.0μlpBI121、1.0μlPrimeSTAR、10μM的引物SEQIDNO:12和SEQIDNO:13各2.0μl、以及31μl双蒸水。PCR反应条件:94℃预变性5min,33个循环(94℃变性30s,58℃退火30s,72℃延伸30s),72℃延伸10min。通过SacI、EcoRI酶切将所得的PCR产物连接(promegaT4连接酶盒)到pCAMBIA2300-1获得pCAMBIA2300-2。SEQIDNO:12:AAGGAGCTCGAATTTCCCCGATCGTTCAAASEQIDNO:13:TCAGAATTCCCAGTGAATTCCCGATCTAGTA用引物SEQIDNO:14和SEQIDNO:15以拟南芥(哥伦比亚型,购自www.arabidopsis.org)DNA为模板扩增拟南芥rd29A启动子(参考ZengJ.,etal.2002,PreparationoftotalDNAfrom“recalcitrantplanttaxa”,ActaBot.Sin.,44(6):694-697中的方法提取拟南芥DNA)。采用TaKaRa的PrimeSTARHSDNA聚合酶。50μlPCR反应体系:10μl5×PSBuffer、3μl2.5mM的dNTP、1.0μl拟南芥DNA、1.0μlPrimeSTAR、10μM的引物SEQIDNO:14和SEQIDNO:15各2.0μl、以及31μl双蒸水。PCR反应条件:94℃预变性5min,33个循环(94℃变性30s,58℃退火30s,72℃延伸30s),72℃延伸10min。通过HindIII、PstI酶切将所得的PCR产物连接(连接方法同上)到pCAMBIA2300-2获得pCAMBIA2300-3。SEQIDNO:14:ACTAAGCTTCCTTCTTGACATCATTCAATTTTASEQIDNO:15:TGACTGCAGTCCAAAGATTTTTTTCTTTCCAATAG用引物SEQIDNO:16和SEQIDNO:17扩增GhCzf6编码基因的全长序列(模板是实施例2所获得GhCzf6全长编码基因),采用TaKaRa的PrimeSTARHSDNA聚合酶。50μlPCR反应体系:10μl5×PSBuffer、3μl2.5mM的dNTP、1.0μlGhCzf6-pGEM、1.0μlPrimeSTAR、10μM的引物SEQIDNO:16和SEQIDNO:17各2.0μl、以及31μl双蒸水。PCR反应条件:94℃预变性5min,33个循环(94℃变性30s,58℃退火30s,72℃延伸2min),72℃延伸10min。通过PstI、SacI酶切将所得的PCR产物连接(连接方法同上)到pCAMBIA2300-3,获得植物表达载体rd29A-GhCzf6-2300。SEQIDNO:16:TGACTGCAGATGGCGGAGGAACATAAATGCCSEQIDNO:17:AAGGAGCTCTCAAATCCTCACGATCTTCTCT实施例4rd29A-GhCzf6-2300表达载体转化农杆菌农杆菌LBA4404(购自BiovectorScienceLab,Inc)感受态细胞的制备:提前1-2天将农杆菌LBA4404在含50μg/ml利福平和50μg/ml链霉素的LB固体培养基上划单斑接种,28℃培养1至2天。挑取单菌落接种于5ml含50μg/ml利福平和50μg/ml链霉素的LB液体培养基中,28℃下摇动培养过夜(约12-16h)至OD600值为0.4,形成种子菌液。取5ml活化后的菌液(1∶20的比例)接种于100ml含50μg/ml利福平和50μg/ml链霉素的LB液体培养基中,28℃摇动培养2-2.5h至OD600=0.8。冰浴菌液10min,每隔3min摇匀一次,令所述细菌均匀进入休眠状态。于4℃下4000g离心10min,弃上清液;加入一定量冰预冷的10%甘油重悬浮菌体,4℃下4000g离心10min,收集沉淀;用冰预冷的10%甘油重复洗3-4次;然后加入适量冰浴预冷的10%甘油重新悬浮细菌沉淀,以40μl/管将其分装,于-70℃保存备用。转化农杆菌:在冰上融化所述的感受态细胞,往40μl的感受态细胞中加入1μl的质粒,混匀后冰浴约10min。将感受态细胞和DNA的混合物用移液枪转移到冰预冷的电击杯(购自bio-rad)中,轻敲使悬浮液到达电击杯底部,注意不要有气泡。将所述电击杯放到电击室的滑道上,推动滑道将电击杯放至电击室基座电极处。使用0.1em规格的电击杯的时候,MicroPulser(购自bio-rad)的程序设置为“Agr”,电击一次。立即取出电击杯,加入28℃预热的LB培养基。快速而轻柔的用移液枪将细胞打匀。将悬浮液转入1.5ml的离心管,在28℃225rpm摇动培养1h。取100-200μl的菌液涂布于相应的抗性筛选培养基平板上(LB固体培养基,含50μg/ml利福平、50μg/ml链霉素、50μg/ml卡那霉素),28℃培养。实施例5利用农杆菌介导的转化法获得转基因烟草用75%酒精浸泡烟草种子(国家烟草中期库,获取单位:中国农科院烟草所,库编号I5A00660)30s,然后用灭菌双蒸水洗两次。再用0.1%升汞浸泡8min,然后用灭菌双蒸水洗两次,完成表面灭菌。