一种由环戊二烯选择性加氢生产环戊烯的方法与流程
本发明涉及一种由环戊二烯选择性加氢生产环戊烯的方法。
背景技术:
环戊烯是一种重要的精细化工原料,可用于制备环戊醇、环戊酮、戊二醛、溴代环戊烷、氯代环戊烷等高附加值产品,而且还是聚环烯烃高分子聚合物的主要原料。
目前,环戊烯主要通过环戊二烯选择性加氢方法制得。环戊二烯主要来自于乙烯裂解副产碳五馏分。因在室温下,环戊二烯容易发生自聚反应生成相对更加稳定的双环戊二烯,因此工业环戊二烯通常以二聚体形式贮存。使用时,仅需将双环戊二烯进行常规的常压蒸馏,收集塔顶温度在40~44℃的馏分,即可得到环戊二烯。
环戊二烯分子中含有两个活泼的双键,在典型加氢条件下,首先一个双键被加氢生成环戊烯,生成的环戊烯分子中的双键,还会发生进一步的加氢反应生成环戊烷。由于环戊二烯加氢生成环戊烯的反应活化能比环戊烯加氢生成环戊烷的反应活化能略低,因此只有在较低的温度下反应,才能更有利地使反应停留在生成环戊烯的阶段。
环戊二烯加氢按反应器型式可分为釜式间歇加氢工艺和固定床连续加氢工艺。例如CN1417179、CN1462734和CN102728386,分别公开了一种采用釜式反应器的环戊二烯间歇加氢工艺。上述间歇加氢工艺由于采用粉末状或小颗粒催化剂,存在催化剂与产品不易分离的难题,不适合于大规模的连续生产。
环戊二烯固定床加氢工艺按反应条件可分为气相加氢工艺和液相加氢工艺。文献(天然气化工,2012,37,20)和文献(河北工业大学学报,2011,40,40)分别报道了一种环戊二烯气相加氢制备环戊烯的方法,该方法反应温度通常高于100℃,因反应温度较高,不利于控制环戊烯进一步加氢反应生成环戊烷的反应,生成的环戊烷势必会增加后续分离工艺的负荷。此外,气相加氢工艺所需的反应器体积较大,设备投资费用较高。
环戊二烯固定床液相加氢是本领域优选的技术方案。例如,US3994986、CN1011877和文献(石化技术与应用,2009,27,218),分别公开了一种采用负载型钯催化剂的环戊二烯固定床加氢生产环戊烯的方法,因贵金属Pd的价格昂贵,现有技术方案存在催化剂成本较高的不足。
技术实现要素:
针对现有环戊二烯固定床液相加氢技术存在的不足,本发明提供了一种采用非贵金属催化剂的由环戊二烯选择性加氢生产环戊烯的方法。
本发明一种由环戊二烯选择性加氢生产环戊烯的方法,包括如下步骤:
(1)含环戊二烯原料与溶剂混合后作为加氢反应原料,再与氢气混合后连续通过填装有选择性加氢催化剂的固定床反应器;所述选择性加氢催化剂由活性组分无定形磷化镍、氧化铝载体和助剂铈组成,所述催化剂的XRD谱图中无Ni2P或Ni12P5的衍射峰,在最终催化剂中,活性组分无定形磷化镍中镍的重量含量以金属镍计,占总催化剂重量的10-15%,磷与镍的摩尔比为2.0-2.4:1,助剂铈与镍的摩尔比为0.01-0.05:1,其余为载体;
(2)将步骤(1)加氢产物分离出氢气后通入第一精馏塔,塔底采出重组分杂质,塔顶馏出物作为第二精馏塔的进料采出;
(3)将步骤(2)第一精馏塔塔顶馏出物通入第二精馏塔,塔底采出环戊烯产品,塔顶采出轻组分作为循环溶剂,返回步骤(1)中循环使用。
本发明方法步骤(1)中,选择性加氢催化剂的制备方法如下:
