抗HIV‑1多肽及其用途的制作方法与工艺
抗HIV-1多肽及其用途技术领域本发明属于生物医学领域,涉及一类多肽,特别是一类抗HIV-1或其它相关包膜类病毒的多肽,以及所述多肽在制备HIV融合抑制剂中的用途,在制备用于治疗或预防HIV感染相关疾病尤其是艾滋病的药物中的用途,以及在制备用于治疗或预防其它相关包膜类病毒感染药物中的用途。
背景技术:
Ⅰ型人免疫缺陷病毒(HIV-1)是艾滋病的病原体,现全球有超过3000万感染者,每年导致约200万人死亡,并且每年还有新增约200万感染者,是一种严重威胁人类健康的全球性感染疾病。HIV-1通过其包膜糖蛋白(Env)介导的病毒-细胞膜融合感染宿主细胞。Env包含表面亚基gp120和跨膜亚基gp41,三个Env形成非共价复合体镶嵌在病毒表面。表面亚基gp120负责病毒感染细胞过程中的分子识别以找到和接近靶细胞,同时起着稳定跨膜亚基gp41的功能,并在适当时机释放出gp41以启动融合;跨膜亚基gp41是病毒-细胞膜融合的直接功能分子。病毒细胞融合过程中有一由gp41N-端螺旋区和C-端螺旋区(NHR和CHR)形成的六螺旋结构;该结构的形成为病毒-细胞膜融合提供能量,对病毒-细胞融合至关重要。阻止六螺旋形成的药物可有效抑制艾滋病毒-细胞膜融合,从而阻止病毒感染和体内传播,用于艾滋病治疗,因此称为融合抑制剂。晶体结构显示在六螺旋中,三个由NHR形成的螺旋结构构成内核,形成三个沟槽,三个CHR反平行结合在沟槽中。外源CHR多肽可结合在NHR靶点中形成无活性的六螺旋结构,阻止内源的活性六螺旋体生成,抑制病毒-细胞融合和病毒感染,从而用作融合抑制剂。典型的C-肽融合抑制剂包括C34(US6,150,088)及其改进多肽、首个上市融合抑制剂T20(US5,464,933)、以及后来发现的CP32(CN1793170,CN1955190)。这些C-肽融合抑制剂通过与其对应NHR靶点结合阻止病毒感染;典型的靶点包括N36(US6,150,088)和DP107(US5,656,480),分别与C34和CP32结合形成六螺旋结构,其中N36和DP107中含有一个共同的结合口袋,与CHR的WaaWaaaI(其中a为氨基酸)模板(又称WWI模板)有关键相互作用,也是小分子融合抑制剂的热门靶点。尽管有上市的融合抑制剂T20和其它临床研究中的融合抑制剂如Sifuvirtide(CN1334122),但由于抗药病毒株的快速出现,使得针对抗性病毒的融合抑制剂研发成为当务之急,尤其是序列来源与已有融合抑制剂明显不同的融合抑制剂。
技术实现要素:
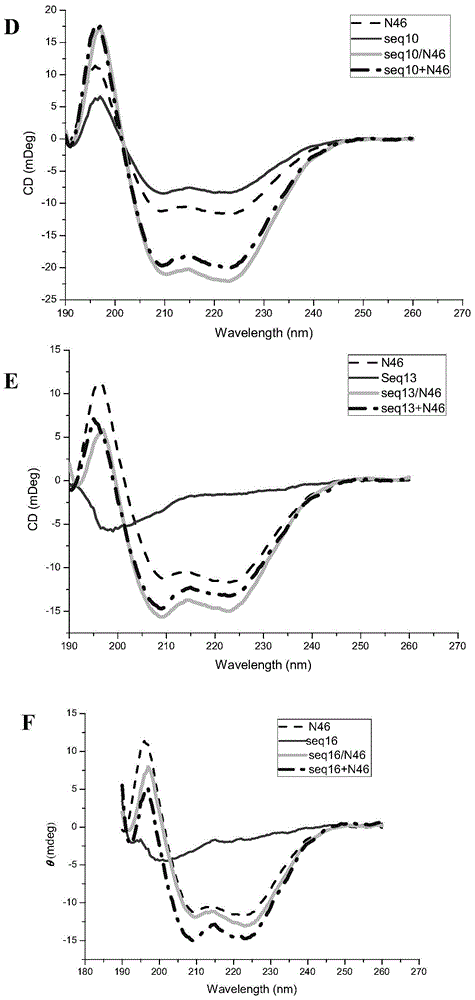
基于以上原因,本发明人提出了从其它类似包膜病毒对应包膜糖蛋白序列设计抗HIV-1活性肽的设计思想。我们根据包膜病毒融合机制的相似性,从其它包膜病毒包膜糖蛋白跨膜亚基的CHR序列连续截取约36个氨基酸的肽序列,设计出抗HIV-1多肽,测定其抗HIV介导的细胞-细胞融合活性,同时研究其与HIV-1gp41NHR的结合,确定其作用靶点,探索研究HIV-1融合抑制剂设计的新思路。由此完成了本发明。本发明的第一方面涉及选自式(1)~式(42)所示的多肽,式(1):X-WIDISIELNKAKSDLEESKEWIERSNGKLDSIGNWH(SEQIDNO:1)-Z;式(2):X-WIDWSIELNKAKSDLEESKEWIERSNGKLDSIGNWH(SEQIDNO:2)-Z;式(3):X-PIDISIELNKAKSDLEESKEWIERSNGKLDSIGNWH(SEQIDNO:3)-Z;式(4):X-WLDISQNLAAVNKSLSDALQHLAQSDTYLSAI(SEQIDNO:4)-Z;式(5):X-WLDWSQNLAAVNKSLSDALQHLAQSDTYLSAI(SEQIDNO:5)-Z;式(6):X-PLDISQNLAAVNKSLSDALQHLAQSDTYLSAI(SEQIDNO:6)-Z;式(7):X-WSGINASVVNIQKEIDRLNEVAKNLNESLIDLQELG(SEQIDNO:7)-Z;式(8):X-WSGWNASVVNIQKEIDRLNEVAKNLNESLIDLQELG(SEQIDNO:8)-Z;式(9):X-ISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELG(SEQIDNO:9)-Z;式(10):X-WEKLNVTLLDLTYEMNRIQDAIKKLNESYINLKE(SEQIDNO:10)-Z;式(11):X-WEKWNVTLLDLTYEMNRIQDAIKKLNESYINLKE(SEQIDNO:11)-Z;式(12):X-FEKLNVTLLDLTYEMNRIQDAIKKLNESYINLKE(SEQIDNO:12)-Z;式(13):X-WGNVNNSISNALNKLEESNRNLDKVNVKLTSTSALI(SEQIDNO:13)-Z;式(14):X-WGNWNNSISNALNKLEESNRNLDKVNVKLTSTSALI(SEQIDNO:14)-Z;式(15):X-LGNVNNSISNALNKLEESNRNLDKVNVKLTSTSALI(SEQIDNO:15)-Z;式(16):X-VKEWEDQFNVALDQVFESIENSQALVDQSNRILSSA(SEQIDNO:16)-Z;式(17):X-WKEWEDQFNVALDQVFESIENSQALVDQSNRILSSA(SEQIDNO:17)-Z;式(18):X-VKFPEDQFNVALDQVFESIENSQALVDQSNRILSSA(SEQIDNO:18)-Z;式(19):X-WDEFDASISQVNEKINQSLAFIRKSDELLHNVNAGK(SEQIDNO:19)-Z;式(20):X-WDEWDASISQVNEKINQSLAFIRKSDELLHNVNAGK(SEQIDNO:20)-Z;式(21):X-SDEFDASISQVNEKINQSLAFIRKSDELLHNVNAGK(SEQIDNO:21)-Z;式(22):X-WYTDKVDISSQISSMNQSLQQSKDYIKEAQKILDTV(SEQIDNO:22)-Z;式(23):X-WYTWKVDISSQISSMNQSLQQSKDYIKEAQKILDTV(SEQIDNO:23)-Z;式(24):X-VYTDKVDISSQISSMNQSLQQSKDYIKEAQKILDTV(SEQIDNO:24)-Z;式(25):X-WFTDKVDISSQISSMNQSLQQSKDYIKEAQRLLDTV(SEQIDNO:25)-Z;式(26):X-WFTDWVDISSQISSMNQSLQQSKDYIKEAQRLLDTV(SEQIDNO:26)-Z;式(27):X-VFTDKVDISSQISSMNQSLQQSKDYIKEAQRLLDTV(SEQIDNO:27)-Z;式(28):X-WSLERLDVGTNLGNAIAKLEDAKELLESSDQILRSM(SEQIDNO:28)-Z;式(29):X-WSLEWLDVGTNLGNAIAKLEDAKELLESSDQILRSM(SEQIDNO:29)-Z;式(30):X-ISLERLDVGTNLGNAIAKLEDAKELLESSDQILRSM(SEQIDNO:30)-Z;式(31):X-WVDISLNLADATNFLQDSKAELEKARKILSEVGRWY(SEQIDNO:31)-Z;式(32):X-WVDWSLNLADATNFLQDSKAELEKARKILSEVGRWY(SEQIDNO:32)-Z;式(33):X-PVDISLNLADATNFLQDSKAELEKARKILSEVGRWY(SEQIDNO:33)-Z;式(34):X-WDQFNVALDQVFESVEKSQNLIDQSNKILDSIEKGN(SEQIDNO:34)-Z;式(35):X-WDQWNVALDQVFESVEKSQNLIDQSNKILDSIEKGN(SEQIDNO:35)-Z;式(36):X-EDQFNVALDQVFESVEKSQNLIDQSNKILDSIEKGN(SEQIDNO:36)-Z;式(37):X-WLNLTGEIDDLEFRSEKLHNTTVELAILIDNINNTL(SEQIDNO:37)-Z;式(38):X-WLNWTGEIDDLEFRSEKLHNTTVELAILIDNINNTL(SEQIDNO:38)-Z;式(39):X-YLNLTGEIDDLEFRSEKLHNTTVELAILIDNINNTL(SEQIDNO:39)-Z;式(40):X-WLNLTSEISTLENKSAELNYTVQKLQTLIDNINSTL(SEQIDNO:40)-Z;式(41):X-WLNWTSEISTLENKSAELNYTVQKLQTLIDNINSTL(SEQIDNO:41)-Z;式(42):X-ILNLTSEISTLENKSAELNYTVQKLQTLIDNINSTL(SEQIDNO:42)-Z;或其衍生物、立体异构体、无生理毒性的盐;其中,X为乙酰基、寡肽序列、亲脂性基团、聚乙二醇或缺失;在本发明的实施方案中,X为乙酰基(CH3-CO-);Z为酰胺基、寡肽序列、亲脂性基团、聚乙二醇或缺失;在本发明的实施方案中,Z为酰胺基(-CO-NH2)。