将表面灭菌的烟草种子置于MS固体培养基(含有18.78mMKNO3、1.25mMKH2PO4、20.6mMNH4NO3、1.5mMMgSO4、3.0mMCaCl2、50μMKI、100μMH3BO3、100μMMnSO4、30μMZnSO4、1μMNa2MoO4、0.1μMCoCl2、100μMNa2EDTA、100μMFeSO4、7.4g/l琼脂、30g/l蔗糖)上于无菌条件下发芽,制备无菌苗。取无菌苗叶片剪成5mm×5mm大小的叶盘,用处于对数生长期的含表达载体rd29A-GhCzf6-2300的农杆菌浸染叶盘10min,吸干菌液,在黑暗条件下共培养2天(MS固体培养基)。将叶片转到分化固体培养基(MS+1mg/l细胞分裂素(BA)+0.1mg/l萘乙酸(NAA)+50mg/l卡那霉素+500mg/l头孢霉素)上,光周期16h/8h(光强2000-3000Lx)条件下培养45天左右,待芽长大后切下转移到生根固体培养基(MS+50mg/l卡那霉素+500mg/l头孢霉素)中培养30天左右,待根系发达后将小苗转入仅加有500mg/L头孢霉素的MS固体培养基上进行编号保存。剪取获得的转基因烟草植株的叶片,提取DNA(同实施例3中拟南芥DNA提取方法),用SEQIDNO:8和SEQIDNO:9行PCR扩增鉴定(50μlPCR反应体系:5μl10×ExBuffer、3μl2.5mM的dNTP、2.0μlDNA、1.0μlExTaq、10μM的引物SEQIDNO:8和SEQIDNO:9各2.0μl、以及35μl双蒸水。PCR反应条件:94℃预变性5min,33个循环(94℃变性30s,58℃退火30s,72℃延伸2min),72℃延伸10min),将PCR鉴定为阳性植株编号为TOF1-TOF20。实施例6过表达GhCzf6转基因烟草T1代植株的抗旱模拟实验灭过菌的蛭石用1/2MS培养基浸透。TOF1-TOF20转基因烟草及对照烟草的种子分别播种在蛭石上,每盆播种15颗种子,25℃、10小时光培养/14小时暗培养循环。每5天浇一次1/2MS,培养25天之后,每盆保留大小较一致的4-5棵苗用于干旱实验,转基因烟草、对照烟草干旱14天(不浇水),25℃、10小时光培养/14小时暗培养循环。T1代转基因植株(TO代转基因植株的种子长成的植株)的抗旱性鉴定表明,对照植株都萎蔫严重,而T1F5、T1F7、T1F9、T1F12、T1F15、T1F16、T1F19七个株系共35棵烟草中有24棵能够正常生长,表现出明显的抗旱性(见图3,以T1F9、T1F12为例,T1F5、T1F7、T1F15、T1F16、T1F19的结果与T1F9、T1F12类似,在此未示出)。实施例7在转录水平上验i正Czf6蛋白表达实施例6中24棵能够在干旱条件下正常生长的T1代转基因植株中随机选取8棵,实施例6中11棵不能在干旱条件下正常生长的T1代转基因植株中随机选取4棵,各剪取干旱14天的叶片0.05g,用植物RNA提取试剂盒(invitrogen)提取总RNA。紫外分光光度测定总RNA在260nm和280nm的吸光度值,计算各个RNA浓度。依照invitrogen反转录试剂盒SuperScriptIIIReverseTranscriptase所示方法进行反转录(1μg总RNA作为模板,反转录引物SEQIDNO:9)。通过SEQIDNO:8和SEQIDNO:9扩增GhCzf6,检测Czf6蛋白相对表达情况。采用TaKaRa的PrimeSTARHSDNA聚合酶,以反转录的cDNA为模板进行PCR反应。50μlPCR反应体系:10μl5×PSBuffer,3μl2.5mM的dNTP,2.0μlcDNA,1.0μlPrimeSTAR、10μM的引物SEQIDNO:10和SEQIDNO:11各2.0μl,以及30μl的双蒸水。PCR反应条件:94℃预变性5min,29个循环(94℃变性30s,58℃退火30s,72℃延伸1min),72℃延伸10min。产物电泳结果如图4所示:M为DNALadderMarker(DL2000,购自深圳瑞真生物技术有限公司),1-8为正常生长植株,9-12为不能正常生长植株。图中所示条带大小与GhCzf6的大小一致。结果表明正常生长植株中GhCzf6的转录较强,不能正常生长植株中没有GhCzf6转录或转录很弱。本领域技术人员可以理解的是本发明也适用于特定描述之外的变化和调整,并且本发明包括所有的这种变化和调整。发明人要求落入下文权利要求范围和精神内的修改和改变的权利。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 新的人原发无精症相关锌指蛋白、编码序列及制法和用途的制作方法
- 一种多肽-人锌指蛋白-73.59和编码这种多肽的多核苷酸的制作方法
- 一种多肽-人锌指蛋白-67.76和编码这种多肽的多核苷酸的制作方法
- 一种多肽-人锌指蛋白-61.27和编码这种多肽的多核苷酸的制作方法
- 一种新的多肽——人锌指蛋白17和编码这种多肽的多核苷酸的制作方法
- 一种新的多肽——锌指蛋白11和编码这种多肽的多核苷酸的制作方法
- 一种多肽——锌指蛋白-61.05和编码这种多肽的多核苷酸的制作方法
- 一种多肽——人锌指蛋白-61.82和编码这种多肽的多核苷酸的制作方法
- 一种多肽——人锌指蛋白-32.56和编码这种多肽的多核苷酸的制作方法
- 一种多肽——锌指蛋白-56.87和编码这种多肽的多核苷酸的制作方法