一种聚苯乙烯类材料的亲水改性方法及其产品与流程
本发明涉及聚合物材料改性技术领域,尤其涉及一种聚苯乙烯类材料的亲水改性方法及其产品。
背景技术:
聚苯乙烯(PSt)类微球具有化学性质稳定,pH耐受,压力耐受等优点,大量应用于生化分子的分离纯化过程。然而,未改性的PSt材料的自身特点严重限制了它的应用领域,例如PSt微球除了用于反相色谱外,不能直接作为色谱介质分离蛋白质生物大分子。其原因在于PSt微球表面缺乏功能基团,很难直接偶联亲水配基;此外PSt微球表面和蛋白质分子之间的强烈疏水作用经常会引起蛋白质不可逆吸附甚至变性。这种疏水作用和非特异性吸附一方面来源于微球基质中的芳环,一方面来源于单体聚合过程中的残余乙烯基双键。要克服这些问题需要对PSt微球进行亲水改性,需要用亲水基团覆盖它的疏水表面,然后再在亲水基团上偶联特定配基以满足不同分离模式要求。如何解决这个问题已成为近年来生化分离领域一个研究热点(He JY et al.Crosslinking effect on the deformation and fracture of monodisperse polystyrene-co-divinylbenzene particles.eXPRESS Polym Lett 2013,7(4):365-374.)。
通过直接在微球表面物理吸附一层亲水性的高分子,然后用交联剂对吸附层进行交联以增加镀层的稳定性,使微球比表面能降低,从而达到降低蛋白质非特异性吸附能力的效果。这类物理镀层的方法都存在最后需要再对镀层进行交联固定,以及镀层容易脱落的问题(Chanda M和Rempel G.L.A New Method of Gel-Coating Polyethyleneimine(PEI)on Organic Resin Beads.High Capacity and Fast Kinetics of PEI Gel-Coated on Polystyrene.Industrial&Engineering Chemistry Research,2001,40:1624-1632)。
相对于物理吸附镀层法,为了获得稳定的亲水镀层,化学共价耦联法进行亲水修饰逐渐受到重视。现有的文献中,已有在聚苯乙烯微球表面共价接枝羟基、PEG、聚乙烯亚胺、胺基、蛋白等亲水基团的报道(Zhang RY et al.Hydrophilic modification gigaporous resins with poly(ethylenimine)for high-throughput proteins ion-exchange chromatography.Journar of Chromatography A,2014,1343:109-118;Wang Z,Huang Y,Li S,Xu H,Linder Markus B.,Qiao M,Biosens.Bioelectron.2010,26,1074–1079)。但是,这类亲水镀层方法主要基于低交联度聚苯乙烯类微球以及大孔超大孔聚苯乙烯类微球作为修饰基材(Wu Y et al.Hybrid nanoporous polystyrene derived from cubic octavinylsilsesquioxane and commercial polystyrene via the Friedel–Crafts reaction.RSC Adv.2015,5,12987–12993;Borujeni K.P.,Tamami B.Polystyrene and silica gel supported AlCl3 as highly chemoselective heterogeneous Lewis acid catalysts for Friedel–Crafts sulfonylation of aromatic compounds.Catal.Comm.2007,8,1191–1196)。CN100360239C、CN101003592A和CN101864019B中公开了低交联度聚苯乙烯类微球容易在有机溶剂中溶胀,大孔超大孔聚苯乙烯类微球有较大的贯穿孔,使得亲水修饰试剂均能够顺利进入微球内部充分反应。