其中,所述寡肽序列为1~10个氨基酸序列,例如可以为EEE、KKK、GKK、或GQAV。其中所述亲脂性基团为脂肪酸和甾醇,例如可以为正辛酸酯(C7H15-CO-O-)、月桂酸酯(C13H27-CO-O-)、棕榈酸酯(C15H31-CO-O-)、或胆固醇(C27H46O)。其中X与Z基团用于改善多肽的水溶性、二级结构及稳定性、增强与细胞膜的结合使之更靠近靶点、或改善药物的药代性质。以上式(1)~(42)的序列均来自于包膜类病毒的融合糖蛋白跨膜亚基的CHR序列,其中一些序列在多肽氨基末端的关键位点引入一个色氨酸(W)残基,首选为1位,或者同时在4位。其中式(1)~(3)来自于人副流感病毒3型HPIV3,式(4)~(6)来自于猴副流感病毒SV5,式(7)~(9)来自于严重急性呼吸综合征SARS病毒,式(10)~(12)来自于小鼠肝炎病毒MHV,式(13)~(15)来自于新城疫病毒NDV,式(16)~(18)来自于人偏肺病毒hMPV,式(19)~(21)来自于呼吸道合胞病毒RSV,式(22)~(24)来自于戊型肝炎病毒HeV,式(25)~(27)来自于尼帕病毒NiV,式(28)~(30)来自于麻疹病毒MeV,式(31)~(33)来自于仙台病毒SeV,式(34)~(36)来自于禽肺病毒APV,式(37)~(39)来自于猫传染性腹膜炎病毒FIPV,式(40)~(42)来自于人冠状病毒229EHCoV-229E。以上病毒株均为具有晶体结构的标准株。根据本发明第一方面的多肽,其选自以下多肽:(1)SEQIDNO:1~42中的任一序列在N端和/或C端有1-10、1-5、1-2个氨基酸的截短;(2)SEQIDNO:1~42中的任一序列在N端和/或C端有1-10、1-5、1-2个氨基酸的延伸;优选地,所述延伸是在该序列来源病毒的融合糖蛋白跨膜亚基的CHR序列基础上的延伸;(3)SEQIDNO:1~42中的任一序列有1-10、1-5、1-2个氨基酸的替换,或上述(1)和(2)的序列中有1-10、1-5、1-2个氨基酸的替换;优选地,所述替换是根据该序列来源病毒的不同病毒株之间由于融合糖蛋白跨膜亚基的CHR序列不同而进行的替换;(4)SEQIDNO:1~42中的任一多肽经过末端和/或侧链化学修饰得到的多肽,或上述(1)~(3)的多肽经过末端和/或侧链化学修饰得到的多肽。(5)上述(1)~(4)的多肽,第一位氨基酸和/或第四位氨基酸用色氨酸残基替换,和/或第八位氨基酸用疏水氨基酸残基替换后得到的多肽。其中所述末端和/或侧链化学修饰包括在氨基末端、羧基末端或侧链修饰,修饰基团包括寡肽、乙酰基、酰胺基,亲脂性基团、亲水性基团等,其作用是提高多肽的稳定性、改善药代动力学或提高与细胞膜的结合。根据本发明第一方面的多肽,其为选自本发明第一方面任一项所述多肽中的一种或数种。在本发明中,所述多肽既包括直接由10-100个氨基酸分子脱水缩合而成的化合物,也包括在该化合物上经过上述修饰后得到的含有修饰基团的化合物。在本发明中,所述多肽的长度优选为含有30-40个氨基酸,例如为30、31、32、33、34、35、36、37、38、39、40。以上所述多肽在经过截短、延伸、替换和/或修饰后仍具有抗HIV-1或其它包膜类病毒与靶细胞例如人体细胞融合的活性。在本发明中,所述疏水氨基酸残基包括色氨酸、苯丙氨酸、异亮氨酸、亮氨酸、缬氨酸、酪氨酸、甲硫氨酸。本发明的多肽可以采用本领域公知的方法直接合成,也可以利用分子生物学手段通过将多肽所对应的核苷酸序列克隆入重组载体中,在重组细胞中表达后得到。因此本发明还涉及核酸分子,其含有编码本发明第一方面所述多肽的核苷酸序列。本发明还涉及重组载体,其含有本发明所述的核酸分子;以及重组细胞,其含有本发明所述的重组载体。本发明的第四方面涉及包含本发明第一方面所述多肽或其衍生物、立体异构体、无生理毒性的盐的融合蛋白,或所述多肽或其衍生物、立体异构体、无生理毒性的盐与其他蛋白或毒素或脂类等载体(如大分子载体或重组载体)偶联或融合而得到的复合物,或者展示上述多肽或其衍生物、立体异构体、无生理毒性的盐的类病毒颗粒。本发明多肽的给药方式以针剂为主,可直接注射,或制成缓释针剂;也可以通过提高代谢稳定性,包括物理的、化学的修饰,使之成为可口服使用的药物;也包括外用或粘膜给药等。本发明的另一方面涉及组合物(例如药物组合物),其含有至少一种本发明第一方面所述的多肽或其衍生物、立体异构体、无生理毒性的盐,或者本发明第四方面所述的融合蛋白、复合物或类病毒颗粒,以及任选的药学上可接受的载体或辅料。