碱式氰尿酸锌粉末的制造方法及防锈颜料组合物的制造方法与流程
本发明涉及碱式氰尿酸锌粉末的制造方法及使用其的防锈颜料组合物的制造方法。
背景技术:
对于钢结构物用涂料、镜子的背面涂料等中使用的防锈颜料而言,需要无铅、铬。其背景是,根据2006年由欧盟实施的作为环境规定的RoHS指令(电子、电气设备中的特定有害物质的使用规定)及WEEE指令(废电气电子设备指令:关于产品的回收、再利用率),不能销售含有铅、汞、镉、六价铬、多溴联苯、多溴联苯醚这6个品种有害物质超过指定值的电子、电气设备。其中,六价铬、铅的情况下,设置1000ppm以下这样的限度。
由于日本经济产业省表明“作为企业应该进行的产品中含有的环境负荷物质的信息公开对象物质,RoHS指令中规定的6个品种是妥当的”,因此,该指令对应在日本也是必须的,另外,存在对象产品也从现在的电气电子群扩大到其他的产品群的趋势。此外,在日本,在1999年“关于特定化学物质向环境的排出量的把握等及管理的改善的促进的法律”(PRTR法、化管法、化学物质排出把握管理促进法)被法制化,政令指定对象物质354种物质被指定。铬化合物和铅化合物成为指定物质。
在日本,2014年度中废除了含有铅丹、碱式铬酸铅、铬酸锌、碱式铬酸锌钾、氰氨化铅、铅酸钙、碱式硫酸铅等铬化合物及铅化合物等防锈颜料的防锈涂料的JIS标准。因此,尽快地强烈需求防锈性能优异、且廉价的无铅、铬的防锈颜料。
作为无铅、铬的防锈颜料,磷酸锌、亚磷酸锌、钼酸锌等锌盐、磷酸铝、钼酸铝等铝盐、磷酸钙、钼酸钙等钙盐、氰氨化锌、氰氨化锌钙等氰氨化系化合物等被使用。
另外,作为其他锌盐,以惯用成分以及有机化合物的锌盐为基础的金属表面的防腐蚀被覆剂中,公开了氰尿酸的锌盐,作为氰尿酸锌的制造法,记载了下述内容:使氧化锌在沸水中与氰尿酸反应,此时,若添加有利的作为催化剂的少量的乙酸,则生成相应的氰尿酸锌(参照专利文献1。)。
作为其他制造方法,公开了下述碱式氰尿酸锌的制造法:以为10~80质量%(相对于糊剂)且成为尽可能少的水含量的能够混炼的糊剂的方式混合氧化锌和氰尿酸,对于得到的糊剂一边加热至50~250℃一边施加剪切作用(参照专利文献2。)。该专利文献2的实施例中,用装有叶片研磨器(blade mill)的桨叶干燥机于105~110℃对混合有脱盐水、氧化锌和氰尿酸的水分量为70质量%的糊剂进行加热,然后在减压下干燥,得到碱式氰尿酸锌。由于反应以糊剂进行,所以之后需要干燥。
另外,作为其他制造方法,公开了下述碱式氰尿酸锌的制造法:将以氰尿酸浓度相对于水为0.1~10.0质量%的方式配合选自氧化锌及碱式碳酸锌中的至少一种、氰尿酸和水而得的混合浆料在5~55℃的温度范围进行使用了分散介质的湿式分散(参照专利文献3。)。