(1)配制酸性镍盐水溶液,然后加入磷酸氢二铵、含助剂X的盐和柠檬酸,使磷/镍摩尔比为2.5-3.0,X/镍摩尔比为0.01-0.05:1,柠檬酸/镍摩尔比为1.0-2.0:1,得到溶液A,向所得溶液A中加入计量的氧化铝或氢氧化铝,形成浆液B,然后缓慢蒸干水分,形成干胶C;
(2)将步骤(1)中所得干胶C置于连续流动的惰性气体氛围下,升温至250-350℃下,并于该温度下处理5-10小时,使干胶中的柠檬酸发生热分解反应,得到催化剂前驱体D;
(3)将步骤(2)中得到的催化剂前驱体D进行成型,成型后催化剂经烘干后,得到成型催化剂前驱体E;
(4)将步骤(3)中得到的成型催化剂前驱体E在氢气氛围中进行还原,还原温度为300-400℃,还原时间为1-10 h,待催化剂还原后,降温至室温,以氧气浓度为0.5-1.0%的钝化气在室温下对催化剂进行钝化0.5-1 h,得到选择性性加氢催化剂。
本发明方法步骤(1)中,所述的溶剂由质量分数为10~80%的甲乙醚和20~90%的乙醚组成。作为本发明方法的进一步优选,步骤(1)中所述的溶剂由质量分数为20~80%的甲乙醚和20~80%的乙醚组成。
本发明方法步骤(1)中,所述的含环戊二烯原料来自于双环戊二烯裂解所或甲基环戊二烯二聚体的裂解,其中环戊二烯或甲基环戊二烯的质量分数为98%以上。
本发明方法步骤(1)中,所述的加氢反应原料中,环戊二烯的质量分数为1~25%,优选10~25%。
本发明方法步骤(1)中,所述的反应条件:反应器入口温度为20~60℃,反应压力为0.5~1.1 MPa,质量空速为1~4 h-1,氢气与环戊二烯的摩尔比为1.1~2.0;优选反应器入口温度为40~60℃,反应压力为0.9~1.1 MPa,质量空速为2~4 h-1,氢气与环戊二烯的摩尔比为1.1~1.3。
本发明方法步骤(2)中塔釜温度为215~255℃,塔顶温度为60~85℃,塔顶压力为0.50~0.65 MPa。
本发明方法步骤(2)中,所述的第一精馏塔的理论塔板数为10~20块。作为本发明方法的进一步优选,所述的第一精馏塔的理论塔板数为15块,第一精馏塔中可以设置塔板,也可以填装填料。
本发明方法步骤(3)中,塔釜温度为95~120℃,塔顶温度为55~80℃,塔顶压力为0.45~0.70 MPa,
本发明方法步骤(3)中,所述的第二精馏塔的理论塔板数为60~70块。作为本发明方法的进一步优选,所述的第二精馏塔的理论塔板数为65块,第二精馏塔中可以设置塔板,也可以填装填料。
环戊二烯加氢生成环戊烯和环戊烯加氢生成环戊烷均是强放热反应,在典型的液相加氢条件下,二者的反应热均高于100 kJ/mol,后者的放热量更大。通常认为,如果反应温度高于120℃,环戊烯加氢生成环戊烷的反应速度会显著加快,一方面导致环戊烯选择性下降,令一方面因环戊烷与产品环戊烯沸点相近,会影响环戊烯产品质量及增加后续产品的分离难度。因此,取走反应热及控制反应在较低的温度下进行,将是高选择性地生产及分离提纯环戊烯的关键。
本发明的关键之一在于采用了由活性组分无定形磷化镍、氧化铝载体和助剂铈组成非贵金属选择性加氢催化剂。同现有技术方案中采用的贵金属Pd催化剂相比,本发明催化剂具有成本低,环戊烯选择性高的优点。
此外,本发明的另一关键之一在于本发明采用的溶剂由甲乙醚和乙醚组成。甲乙醚在本发明所采用反应压力下沸点为79.0~87.9℃。在本发明采用的反应条件下,环戊二烯加氢放热使甲乙醚气化,利用二甲醚的气化潜热来控制催化剂床层温度。通过调节溶剂中甲乙醚的含量,可使催化剂床层温度稳定地控制在80~90℃,保证了本发明采用的选择性加氢催化剂充分发挥了自身优越的催化性能,使环戊烷副产物选择性控制在0.5%以下。