在本发明中,所述组合物中可以包含至少一种多肽或其衍生物、立体异构体、无生理毒性的盐,或者本发明第四方面所述的融合蛋白、复合物或类病毒颗粒,例如可以为两种或两种以上,以进一步提高融合抑制效果;或者所述组合物中可以包含本发明的多肽或其衍生物、立体异构体、无生理毒性的盐、本发明所述的融合蛋白、复合物或类病毒颗粒,以及其它融合抑制剂,例如C34、T20或CP32。本发明的另一方面涉及本发明第一方面所述的多肽或其衍生物、立体异构体、无生理毒性的盐、第四方面所述的融合蛋白、复合物或类病毒颗粒在制备HIV融合抑制剂中的用途。在本发明中,所述融合抑制剂是指可以阻止HIV-1自身的活性六螺旋结构的形成进而抑制病毒-细胞膜融合的化合物或组合物,即是指通过形成无活性的六螺旋结构阻止病毒-细胞膜融合的化合物或组合物。在本发明的实施方案中,所述融合抑制剂是指多肽或经过修饰的多肽,或者包含多肽或经过修饰的多肽的组合物。本发明还涉及本发明第一方面所述的多肽或其衍生物、立体异构体、无生理毒性的盐、第四方面所述的融合蛋白、复合物或类病毒颗粒在制备用于治疗或预防HIV感染相关疾病例如艾滋病的药物或者其它包膜类病毒感染引起的疾病中的用途。在本发明中,所述HIV包括HIV-1和HIV-2型病毒,在本发明的实施方案中,所述HIV为HIV-1型病毒。在本发明的实施方案中,所述其它包膜类病毒是指利用第一类融合蛋白作为融合工具的病毒,包括但不限于人副流感病毒3型HPIV3,猴副流感病毒SV5,严重急性呼吸综合征SARS病毒,小鼠肝炎病毒MHV,新城疫病毒NDV,人偏肺病毒hMPV,呼吸道合胞病毒RSV,戊型肝炎病毒HeV,尼帕病毒NiV,麻疹病毒MeV,仙台病毒SeV,禽肺病毒APV,猫传染性腹膜炎病毒FIPV或人冠状病毒229EHCoV-229E。本发明还涉及一种预防或治疗HIV感染相关疾病例如艾滋病或者其它包膜类病毒感染引起的疾病的方法,所述方法包括给有需要的受试者以预防或治疗有效量的本发明第一方面所述的多肽或其衍生物、立体异构体、无生理毒性的盐、第四方面所述的融合蛋白、复合物或类病毒颗粒的步骤。根据融合机理,HIV用第一类融合蛋白(ClassIfusionProtein)作为病毒-细胞膜融合的工具分子,利用这类融合蛋白的还有其它多种病毒,包括一些严重威胁人类健康的病毒。第一类融合蛋白有下列共性:都含有跨膜亚基和表面亚基,其中跨膜亚基直接参与融合;跨膜亚基细胞膜外部分一般含有融合肽、NHR和CHR等功能区;融合过程中NHR三聚形成内核,CHR反平行折叠到NHR形成的沟槽中,形成六螺旋结构,形成过程中放出的能量促使病毒和细胞膜融合使病毒完成感染[1]。根据这些原理,发明人针对利用第一类融合蛋白作为融合工具的病毒,根据其融合糖蛋白跨膜亚基的CHR,设计HIV融合抑制剂。这类病毒的包膜糖蛋白的氨基酸序列已知,功能区也得到了确定,发明人从其中CHR序列中截取适当的多肽片段,根据融合机理的相似性,预测这些多肽片段可以反平行折叠到NHR形成的沟槽中,形成无活性的六螺旋结构,并用融合抑制活性实验进行验证。因此本发明还涉及一种获得抑制HIV与靶细胞融合的融合抑制剂的方法,该方法包括从除HIV以外的病毒的CHR功能区序列上截取适当长度的多肽片段,所述多肽片段能够和NHR功能区结合形成无活性的六螺旋结构。需要加以说明的是,由于CHR一般的长度约为40个氨基酸残基,因此截取30-40个残基片段用作HIV融合抑制剂相当直观,无需特别地技术,但不排除进行计算机模拟以求得最好截取效果。另外,根据直观的逻辑,将这些截取片段用于HIV以外的与所截取病毒相同或不同病毒的融合抑制剂也应在本发明的保护范围内。其中所述除HIV以外的病毒是指利用第一类融合蛋白作为融合工具的病毒,例如为包膜类病毒;在本发明的一个实施方案中,所述病毒为人副流感病毒3型HPIV3[2],猴副流感病毒SV5[3],严重急性呼吸综合征SARS病毒[4],小鼠肝炎病毒MHV[4],新城疫病毒NDV[5],人偏肺病毒hMPV[6],呼吸道合胞病毒RSV[3],戊型肝炎病毒HeV[2],尼帕病毒NiV[7],麻疹病毒MeV[7],仙台病毒SeV[7],禽肺病毒APV[8],猫传染性腹膜炎病毒FIPV[9]或人冠状病毒229EHCoV-229E[9]。其中所述适当长度是指包含30-40个氨基酸,例如为30、31、32、33、34、35、36、37、38、39、40。本发明还涉及根据本发明所述的上述方法获得的抑制HIV与靶细胞融合的融合抑制剂。在本发明的实施方案中,所述融合抑制剂选自上述式(1)~(42)所示的多肽。在本发明的实施方案中,所述融合抑制剂是指多肽或经过修饰的多肽,或者包含多肽或经过修饰的多肽的组合物。