针对交联剂大于20%,特别是交联剂大于55%的高交联度甚至超高交联度聚苯乙烯类微球,因为难以溶胀和由于孔径小,苯环上Friedel-Crafts反应的取代位置仅能在微球表面进行,空间位阻效应使得修饰与进一步接枝很难深入到微球内部,亲水改性存在困难(Li Q et al.In situ inhibitor removal promoted heterogeneous Friedel-Crafts reaction of polystyrene microsphere with Lewis acids catalysts.Journal of Molecular Catalysis A Chemical.2013,370:56-63),非特异性吸附程度屏蔽得不明显。
因此,在本领域中期望能够开发一种可以实现对高交联度聚苯乙烯类材料的内部和表面的疏水基团进行改性的方法。
技术实现要素:
针对现有技术的不足,本发明的目的在于提供一种聚苯乙烯类材料的亲水改性方法及其产品,该方法可以实现对聚苯乙烯类材料内部和表面的疏水芳香环和残留乙烯基双键的亲水改性,从而大幅度降低聚苯乙烯类材料的疏水性。
为达到此发明目的,本发明采用以下技术方案:
第一方面,本发明提供一种聚苯乙烯类材料的亲水改性方法,所述方法为:对交联度在20%以上的聚苯乙烯类材料的芳香环和乙烯基双键进行亲水改性,得到亲水改性的苯乙烯类材料。
本发明通过对交联度在20%以上的聚苯乙烯类材料所含的芳香环和乙烯基双键进行亲水改性,来克服现有技术中对高交联度聚苯乙烯类材料难以实现亲水改性的困难,对疏水芳环和未完全聚合的疏水残余乙烯基双键进行亲水改性,大幅度降低了材料的疏水性。
在本发明所述聚苯乙烯类材料的亲水改性方法中,所述聚苯乙烯类材料的交联度为20%以上,例如20%、22%、25%、28%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%或99%等,优选为20%-80%,进一步优选为50-60%。
优选地,所述聚苯乙烯类材料由苯乙烯类单体与二乙烯基苯单体通过共聚制备得到,优选为聚苯乙烯类微球。
优选地,所述苯乙烯类单体为取代的或未取代的苯乙烯类单体,优选为苯乙烯、α-甲基苯乙烯、α-乙基苯乙烯、对甲基苯乙烯或邻甲基苯乙烯中的任意一种或至少两种的组合。
优选地,步骤(1)所述二乙烯基苯单体占苯乙烯类单体与二乙烯基苯单体总质量的20%以上,例如20%、22%、25%、28%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%或99%等,优选20-80%,进一步优选50-70%。
在本发明中,交联度在20%以上的聚苯乙烯类材料可以采用共聚方法来制备,例如,在本发明中可以采用以下方法来制备聚苯乙烯类微球:
(i)将苯乙烯类单体、二乙烯基苯类单体和添加剂混合溶于溶剂中作为分散相;所述添加剂包括引发剂,任选地包括致孔剂或助表面活性剂中的任意一种或至少两种的组合;
优选地,所述引发剂为过氧化苯甲酰(BPO)、过氧化十二酰(LPO)、过氧化二碳酸二乙基己酯(EHP)、偶氮二异丁氰(AIBN)或偶氮二异庚腈(ADVN)中的任意一种或至少两种的组合;
优选地,所述致孔剂为疏水性的C4-C22烷烃类、C4-C22烷链醇类、饱和芳香族化合物或线形苯乙烯低聚物中的任意一种或至少两种的组合;
优选地,所述助表面活性剂为十二醇(LA)、1-辛醇或2-辛醇中的任意一种或至少两种的组合;
优选地,所述溶剂为去离子水(H2O)、明胶(Gelatin)、纤维素(Cellulose)、羟烷基纤维素、聚乙烯吡咯烷酮(PVP)、无水硫酸钠(Na2SO4)、十二烷基硫酸钠(SDS)或十二烷基磺酸钠(SLS)中的任意一种或至少两种的组合,进一步优选为两种溶剂以1:100-100:1,优选10:90-90:1的体积比混合的混合溶剂;
(ii)将分散剂或乳化剂以及任选的阻聚剂加入去离子水中,混合后作为连续相;
优选地,所述乳化剂和分散剂选自聚乙烯醇(PVA)、明胶(Gelatin)、纤维素(Cellulose)、羟烷基纤维素、聚乙烯吡咯烷酮(PVP)、无水硫酸钠(Na2SO4)、十二烷基硫酸钠(SDS)或十二烷基磺酸钠(SLS)中的任意一种或至少两种的组合;
优选地,所述阻聚剂为对苯二酚(HQ)、叔丁基邻苯二酚或对苯酚单丁醚中的任意一种或至少两种的组合;