由于本发明采用的溶剂由甲乙醚和乙醚组成,二者在常压下的沸点均低于环戊二烯原料和环戊烯产品,使得产品分离流程大大简化。同现有技术相比,本发明方法省去了溶剂分离步骤,溶剂和未反应的环戊二烯原料共同以第二精馏塔的塔顶轻组分采出,一方面可作为本发明的循环溶剂,另一方面也可将未反应的环戊二烯原料带回加氢反应器继续反应。待装置达到稳态后,溶剂和部分未反应的环戊二烯原料在装置中循环,可提高新鲜原料中环戊二烯的转化率,且无需补充新鲜溶剂。
附图说明
图1为本发明流程示意图。
图1中,1-原料储罐,2-进料泵,3-加氢反应器,4-气液分离器,5-进料泵,6-第一精馏塔,7-第二精馏塔。
图1中101~111为物流,其中101-含环戊二烯原料,102-新鲜溶剂,103-加氢反应原料,104-氢气,105-加氢反应流出物,106-循环氢气,107-第一精馏塔进料,108-重组分杂质,109-第二精馏塔进料,110-环戊烯产品,111-循环溶剂。
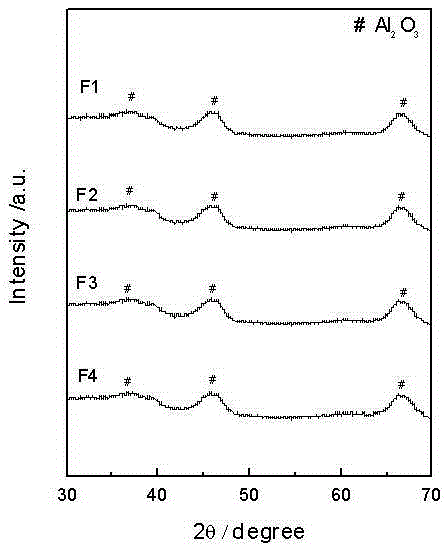
图2为本发明采用的选择性加氢催化剂的XRD谱图。
具体实施方式
下面结合附图及实施例对本发明作进一步描述。以下仅为本发明较佳的具体实施方式,但本发明的保护范围不局限于此,任何熟悉本技术领域的技术人员可轻易想到变化或替换,都应涵盖在本专利的保护范围内。
如图1所示含环戊二烯原料(101)与新鲜溶剂(102)和循环溶剂(111)在原料储罐1中混合后,组成加氢反应原料(103);调节各股物料的流量,使加氢原料中环戊二烯的质量分数为10~25%,溶剂中甲乙醚的质量分数为20~80%;加氢反应原料(103)经与氢气(104)混合后进入加氢反应器2,在选择性加氢催化剂的作用下,在反应器入口温度为40~60℃,反应压力为0.9~1.1 MPa,质量空速为2~4 h-1,氢气与环戊二烯的摩尔比为1.1~1.3的条件下,发生环戊二烯选择性加氢反应;生成的加氢反应流出物(105)在气液分离罐4中进行气液分离,分离出氢气(106)可循环使用;分离出液相加氢产物作为第一精馏塔6的进料(107),塔釜温度为215~255℃,塔顶温度为60~85℃,塔顶压力为0.50~0.65 MPa的条件下操作,塔底采出重组分杂质(108);第一精馏塔塔顶馏出物作为第二精馏塔的进料(109),塔釜温度为95~120℃,塔顶温度为55~80℃,塔顶压力为0.45~0.70 MPa的条件下操作,塔底采出环戊烯产品(110),塔顶馏出轻组分作为循环溶剂(111),返回原料储罐1中循环使用。
以下结合实施例进一步说明本发明的效果。
实施例1-4
催化剂制备步骤(1)
按表1所示重量称取六水硝酸镍加入500 ml去离子水,然后加入0.05 mol/L硝酸调节pH值至2-3,然后再按表1所示重量加入磷酸氢二铵、六水硝酸铈、一水柠檬酸,然后在室温下搅拌1小时,分别得到溶液A1-A4。
表 1
分别向溶液A1-A4中加入氢氧化铝粉77g、72g、67g和78g,使之分散在溶液中,分别得到浆液B1-B4,然后将所得浆液移入旋转蒸发仪中在85℃下蒸干水分,然后于120℃下烘干24小时,得到干胶C1-C4。
催化剂制备步骤(2)