在本发明中,X为乙酰基、Z为酰胺基的式(1)~式(42)所示的多肽也分别称为多肽1~42。发明的有益效果本发明的多肽显示出明显的抗HIV活性,靶点作用实验显示,所述多肽作用于gp41NHR区,通过与NHR形成无活性的六螺旋结构,破坏HIVNHR结构,抑制HIV与靶细胞的融合。同时,活性实验结果表明,本发明多肽的作用机制与含有WWI结构模板口袋结合区的已知多肽融合抑制剂不同。说明书附图图1显示了本发明多肽与N46的相互作用其中横坐标wave为扫描波长,单位为nm;纵坐标为CD信号,单位为mdeg。每个小图中有四条曲线,例如seq1、N46分别为多肽1,N46单独的CD谱,Seq1/N46为多肽1和N46混合后测得的CD谱,Seq1+N46为二者单独CD谱的叠加,用于和Seq1/N46比较以确定二者是否有相互作用。其它小图标注的含义以此类推;其中A为多肽1,B为多肽4,C为多肽7,D为多肽10,E为多肽13,F为多肽16,G为多肽19,H为多肽22。具体实施方式下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。在本发明中使用的缩写具有下面的意义:AIDS(AcquiredImmureDeficiencySyndrome)艾滋病,获得性免疫缺陷综合症。Ala(Alanine,A)丙氨酸Arg(Arginine,R)精氨酸Asn(Asparagine,N)天冬酰胺Asp(Asparticacid,D)天冬氨酸DCM(Dichloromethane)二氯甲烷DMF(N,N-Dimethylmalonate)二甲基甲酰胺Env(Envelopeglycoprotein)包膜糖蛋白ESI-MS(Electronicsprayionmassspectroscopy)电喷质谱Fmoc(Fluorenylmethoxycarbonyl)芴甲氧羰基Gly(Glycine,G)甘氨酸Gln(Glutamine,Q)谷酰胺Glu(Glutamicacid,E)谷氨酸6-HB(six-helixbundle)六螺旋体HBTU2-(1H-1-羟基苯并三唑)-1,1,3,3-四甲基六氟磷酸His(Histidine,H)组氨酸HoBt(1-Hydroxylbenzotiazoleanhydrous)1-羟基苯并三氮唑NHR(N-terminalheptadrepeat)N-端七联重复序列CHR(C-terminalheptadrepeat)C-端七联重复序列HIV(Humanimmunodeficiencyvirus)人免疫缺陷病毒HIV-11型人免疫缺陷病毒HPLC(highperformanceliquidchromatography)高效液相色谱Ile(Isoleucine,I)异亮氨酸Leu(Leucine,L)亮氨酸Met(Methionine,M)甲硫氨酸Nal正亮氨酸Lys(Lysine,K)赖氨酸Phe(Phenylalanine,F)苯丙氨酸Ser(Serine,S)丝氨酸TFA(Trifluoroaceticacid)三氟乙酸Thr(Threonie,T)苏氨酸Tyr(Tyrosine,Y)酪氨酸Val(Valine,V)缬氨酸W为色氨酸、N为天冬酰胺、A为丙氨酸、S为丝氨酸、K为赖氨酸、L为亮氨酸、E为谷氨酸、Q为谷氨酰胺、I为异亮氨酸、H为组氨酸、M为甲硫氨酸、T为苏氨酸、D为天冬氨酸、R为精氨酸、Y为酪氨酸、F为苯丙氨酸,V为缬氨酸,P为脯氨酸,G为甘氨酸。实施例所用固相合成载体Rink酰胺树脂为天津南开合成责任有限公司产品;HBTU、HOBt、DIEA及Fmoc保护的天然氨基酸和D型的非天然氨基酸为上海吉尔生化公司及成都诺新技术责任公司产品。N-甲基吡咯烷酮(NMP)为ACROS公司产品;TFA为北京博迈科技有限公司产品;DMF、DCM为韩国三星公司产品;色谱纯乙腈为Fisher公司产品。其它试剂如无说明均为国产分析纯产品。实施例1抗HIV-1多肽1-42的设计发明人系统考察了包膜类病毒的融合原理及其抑制特性,从多个包膜类病毒(人副流感病毒3型HPIV3[2],猴副流感病毒SV5[3],严重急性呼吸综合征SARS病毒[4],小鼠肝炎病毒MHV[4],新城疫病毒NDV[5],人偏肺病毒hMPV[6],呼吸道合胞病毒RSV[3],戊型肝炎病毒HeV[2],尼帕病毒NiV[7],麻疹病毒MeV[7],仙台病毒SeV[7],禽肺病毒APV[8],猫传染性腹膜炎病毒FIPV[9]和人冠状病毒229EHCoV-229E[9])的融合糖蛋白跨膜亚基的CHR序列中,截取对应的长度约为36个氨基酸的多肽。设计的多肽序列SEQIDNO:1-42见表1,同时标明了其来源病毒。