(iii)将步骤(i)得到的分散相均匀分散于步骤(ii)所得连续相中,形成油/水乳液,将该乳液移入带有冷凝管、搅拌棒、氮气导管的反应器内,通氮气置换氧气后,在30-150℃、优选50-120℃、更优选60-100℃下聚合反应1-48h,优选5-24h,得到聚苯乙烯类微球。
由以上方法制备得到的聚苯乙烯类微球粒径在0.1~1000μm可控;孔径在0~1000nm可控;交联度在20%~80%可控,优选为40%~80%;比表面积为20~1000m2/g,孔体积为100~1000mm3/g;微球结构可为实心、多孔、大孔、空心、单眼或凹陷等形态。
作为本发明的优选技术方案,本发明所述聚苯乙烯类材料的亲水改性方法包括以下步骤:
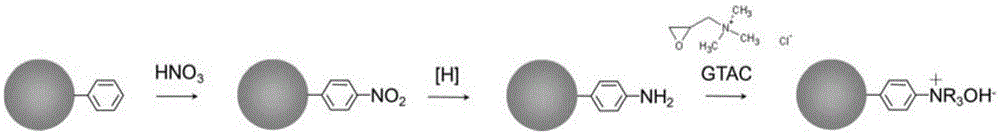
(1)将交联度在20%以上的聚苯乙烯类材料经硝化反应得到芳香环上氢原子被硝基取代的聚苯乙烯类材料,而后在还原剂的作用下发生还原反应,将硝基还原成氨基,之后氨基与亲水性基团供体反应得到芳香环亲水改性的聚苯乙烯类材料;
(2)将步骤(1)得到的芳香环亲水改性的聚苯乙烯类材料在氧化剂的作用下将乙烯基双键氧化成活性环氧基团,而后与亲水改性基团供体反应得到对芳香环和乙烯基双键实现亲水改性的聚苯乙烯类材料。
本发明通过使用硝化反应将聚苯乙烯类材料的芳香环上的氢取代为硝基,进而通过将硝基还原为氨基,使氨基与亲水性基团供体反应来完成对聚苯乙烯类材料中芳香环的亲水改性。
对交联度较高的聚苯乙烯类材料上芳香环的改性现有技术中使用Friedel-Crafts反应对聚苯乙烯类微球表面的芳香环进行改性,由于空间位阻效应使得修饰与进一步接枝很难深入到微球内部,亲水改性存在困难(Journal of Molecular Catalysis A:Chemical,2013,370:56-63),而本发明应用硝化反应,能够很好地完成聚苯乙烯类材料表面和内部的芳香环的亲水改性,例如当聚苯乙烯类材料为聚苯乙烯类微球时,硝基化试剂由于分子很小,很容易进入微球内部,从而完成内部芳香环的亲水改性。
本发明所述聚苯乙烯类材料的亲水改性方法中,步骤(1)所述硝化反应中所用的硝化试剂为浓硝酸或浓硝酸与浓硫酸的混合物。本发明所述浓硝酸为浓度为65%的浓硝酸,浓硫酸为浓度大于或等于70%的硫酸溶液。
优选地,相对于1g交联度在20%以上的聚苯乙烯类材料,硝化试剂的用量为5~20mL,例如5mL、6mL、8mL、10mL、12mL、14mL、16mL、18mL、19mL或20mL,优选为10mL。
本发明所述聚苯乙烯类材料的亲水改性方法中,步骤(1)所述硝化反应如下进行:在冰浴下,向溶胀后的交联度在20%以上的聚苯乙烯类材料中加入硝化试剂,而后升至室温,之后再升温至70-90℃,继续反应,得到硝化产物;
例如,所述再升温至70-90℃,可以为升温至70℃、71℃、73℃、75℃、77℃、79℃、80℃、82℃、84℃、85℃、86℃、88℃或90℃。
优选地,所述继续反应的时间为0.5-4小时,例如0.5小时、0.8小时、1小时、1.3小时、1.5小时、1.8小时、2小时、2.3小时、2.5小时、2.8小时、3小时、3.2小时、3.5小时、3.8小时或4小时。
本发明所述聚苯乙烯类材料的亲水改性方法中,步骤(1)所述还原剂为氯化亚锡或多硫化钠。
优选地,相对于1g交联度在20%以上的聚苯乙烯类材料,步骤(1)所述还原剂的用量为1~10g,例如1.3g、1.5g、1.8g、2g、2.5g、3g、3.5g、4g、4.5g、5g、5.5g、6g、6.5g、7g、7.5g、8g、8.5g、9g、9.5g或9.8g。
优选地,步骤(1)所述还原反应在酸性溶液中进行,优选在盐酸溶液中进行。
优选地,步骤(1)所述还原反应的温度为50-110℃,例如50℃、55℃、60℃、65℃、70℃、75℃、80℃、85℃、90℃、95℃、100℃、105℃或110℃,优选为80-110℃。
优选地,步骤(1)所述还原反应的时间为0.5~8小时,例如0.5小时、1小时、1.3小时、1.5小时、1.8小时、2小时、2.5小时、3小时、3.5小时、4小时、小4.5时、5小时、5.5小时、6小时、6.5小时、7小时、7.5小时或8小时,优选2-5小时。
优选地,在进行还原反应前对芳香环上氢原子被硝基取代的聚苯乙烯类材料进行溶胀处理。
本发明所述聚苯乙烯类材料的亲水改性方法中,步骤(1)所述亲水性基团供体为季铵化试剂、聚乙二醇或壳聚糖中的任意一种或至少两种的组合,优选季铵化试剂。