这些碱式氰尿酸锌的制造法均是以含有水和碱式氰尿酸锌粒子的浆料或糊剂的状态进行反应。因此,在使用碱式氰尿酸锌作为防锈颜料时,必须将浆料或糊剂干燥后,进行干式粉碎而微粉末化。但是,浆料或糊剂由于水分量多,因此,干燥成本花费多。另外,由于干燥物成为大块,并且非常硬,因此进行微粉末化时耗费巨大的劳动力,具有粉碎成本非常高的缺点。
关于在防锈颜料中使用了氰尿酸锌的铁等的防锈涂料或防腐蚀被覆剂,以环氧树脂酯为基础的焙烧底漆的组成公开了在由脂肪酸含量为42%的环氧树脂-蓖麻油酸酯(混合二甲苯中为60%)400质量份、磷酸锌110质量份、微细滑石120质量份、二氧化钛(金红石)80质量份、重晶石193.5质量份、乙二醇15质量份、正丁醇15质量份、四氢化萘30质量份、高级芳香族化合物110质量份、非塑化脲树脂(丁醇中为65%)110质量份及混合二甲苯200质量份组成的清漆1373.5质量份中,添加26.5质量份氰尿酸锌(Zn31.7%)作为防腐蚀剂而得的组成。(参照专利文献1。)。
现有技术文献
专利文献
专利文献1:日本特公昭62-5194号公报
专利文献2:美国专利第4,507,270号说明书
专利文献3:日本再公表专利第2011/162353号小册子
技术实现要素:
发明所要解决的课题
本发明的课题在于解决上述现有技术的问题。即,本发明提供粉末状的碱式氰尿酸锌的高效且廉价的制造方法,所述制造方法通过不经由浆料或糊剂状态的反应,从而不需要粉碎工序,并且,提供使用了通过本发明得到的碱式氰尿酸锌粉末的防锈颜料组合物的制造方法。
用于解决课题的手段
为了解决上述课题,本申请的发明人们进行了深入研究,结果发现了通过将混合粉末在密闭或开放的条件下在30~300℃进行加热处理,来高效制造筛孔1000μm的筛余物小于1质量%的碱式氰尿酸锌粒子粉末的方法,所述混合粉末是包含氧化锌、氰尿酸及水的混合粉末,氧化锌相对于氰尿酸的摩尔比为2~3,且上述混合粉末的水分量为9~18质量%。
即,作为本发明的第1观点,为筛孔1000μm的筛余物小于1质量%的碱式氰尿酸锌粉末的制造方法,其特征在于,将混合粉末在密闭或开放的条件下在30~300℃的范围进行加热处理,所述混合粉末是包含氧化锌、氰尿酸及水的混合粉末,氧化锌相对于氰尿酸的摩尔比为2~3,且上述混合粉末的水分量为9~18质量%,
作为第2观点,为根据第1观点所述的筛孔1000μm的筛余物小于1质量%的碱式氰尿酸锌粉末的制造方法,其中,上述混合粉末是筛孔1000μm的筛余物小于1质量%、且筛孔400μm的筛余物小于10质量%的混合粉末,