为方便浓度测定,我们首先在多肽氨基末端关键位点引入一个色氨酸(W)残基,首选为1位,或者4位。然后我们根据HIV-1gp41CHR多肽序列,在1、4位点再引入一个色氨酸,形成WaaWaaaX(其中a为氨基酸)模板,其中X为疏水氨基酸残基,使其具有和gp41CHR相同的结合区模板,考察活性。在Seq16,Seq17中4位引入W时,我们同时将3位F突变为E,以保证该段序列的整体疏水性保持不变。活性验证结果参见实施例3。实施例2抗HIV-1多肽1-42的制备采用标准的Fmoc固相多肽合成方法。将实施例1设计的抗HIV-1多肽序列SEQIDNO:1-42的C-端酰胺化,N-端乙酰化,得到多肽1-42。选用RinkAmide树脂,肽类由C-端向N-端延长。缩合剂为HBTU/HOBt/DIEA。脱保护剂为哌啶/DMF溶液。裂解剂为TFA,粗肽水溶解后冻干保存。用中压液相色谱法或HPLC进行分离纯化,纯肽含量>95%。基质辅助激光解析飞行时间质谱(MALDI-TOF-MS)确定多肽分子量。具体微波多肽合成条件如下:氨基酸:0.2M的DMF溶液;活化剂:0.45MHBTU/HOBt的DMF溶液;活化碱:2MDIEA的NMP溶液;脱保护剂:20%v/v哌啶的DMF溶液;封闭试剂:20%v/v乙酸酐的DMF溶液。称取RinkAmide树脂0.5g(0.25mmol)置入CEM微波多肽合成仪的反应器中,然后将氨基酸、活化剂、活化碱、脱保护试剂、封闭试剂按上述条件配好后,用CEM微波全自动多肽合成仪进行合成。完成后肽树脂用DMF洗涤3遍后用无水甲醇收缩,室温真空干燥,得肽树脂2.05g。肽树脂的裂解:将上述合成好的肽树脂称量后放入250ml茄形瓶中,冰浴,电磁搅拌。按1g肽树脂加入10ml的量配置裂解液(体积比:三氟乙酸:乙二硫醇:间苯酚:水=82.8:10:5:2.5)。TFA需预先冰浴降温30min或者预先放于冰箱中使用。将配制好的裂解液加入到冰浴条件下的肽树脂中,电磁搅拌,树脂变橙红色,冰浴条件下反应30min,然后撤冰浴,室温下再继续反应90min使反应完成。剧烈搅拌下向反应器中加入冷乙醚200ml,析出白色沉淀,继续搅拌30min;用G4的砂芯抽滤漏斗滤出析出物,用冷乙醚反复洗涤3遍,晾干。加入双蒸水50ml,乙腈5ml使固体充分溶解,抽滤,滤液冻干得粗肽1.03g。所得粗肽用中压或高压色谱进行纯化。色谱柱为C18柱,洗脱液为乙腈,水及少量乙酸。具体操作步骤:称取粗肽1g,加水20ml,乙腈5ml溶解,3000转/分钟下离心10min,取上清液上样。色谱柱预先用15%乙腈/水/0.1%冰乙酸溶液200ml平衡,上样后继续用200ml同样洗脱液平衡,高效液相检测洗脱液成分。根据检测结果逐步升高乙腈含量,直至所纯化的多肽峰被洗脱出来。合并同组分洗脱液,旋转蒸发除去大部分溶剂,冻干得纯肽,HPLC检测含量>90%。纯肽经MALDI-TOF-MS质谱确定其分子量。采用以上方法分别合成多肽1-42(见表1)。表1:多肽序列、来源及分子量实施例3:多肽的抗HIV-1融合活性检测。我们用HIV-1Env介导的细胞-细胞融合模型对实施例2制备的多肽1-42进行活性测定。靶细胞为TZM-bl细胞(美国NIH艾滋病试剂和参照物项目提供,目录号为8129),其表面表达CD4T-细胞受体和趋化因子辅助受体CCR5和CXCR4,可被HIV-1Env识别,同时细胞内还转录荧光素酶报告基因,但不含该基因的启动子,因此单独细胞的荧光素酶背景表达量很低。效应细胞为HL2/3细胞(美国NIH艾滋病试剂和参照物项目提供,目录号为1294),其表面表达HIV-1Env,由Env进攻靶细胞,完成细胞融合,同时细胞内还转录荧光素酶报告基因的启动子。两种细胞先在含有氨苄/链霉素双抗的含10%胎牛血清的DMEM中,37℃下在含有5%CO2的培养箱中单独培养。两种细胞均为贴壁细胞,细胞胰酶/EDTA消化后收获传代。细胞用细胞计数板计数。将TZM-bl靶细胞用培养基调整到浓度为75万/ml,以每孔50μl加入96孔细胞培养板中(3.75万/孔),5%CO2,37度下培养24小时。将多肽1-42或阳性对照样品T20溶于磷酸缓冲溶液生理盐水(PBS)中,或加入适量DMSO使充分溶解,用紫外光谱仪在280nm处测定多肽浓度。然后将多肽溶液稀释到适当的浓度,在96孔酶标板(Corning)中等比稀释(浓度为1200、300、75、19、5、1.25μM)。配制150万/ml的HL2/3效应细胞。