优选地,所述季铵化试剂为缩水甘油三甲基氯化铵(GTA)或缩水甘油基三丙基氯化铵(GTPA)。
优选地,步骤(1)所述亲水性基团供体与聚苯乙烯类材料的质量比为3-6:1,例如3:1、3.3:1、3.5:1、3.8:1、4:1、4.2:1、4.5:1、4.8:1、5:1、5.3:1、5.5:1、5.8:1或6:1。
优选地,步骤(1)所述氨基与亲水性基团供体反应的温度为0-90℃,例如0℃、5℃、10℃、15℃、20℃、25℃、30℃、35℃、40℃、45℃、50℃、55℃、60℃、65℃、70℃、75℃、80℃、85℃或90℃,优选为50-70℃。
优选地,步骤(1)所述氨基与亲水性基团供体反应的时间为0.5-4小时,例如0.5小时、1小时、1.3小时、1.5小时、1.8小时、2小时、2.2小时、2.5小时、2.8小时、3小时、3.2小时、3.5小时、3.8小时或4小时。
优选地,在步骤(1)所述氨基与亲水性基团供体反应之前,对硝基还原为氨基的聚苯乙烯类材料进行溶胀处理。
本发明所述聚苯乙烯类材料的亲水改性方法中,步骤(2)所述氧化剂的为间氯过氧苯甲酸、2-氯过氧苯甲酸或对氯过氧苯甲酸中的任意一种或至少两种的组合。
优选地,步骤(2)所述芳香环亲水改性的聚苯乙烯类材料与氧化剂的质量比为1-10:1,例如1.3:1、1.5:1、1.8:1、2:1、2.3:1、2.5:1、2.8:1、3:1、3.3:1、3.5:1、3.8:1、4:1、4.2:1、4.5:1、4.8:1、5:1、5.3:1、5.8:1、6:1、6.5:1、7:1、7.5:1、7.8:1、8:1、8.5:1、9:1、9.5:1或9.8:1。
优选地,步骤(2)所述氧化反应的温度为0-90℃,例如0℃、5℃、10℃、15℃、20℃、25℃、30℃、35℃、40℃、45℃、50℃、55℃、60℃、65℃、70℃、75℃、80℃、85℃或90℃,优选为25-50℃。
优选地,步骤(2)所述氧化反应的时间为0.5-10小时,例如0.5小时、1小时、1.5小时、2小时、2.5小时、3小时、3.5小时、4小时、4.5小时、5小时、5.5小时、6小时、6.5小时、7小时、7.5小时、8小时、8.5小时、9小时、9.5小时或10小时,优选为2-5小时。
优选地,步骤(2)所述活性环氧基团为环氧乙烷基。
本发明所述聚苯乙烯类材料的亲水改性方法中,步骤(2)所述亲水改性基团供体为胺化试剂、聚乙二醇或壳聚糖中的任意一种或至少两种的组合,优选胺化试剂。
优选地,所述胺化试剂为二甲胺、二乙胺、三乙烯四胺或四乙烯五胺中的任意一种或至少两种的组合。
优选地,相对于1g交联度在20%以上的聚苯乙烯类材料,步骤(2)所述胺化试剂的用量为1~10g,例如1.3g、1.5g、1.8g、2g、2.5g、3g、3.5g、4g、4.5g、5g、5.5g、6g、6.5g、7g、7.5g、8g、8.5g、9g、9.5g或9.8g。
优选地,步骤(2)所述与亲水改性基团供体反应的温度为0-90℃,例如0℃、5℃、10℃、15℃、20℃、25℃、30℃、35℃、40℃、45℃、50℃、55℃、60℃、65℃、70℃、75℃、80℃、85℃或90℃,优选为40-60℃。
优选地,步骤(2)所述与亲水改性基团供体反应的时间为0.5-4小时,例如0.5小时、1小时、1.3小时、1.5小时、1.8小时、2小时、2.2小时、2.5小时、2.8小时、3小时、3.2小时、3.5小时、3.8小时或4小时。
优选地,在步骤(2)所述氧化之前将步骤(1)得到的芳香环亲水改性的聚苯乙烯类材料进行溶胀处理。
本发明所述聚苯乙烯类材料的亲水改性方法中,包括在亲水改性前将聚苯乙烯类材料进行溶胀处理。例如本发明的步骤(1)中在进行硝化反应前对交联度在20%以上的聚苯乙烯类材料进行溶胀处理;在步骤(1)所述还原反应前对硝基化的聚苯乙烯类材料进行溶胀处理;在步骤(1)所述氨基与亲水性基团供体反应之前,对硝基还原为氨基的聚苯乙烯类材料进行溶胀处理;以及在步骤(2)所述氧化之前将步骤(1)得到的芳香环亲水改性的聚苯乙烯类材料进行溶胀处理。
优选地,所述溶胀处理为:将聚苯乙烯类材料放入有机溶剂中,密封振荡,完全溶胀后,抽滤,去除有机溶剂。