将干胶C1-C4置于管式加热炉中,在连续流动的N2氛围中,于250-300℃下处理3-10小时,使干胶中的柠檬酸发生热分解反应,待达到所需的处理时间后,使温度降低至室温(25℃)取出,分别得到催化剂前驱体D1-D4。处理条件见表2。
表2
催化剂制备步骤(3)
将催化剂前驱体D1-D4与适量的硝酸、水、田菁粉混合捏合,挤条成型。挤条成型后,所得挤出物先于室温下干燥24小时,再于120℃烘干24小时,分别得到成型催化剂前驱体E1-E4。
催化剂制备步骤(4)
按表3所列的条件,将成型催化剂前驱体E1-E4分别置于管式加热炉中,在连续流动的H2氛围中,在350-400℃下还原5-10小时。待达到所需的还原时间后,使温度降低至室温(25℃),然后以氧气体积浓度为0.75%的O2/N2钝化气对催化剂进行钝化处理0.8小时,得到最终钝化态的催化剂F1-F4。
表3
采用X射线衍射(XRD)对钝化态催化剂F1-F4进行表征,结果表明催化剂上磷化镍处于无定形状态。图2示出了F1-F4催化剂的XRD谱图。
采用电感耦合等离子体发射光谱(ICP)测定钝化态催化剂组成。Ni担载量以Ni金属重量占总催化剂重量百分比计,P含量和Ce含量分别以P/Ni、Ce/Ni摩尔比表示。F1催化剂:Ni含量10.12%,P/Ni=2.35,Ce/Ni=0.01;F2催化剂:Ni含量12.51%,P/Ni=2.24,Ce/Ni=0.03;F3催化剂:Ni含量14.96%,P/Ni=2.06,Ce/Ni=0.05;F4催化剂:Ni含量10.23%,P/Ni=2.14,Ce/Ni=0.02。
实施例5-9
本发明各实施例所用的含环戊二烯原料来自于双环戊二烯裂解,其中环戊二烯的质量分数为98%,其余为双环戊二烯。
本发明各实施例采用的工艺流程如图1所示。表4列出了各实施例的加氢反应原料(103)的组成。
表4
表5列出了本发明各实施例加氢反应器的操作条件。
表5
为了说明本发明方法的效果,采用环戊二烯单程转化率、环戊烯的单程选择性和环戊烷的单程选择性表示本发明方法对环戊二烯选择性加氢的有益作用。
表6列出了本发明各实施例环戊二烯选择性加氢单程反应结果。本发明方法通过控制溶剂中甲乙醚的质量分数,使甲乙醚在本发明反应条件下吸收环戊二烯加氢反应放出的热量而气化,利用甲乙醚的气化潜热来控制催化剂床层温度,使加氢反应维持在较低的温度下进行。如表6所示,采用本发明方法可使加氢反应器出口温度控制在80~90℃,有利于本发明采用的选择性加氢催化剂充分发挥了自身优越的催化性能,因此将环戊二烯深度加氢生成环戊烷的单程选择性控制在0.5%以下,获得较高的环戊烯产品选择性。
表6
表7列出了本发明各实施例的加氢产物分离与精制工艺条件,工艺流程如图1所示。第一精馏塔的理论塔板数为15块,第二精馏塔的理论塔板数为65块。本发明各实施例所采用的精馏塔可以设置蒸馏塔板,也可以填装填料。板式塔和填料塔均为本领域技术人员所熟知的技术内容。本发明各实施例均采用填料塔,所用填料为金属鲍尔环填料。
表7
表8列出了环戊烯产品(110)和循环溶剂(111)的组成。采用本发明方法投建的装置达到稳态后,无需再补充新鲜溶剂。如表9所示,本发明的另一有益效果是,未反应的环戊二烯随循环溶剂返回了加氢反应器继续反应,可将环戊二烯的总转化率提高99%以上,环戊烯产品总收率提高至97%以上。
表8
表9
- 还没有人留言评论。精彩留言会获得点赞!