将20μl/孔的等比稀释的多肽1-42加入前一天培养的TZM-bl细胞中,然后加入50μl/孔的配制好的HL2/3效应细胞;96孔细胞培养板的其中一排用PBS替代抑制剂用于测定饱和融合信号,另一排用DMEM培养基替代HL2/3细胞用于测定背景信号。5%CO2,37℃下培养6-8小时使之充分融合。将荧光素酶报告基因的试剂盒(Promega)从冰箱中取出,将5x细胞裂解液根据用量用双蒸水稀释至1x裂解液,室温下放置;用底物缓冲液溶解底物,室温下放置;同时将酶标仪(MolcularDevicesM5多功能酶标仪)检测条件设置好备用。将融合好的细胞取出,弃去培养基,用200μl/孔PBS洗两次,尽量去除清洗液;然后以50μl/孔加入平衡到室温的裂解液,轻轻震动下反应5min使细胞充分裂解;将裂解液以40μl/孔加入96孔化学发光检测用酶标板板(Corning)中,加样时尽量避免引入气泡;避光下将底物以40μl/孔迅速加入化学发光用酶标板中,立即在酶标仪上测定化学发光。根据饱和融合信号和背景信号的比值确定靶细胞和效应细胞的有效融合,比值>5表明有效融合。根据样品的浓度-化学发光信号曲线确定其半抑制剂浓度(IC50),阳性对照样品的IC50值应稳定在一定范围;理想的抑制曲线中高浓度抑制剂下信号应接近背景信号,最低浓度抑制剂下信号应接近饱和融合信号。多肽1-42的细胞融合抑制活性列于表2,阳性对照T20的IC50为2±0.5nM,与文献报道相符[10]。表2:多肽1-42的细胞融合抑制活性根据表2的活性检测结果,多肽1-42除3、15、21、42外均显示出明显抗HIV活性,半抑制浓度IC50从1到数十μM不等,而所有研究的病毒中都含有活性多肽序列,且活性都有改进空间,符合药物先导化合物的活性标准。同时还可看出,活性与我们引入的色氨酸残基有些关系,但没有明显的规律,因此其作用机制与含有WWI结构模板口袋结合区的已知多肽融合抑制剂不同。但以上结果表明我们可以通过改变氨基酸残基进行活性改进。例如化合物21仅通过引入一个色氨酸残基,活性得到约7倍提高。实施例4:多肽的实验室适应病毒株HIV-1IIIB感染抑制实验1.实验用品实验室适应病毒株HIV-1IIIB、MT-2细胞(通过美国国立健康研究院(NIH)艾滋病研究参考试剂计划获得)。2.实验方法用实验室适应病毒株HIV-1IIIB感染抑制实验考察了部分活性较好多肽的活性。在200ul的RPMI1640培养基中,用100TCID50(50%组织培养感染量)的HIVIIIB毒株去感染104/mlMT-2细胞,同时加入呈浓度梯度的不同抑制剂,过夜培养之后弃去培养上清液,换上新鲜的培养基。感染后第四天,从每个孔中取出100ul培养上清液,加入等体积的5%TritonX-100,用ELISA方法定量检测p24抗原。具体实验方法亦可参考Jiang,S.,etal.,AntimicrobialAgentandChemotherapy,2004.48(11):p.4349-4359.3.实验结果如下面的表3所示。表3:多肽的HIV病毒感染抑制活性我们选择细胞融合活性中活性较好的多肽,测定其抗病毒感染活性。结果显示,这些测试多肽显示与细胞融合活性相似的IC50值,可以用作先导化合物开发新型融合抑制剂。实施例5:抗HIV-1多肽1-42与NHR靶点的作用我们用圆二色谱(CD)研究实施例2制备的多肽1、4、7、10、13、16、19、22与gp41NHR靶点的相互作用,我们选用N46(序列:Ac-TLTVQARQLLSGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARIL-CONH2)作为gp41NHR靶点,其中序列TLTVQARQLLSGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARIL为SEQIDNO:43。圆二色谱仪为BiologicMOS450谱仪。将待测定CHR多肽1、4、7、10、13、16、19、22分别溶于PBS中,N46溶于双蒸水中,根据280nm下紫外吸收确定浓度;然后配制20μM的多肽PBS溶液。配制要检测的多肽样品:将N46和多肽1、4、7、10、13、16、19、22分别以1:1体积比混合得二者混合样品;如果是单独多肽样品,将20μM的样品与缓冲溶液以1:1混合。样品在37℃下放置30min使充分反应。上述实验步骤可确保样品中的多肽浓度保持一致。将配制好的样品在圆二色谱仪上测定,仪器扫描波长范围为190-260nm,波长间隔为1nm,扫描速度为100nm/min,扫描4次进行平均。先用缓冲溶液扫描得到空白,然后扫描样品信号,将空白信号从样品信号中扣除得到CD信号。