优选地,所述溶胀处理所用的有机溶剂可以为但不限于二甲基亚砜、二氯甲烷、二甲基甲酰胺(DMF)、乙醇或二氧六环中的任意一种或至少两种的组合;只要与水混溶的极性有机溶剂均可。
优选地,相对于1g待改性的聚苯乙烯类材料,所述溶胀处理所用的有机溶剂的体积为3-15mL,例如3mL、4mL、5mL、6mL、7mL、8mL、9mL、10mL、11mL、12mL、13mL、14mL或15mL。
优选地,所述溶胀的时间为1-24小时,例如1小时、2小时、3小时、5小时、7小时、9小时、10小时、12小时、14小时、15小时、16小时、18小时、20小时、21小时、22小时、23小时或24小时,优选5-12小时。
在改性前对于待改性的聚苯乙烯类材料进行溶胀处理,目的在于使得聚苯乙烯类材料充分溶胀,以更加有利于亲水物质进入聚苯乙烯类材料内部(例如微球内部),以实现对表面和内部的芳香环和乙烯基双键的亲水改性。
上述亲水改性方法,对于所述聚苯乙烯类材料的结构没有限制,例如可以为实心、微孔、小孔、大孔、超大孔、空心、凹陷等多种结构的微球,以及结合膜乳化技术、快速膜乳化技术、种子溶胀聚合技术、微流控技术等制备尺寸均一可控的微球;也可以为膜等其它结构。
另一方面,本发明提供了由本发明所述的方法制备得到的亲水改性的聚苯乙烯类材料。
本发明改性之后的聚苯乙烯类材料的疏水性大大降低,通过不同胺化试剂的取代反应,其表面可以进一步衍生成伯、仲、叔胺类的阴离子交换功能基团,用作多种分离模式下的生化分离用介质,因此在生化分离纯化领域有很好的应用前景和优势。
相对于现有技术,本发明具有以下有益效果:
本发明通过对聚苯乙烯类材料中的疏水芳环和未完全聚合的疏水残余乙烯基双键进行亲水改性,可以实现对聚苯乙烯类材料内部和表面的疏水芳香环和乙烯基双键的亲水改性,从而大幅度降低聚苯乙烯类材料的疏水性。由于利用硝化反应和还原反应来对芳香环进行亲水改性,从而克服了采用Friedel-Crafts反应对高交联度聚苯乙烯类材料改性时,由于空间位阻效应使得改性仅能在材料表面进行而不能深入到材料内部的缺陷,实现了对聚苯乙烯类材料内部和表面的疏水芳环和乙烯基双键的亲水改性,亲水改性后所得产品可作为用作多种分离模式下的生化分离用介质,因此,在生化分离纯化领域有很好的应用前景和优势。
附图说明
图1为聚苯乙烯类微球基材中疏水芳环季胺化反应示意图;
图2为聚苯乙烯类微球基材中未完全聚合的残余乙烯基双键季铵化反应示意图;
图3A为实施例4中聚苯乙烯类微球亲水改性前的SEM图;
图3B为实施例4中亲水改性后聚苯乙烯类微球的SEM图;
图4A为实施例4中亲水改性前聚苯乙烯类微球的红外光谱图;
图4B为实施例4中亲水改性后聚苯乙烯类微球的红外光谱图;
图5为实施例4的亲水改性后的聚苯乙烯类微球填充柱的压力流速曲线图;
图6为实施例3所得聚苯乙烯类微球与实施例4所得亲水改性的聚苯乙烯类微球对CO2吸附结果图。
具体实施方式
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
实施例1聚苯乙烯类(PSt)微球的制备
将2g甲基苯乙烯(MST)和2g苯乙烯、1g二乙烯基苯(DVB)混合后,加入制孔剂2g正庚烷(HP)、0.3g过氧化二苯甲酰(BPO),溶解后作为分散相。在100mL去离子水中加入2g聚乙烯醇(PVA)和0.0015g十二烷基硫酸(SDS)溶解后作为连续相。利用常规膜乳化技术(3.5μm的膜孔)将分散相均匀分散于连续相中形成均一的油/水型乳液。然后将制备好的乳液移入带有冷凝管、搅拌棒、氮气导管的反应器内,通氮气置换氧气后,在70℃下聚合反应10h后得到微球聚合悬浮液。聚合结束后,用布氏漏斗抽滤得到微球产品。产品经去离子水和乙醇反复洗涤后,放入真空干燥箱内干燥。经测试所得微球的平均粒径为24μm、表面孔径为40nm,比表面积为395m2/g。
实施例2聚苯乙烯类(PSt)微球的制备
称取10g乙基苯乙烯(EST)、10g二乙烯基苯(DVB)、0.5g过硫酸钾(KPS)混合溶解后作为油相;在150mL去离子水加入1.9g PVA,0.0001g SDS以及0.0045g Na2SO4完全溶解后作为水相。利用快速膜乳化技术(2.8μm的膜孔)将油水相混合液快速压过微孔膜管并循环乳化4次后得到均一的乳液。然后将制备好的乳液移入带有冷凝管、搅拌棒、氮气导管的反应器内,通氮气置换氧气后,在85℃下聚合反应20h后得到微球聚合悬浮液。聚合结束后,用离心机在8000rpm下离心得到微球产品。产品经去离子水和乙醇反复离心洗涤后,放入真空干燥箱内干燥。经测试所得微球的平均粒径为15μm、表面孔径为20nm,比表面积为524m2/g。
实施例3聚苯乙烯类(PSt)微球的制备
称取4.5g苯乙烯(St)、5.5g DVB以及0.28g偶氮脒(V65)、3g十六烷(HD)及3g正己醇(HA)混合溶解后作为油相;在300mL去离子水加入3.5g PVA,0.0008g SDS以及0.009g Na2SO4完全溶解后作为水相。利用常规膜乳化技术(5.2μm的膜孔)将油相均匀分散于水相中形成均一的油/水型乳液。然后将制备好的乳液移入带有冷凝管、搅拌棒、氮气导管的反应器内,通氮气置换氧气后,在80℃下聚合反应20h后得到微球聚合悬浮液。聚合结束后,用布氏漏斗抽滤得到微球产品。产品经去离子水和乙醇反复洗涤后,放入真空干燥箱内干燥,最终获得高交联度聚苯乙烯微球。经测试所得微球的平均粒径为30μm、表面孔径为60nm,比表面积为390m2/g。
实施例4对实施例3制备的聚苯乙烯类(PSt)微球的季铵化
如图1所示,取12g实施例3所得微球,加入40mL DMF溶胀,室温过夜,低转速搅拌,过滤除去DMF。冰水浴冷却下将25mL浓硫酸和77mL浓硝酸混匀(硫酸和硝酸提前存于4℃冰箱待用),将溶胀好的上述微球加入所述混合酸中,去掉冰浴,使温度逐渐升温到室温,再于1.5小时内渐渐加热升温到75℃,并保持这个温度反应2h,反应结束后,冷却至室温,用去离子水缓慢逐渐稀释,玻璃砂芯漏斗过滤后,用大量去离子水洗涤至中性,干燥。加入40mL DMF溶胀过夜,在DMF中充分溶胀后,加入99g SnCI2·2H2O(C.P.)及66mL HCI(35.6~38%,A.R.)的混合溶液中,快速搅拌,均匀分散后,于1小时内升温至100±2℃,保持该温度反应8小时。而后将得到的微球用DMSO或者二氧六环进行溶胀,溶胀后抽滤掉有机溶剂,将微球加入缩水甘油三甲基氯化铵(GTA)的水溶液中(微球与缩水甘油三甲基氯化铵的质量比为1:3),60℃,反应4h,水洗至中性,乙醇洗涤,l mol/L的NaOH水溶液淋洗,再水洗至流出水酚酞不变色(pH值为中性),存于50%乙醇水溶液备用。
如图2所示,取10g以上所得微球与130mL二氯甲烷溶胀,室温混合过夜,加入10g间氯过氧苯甲酸,25℃反应1.5小时后,去离子水和乙醇交替洗涤、干燥。加入100mL二甲胺水溶液(含二甲胺50g),60℃,反应4h;依次用去离子水、甲醇、二氯甲烷、甲醇洗涤,真空干燥,得到季铵化的PSt。经检测改性后的微球表面亲水程度>99%。
利用电子扫描显微镜(JEM-6700F,Japan)对改性前后的聚苯乙烯类微球进行表征,结果如图3所示,其中图3A为改性前聚苯乙烯类微球的SEM图,图3B为改性后聚苯乙烯类微球的SEM图,由图3可知,经历多步修饰反应,微球表面呈现腐蚀形貌,粗糙度增强。
利用红外光谱仪(FT/IR-400/600,JASCO,USA)对改性前后的聚苯乙烯类微球进行表征,结果如图4所示,图4A为亲水改性前聚苯乙烯类微球的红外光谱图,图4B为亲水改性后聚苯乙烯类微球的红外光谱图,由图4可知,亲水改性后聚苯乙烯类微球的红外光谱图出现OH/NH(3300–3700cm-1)、NH2(1120cm-1)等亲水基团的特征吸收峰,可以进一步验证成功对聚苯乙烯类微球进行了亲水改性。
对季铵化的PSt微球进行阴离子交换容量测定,测试方法如下:
将季铵化的PSt微球装入交换柱中缓慢沉降,用去离子水流经柱体至流出液体对酚酞不显色为止。先用酸式滴定管准确量取25mL浓度为0.01mol/L的盐酸溶液进行淋洗并收集到250mL三角瓶中;再用1mol/L的氯化钠溶液滴加入柱体进行淋洗并同样收集到250mL三角瓶中。在收集液中加入3滴酚酞指示剂,用0.01mol/L的氢氧化钠溶液滴定至收集液呈现微红色并保持15s内不褪色为终点,记录所耗碱液体积。该介质产品的离子交换容量按照下式进行计算:
其中,C1为0.01mol/L盐酸溶液的浓度,V1为盐酸溶液的消耗体积(mL);C2为0.01mol/L氢氧化钠溶液的浓度,V2为氢氧化钠溶液的消耗体积(mL);V为柱体中介质的体积。
按上述测定及计算方法,本实施例得到的季铵化的PSt微球的阴离子交换容量为0.4mmol/mL介质。
实施例5对实施例3制备的聚苯乙烯类(PSt)微球的季铵化