通过比较待测多肽和N46多肽混合前和混合后的CD信号变化来确定二者的相互作用。CD信号变化表明两者具有相互作用。多肽1、4、7、10、13、16、19、22分别与N46混合后都出现CD信号变化,表明它们均作用于gp41NHR,通过与NHR形成无活性的六螺旋结构抑制HIV与人体细胞的融合(图1)。尽管本发明的具体实施方式已经得到详细的描述,本领域技术人员将会理解。根据已经公开的所有教导,可以对那些细节进行各种修改和替换,这些改变均在本发明的保护范围之内。本发明的全部范围由所附权利要求及其任何等同物给出。参考文献1.Schibli,D.J.andW.Weissenhorn,ClassIandclassIIviralfusionproteinstructuresrevealsimilarprinciplesinmembranefusion(Review).MolecularMembraneBiology,2004.21(6):p.361-371.2.Porotto,M.,etal.,InhibitionofHendraVirusFusion.JVirol,2006.80(19):p.9837-9849.3.Mathews,J.M.,etal.,Thecoreoftherespiratorysyncytialvirusfusionproteinisatrimericcoiledcoil.JournalofVirology,2000.74(13):p.5911-5920.4.Bosch,B.J.,etal.,Severeacuterespiratorysyndromecoroavirus(SARS-CoV)infectioninhibitionusingspikeproteinheptadrepeat-derivedpeptides.ProceedingsoftheNationalAcademyofSciencesoftheUnitedStatesofAmerica,2004.101(22):p.8455-8460.5.Young,J.K.,etal.,InteractionofpeptideswithsequencesfromtheNewcastlediseasevirusfusionproteinheptadrepeatregions.JournalofVirology,1999.73(7):p.5945-5956.6.Miller,S.A.,etal.,Examinationofafusogenichexamericcorefromhumanmetapneumovirusandidentificationofapotentsyntheticpeptideinhibitorfromtheheptadrepeat1region.JournalofVirology,2007.81(1):p.141-149.7.Lamb,R.A.,R.G.Paterson,andT.S.Jardetzky,Paramyxovirusmembranefusion:LessonsfromtheFandHNatomicstructures.Virology,2006.344(1):p.30-37.8.Zhu,J.Q.,etal.,BiochemicalandbiophysicalanalysisofheptadrepeatregionsfromthefusionproteinofMenanglevirus,anewlyemergentparamyxovirus.ArchivesofVirology,2003.148(7):p.1301-1316.9.Xu,Y.H.,etal.,Crystalstructureofsevereacuterespiratorysyndromecoronavirusspikeproteinfusioncore.JournalofBiologicalChemistry,2004.279(47):p.49414-49419.10.Wild,C.T.,etal.,Peptidescorrespondingtoapredictivealpha-helicaldomainofhuman-immunodeficiency-virustype-1gp41arepotentinhibitorsofvirus-infection.ProceedingsoftheNationalAcademyofSciencesoftheUnitedStatesofAmerica,1994.91(21):p.9770-9774.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1