取10g实施例3所得微球,加入50mL二甲基亚砜(DMSO)溶胀,室温过夜,低转速搅拌,过滤除去DMSO。冰水浴冷却下,将微球缓慢加入100mL浓硝酸(浓硝酸提前存于4℃冰箱待用)中,去掉冰浴,使温度逐渐升温到室温,再于1小时内渐渐加热升温到70℃,并保持这个温度反应0.5h,反应结束后,冷却至室温,用去离子水缓慢逐渐稀释,玻璃砂芯漏斗过滤后,用大量去离子水洗涤至中性,干燥。加入80mL DMF溶胀过夜,在DMF中充分溶胀后,加入10g SnCI2·2H2O(C.P.)及20mL HCl(35.6~38%,A.R.)的混合溶液中,快速搅拌,均匀分散后,于1小时内升温至110℃,保持该温度反应0.5小时。而后将得到的微球用30mLDMSO或者二氧六环进行溶胀,溶胀后抽滤掉有机溶剂,将微球加入缩水甘油三甲基氯化铵(GTA)的水溶液中(微球与缩水甘油三甲基氯化铵的质量比为1:6),60℃,反应0.5h,水洗至中性,乙醇洗涤,l mol/L的NaOH水溶液淋洗,再水洗至流出水酚酞不变色(pH值为中性),存于50%乙醇水溶液备用,反应示意图如图1所示。
取10g以上所得微球与130mL二氯甲烷室温混合过夜,加入1g间氯过氧苯甲酸,90℃反应0.5小时后,去离子水和乙醇交替洗涤、干燥。加入100mL二甲胺水溶液,90℃,反应0.5h;依次用去离子水、甲醇、二氯甲烷、甲醇洗涤,真空干燥,得到季铵化的PSt。经检测改性后的微球表面亲水程度>99%。反应示意图如图2所示。
利用电子扫描显微镜对改性前后的聚苯乙烯类微球进行表征,所得结果与图3相似,证明经历多步修饰反应,微球表面呈现腐蚀形貌,粗糙度增强。同样利用红外光谱谱上出现OH、NH、NH2等亲水基团的特征吸收峰,可以进一步验证成功对聚苯乙烯类微球进行了亲水改性。并且利用与实施例4相同的测试得到本实施例得到的季铵化的PSt微球的阴离子交换容量为0.42mmol/mL介质。
实施例6对实施例1制备的聚苯乙烯类(PSt)微球的季铵化
取10g实施例1所得微球,加入100mLDMF溶胀,室温过夜,低转速搅拌,过滤除去DMF。冰水浴冷却下,25mL浓硫酸和77mL浓硝酸混匀(硫酸和硝酸提前存于4℃冰箱待用),将溶胀好的上述微球加入所述混合酸中,去掉冰浴,使温度逐渐升温到室温,再于1小时内渐渐加热升温到90℃,并保持这个温度反应4h,反应结束后,冷却至室温,用去离子水缓慢逐渐稀释,玻璃砂芯漏斗过滤后,用大量去离子水洗涤至中性,干燥。加入30mL DMF溶胀过夜,在DMF中充分溶胀后,加入100g SnCI2·2H2O(C.P.)及68mL HCl(35.6~38%,A.R.)的混合溶液中,快速搅拌,均匀分散后,于0.5小时内升温至50℃,保持该温度反应5小时。而后将得到的微球用30mLDMSO或者二氧六环进行溶胀,溶胀后抽滤掉有机溶剂,将微球加入缩水甘油三甲基氯化铵(GTA)的水溶液中(微球与缩水甘油三甲基氯化铵的质量比为1:4),0℃,反应4h,水洗至中性,乙醇洗涤,l mol/L的NaOH水溶液淋洗,再水洗至流出水酚酞不变色(pH值为中性),存于50%乙醇水溶液备用,反应示意图如图1所示。
取10g以上所得微球与150mL二氯甲烷室温混合过夜,加入10g间氯过氧苯甲酸,50℃反应10小时后,去离子水和乙醇交替洗涤、干燥。加入20mL二甲胺水溶液(含二甲胺10g),40℃,反应4h;依次用去离子水、甲醇、二氯甲烷、甲醇洗涤,真空干燥,得到季铵化的PSt。经检测改性后的微球表面亲水程度>99%。反应示意图如图2所示。
利用电子扫描显微镜对改性前后的聚苯乙烯类微球进行表征,所得结果与图3相似,证明经历多步修饰反应,微球表面呈现腐蚀形貌,粗糙度增强。同样利用红外光谱谱上出现OH、NH、NH2等亲水基团的特征吸收峰,可以进一步验证成功对聚苯乙烯类微球进行了亲水改性。并且利用与实施例4相同的测试得到本实施例得到的季铵化的PSt微球的阴离子交换容量为0.41mmol/mL介质。
实施例7对实施例2制备的聚苯乙烯类(PSt)微球的季铵化
取10g实施例2所得微球,加入60mL二甲基亚砜(DMSO)溶胀,室温过夜,低转速搅拌,过滤除去DMSO。冰水浴冷却下,将微球缓慢加入100mL浓硝酸(浓硝酸提前存于4℃冰箱待用)中,去掉冰浴,使温度逐渐升温到室温,再于1小时内渐渐加热升温到80℃,并保持这个温度反应3h,反应结束后,冷却至室温,用去离子水缓慢逐渐稀释,玻璃砂芯漏斗过滤后,用大量去离子水洗涤至中性,干燥。加入80mL DMF溶胀过夜,在DMF中充分溶胀后,加入50g SnCI2·2H2O(C.P.)及55mL HCl(35.6~38%,A.R.)的混合溶液中,快速搅拌,均匀分散后,于1小时内升温至90℃,保持该温度反应8小时。而后将得到的微球用30mLDMSO或者二氧六环进行溶胀,溶胀后抽滤掉有机溶剂,将微球加入缩水甘油三甲基氯化铵(GTA)的水溶液中(微球与缩水甘油三甲基氯化铵的质量比为1:6),20℃,反应4h,水洗至中性,乙醇洗涤,l mol/L的NaOH水溶液淋洗,再水洗至流出水酚酞不变色(pH值为中性),存于50%乙醇水溶液备用,反应示意图如图1所示。
取10g上述所得微球与100mL二氯甲烷室温混合过夜,加入2g间氯过氧苯甲酸,0℃反应10小时后,去离子水和乙醇交替洗涤、干燥。加入200mL二甲胺水溶液(含二甲胺100g),20℃,反应4h;依次用去离子水、甲醇、二氯甲烷、甲醇洗涤,真空干燥,得到季铵化的PSt。经检测改性后的微球表面亲水程度>99%。反应示意图如图2所示。
利用电子扫描显微镜对改性前后的聚苯乙烯类微球进行表征,所得结果与图3相似,证明经历多步修饰反应,微球表面呈现腐蚀形貌,粗糙度增强。同样利用红外光谱谱上出现OH、NH、NH2等亲水基团的特征吸收峰,可以进一步验证成功对聚苯乙烯类微球进行了亲水改性。并且利用与实施例4相同的测试得到本实施例得到的季铵化的PSt微球的阴离子交换容量为0.4mmol/mL介质。
实施例8使用实施例4所得的季铵化的PSt微球分离溶菌酶
采用实施例4所得微球分离样品溶菌酶,岛津高效液相仪检测分离曲线。
洗脱条件(检测波长280nm):0-10min,B相,PBS,100%;10.01-40min,A相,2M NaCl+PBS,100%(即B相,0%);40.01-55min,B相,PBS,100%;55.01-85min,A相,1M NaOH+PBS,100%(即B相,0%);85.01-100min,B相,PBS,100%。
流动相A1(10.01-40min)是2M NaCl+PBS,流动相B是pH=7.0,50mM PBS;
流动相A2(55.01-85min)是1M NaOH+PBS,流动相B是pH=7.0,50mM PB。
不锈钢柱长度为25cm,填充微球为实施例五中所制备的阴离子交换介质。柱体积V=4.15mL,进样量为20μL,流速1mL/min。
实验发现溶菌酶能完全通过离子交换作用保留在层析介质上,采用A1流动相能够完全实现100%蛋白洗脱,出峰时间<1min,分离度R>1.6。图5为层析过程的压力流速曲线图。由图5可知,制备的微球介质能够耐受至少75个大气压力而没有碎球现象,且柱内压力在较大流速范围内随着流速增加保持线性增加。
实施例9实施例3制备的未亲水改性的PSt微球和实施例4制备的季铵化的PSt微球对CO2气体的吸附效果
自然科学基金(No.21206175)相关研究对本实施例的开展提供了基础,实验方法如下:将实施例3与实施例4新制备的微球,放置于70℃烘箱干燥过夜后,分别用于装填文献[D.An,L.B.Wu,B.G.Li,S.P.Zhu,Synthesis and SO2absorption/desorption properties of poly(1,1,3,3-tetramethylguanidine acrylate).Macromol.40(2007)3388-3393]所述U型气体吸附装置()。用氮气(N2,纯度≥99.995%)充分填充U型管后,以1~100mL/min流速通入二氧化碳气体(CO2,≧99.999%),通过称量装置系统重量变化计算单位微球介质对气体的吸附载量,图6为微球产品对CO2气体的吸附结果图。由图6可知,实施例4新制备的季铵化的PSt微球产品可以作为一款对CO2高效捕获的聚合物微球。
申请人声明,本发明通过上述实施例来说明本发明的工艺方法,但本发明并不局限于上述工艺步骤,即不意味着本发明必须依赖上述工艺步骤才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所选用原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!