作为第3观点,为根据第1观点或第2观点所述的筛孔1000μm的筛余物小于1质量%的碱式氰尿酸锌粉末的制造方法,其中,所得的碱式氰尿酸锌粒子的利用透射型电子显微镜观察所观察到的一次粒子的长轴为50~2000nm,且短轴为20~300nm,
作为第4观点,为根据第3观点所述的筛孔1000μm的筛余物小于1质量%的碱式氰尿酸锌粉末的制造方法,其中,上述氰尿酸锌粒子的长轴与上述短轴之比为2~10,
作为第5观点,为根据第1观点~第4观点的任一项所述的筛孔1000μm的筛余物小于1质量%的碱式氰尿酸锌粉末的制造方法,其中,上述加热处理利用具有混合机构和加热机构的粉体混合机进行,
作为第6观点,为防锈颜料组合物的制造方法,其具有下述(a)工序及(b)工序:
(a)工序:将混合粉末在密闭或开放的条件下在50~300℃的范围进行加热处理从而得到筛孔1000μm的筛余物小于1质量%的碱式氰尿酸锌粉末的工序,所述混合粉末是包含氧化锌、氰尿酸及水的混合粉末,氧化锌相对于氰尿酸的摩尔比为2~3,且上述混合粉末的水分量为9~18质量%;
(b)工序:将(a)工序中得到的筛孔1000μm的筛余物小于1质量%的氰尿酸锌粉末、粘合剂成分和稀释剂混合的工序,
作为第7观点,为根据第6观点所述的防锈颜料组合物的制造方法,其中,上述粘合剂为油性粘合剂或合成树脂粘合剂,
作为第8观点,为根据第6观点所述的防锈颜料组合物的制造方法,其中,上述稀释剂为选自水、松节油、矿物油精(Mineral Spirit)、甲苯、二甲苯、甲基乙基酮、甲基异丁基酮、乙酸溶纤剂、乙醇、丁醇、异丙醇、环己烷、乙酸乙酯、乙酸丁酯中的至少一种。
发明的效果
通过本发明得到的筛孔1000μm的筛余物小于1质量%的碱式氰尿酸锌粒子粉末的防锈性能优异,能够作为无铅、铬的无公害的防锈颜料使用。另外,由于所得的碱式氰尿酸锌粉末为白色,所以在用于防锈颜料组合物的情况下,几乎不对其他着色颜料造成影响,能够制备各种色调的防锈颜料组合物。
附图说明
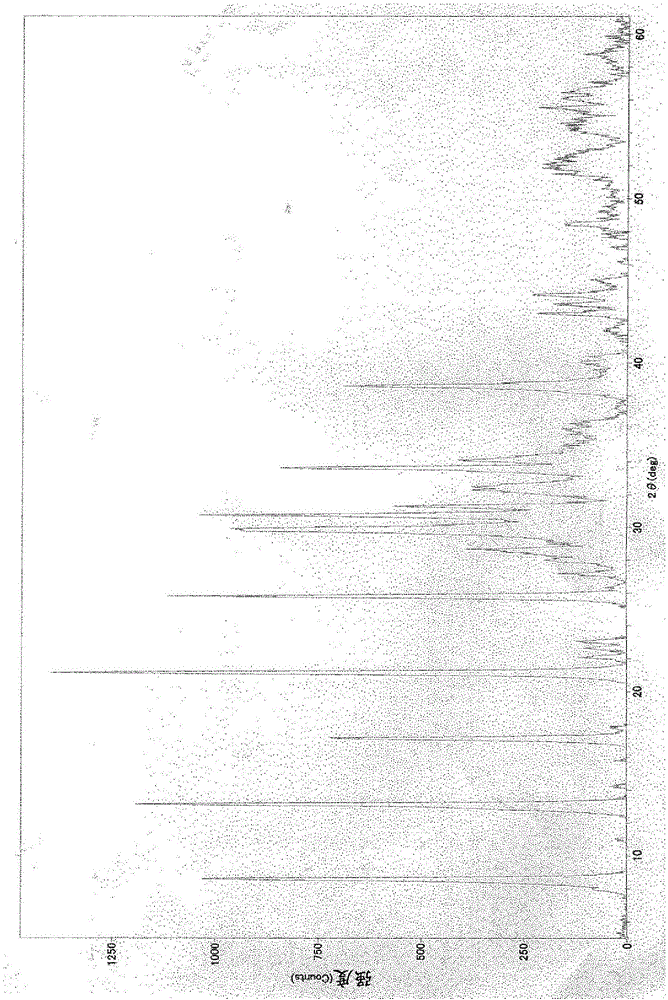
图1为实施例1的XRD衍射图案
图2为实施例1的透射型电子显微镜照片
图3为实施例2的透射型电子显微镜照片
图4为实施例12的激光法粒径测定图
图5为比较例5的激光法粒径测定图
具体实施方式
本发明为筛孔1000μm的筛余物小于1质量%的氰尿酸锌粉末的制造方法,其特征在于,将混合粉末在密闭或开放的条件下在30~300℃的范围进行加热处理,所述混合粉末是含有氧化锌、氰尿酸及水的混合粉末,氧化锌相对于氰尿酸的摩尔比为2~3,且上述混合粉末的水分量为9~18质量%。
本发明中,氧化锌可以使用市售的1种、2种及3种的任一者的氧化锌的粉末。另外,氰尿酸可以使用例如日产化学工业株式会社制的氰尿酸粉末。
本发明中,为包含氧化锌、氰尿酸及水的混合粉末,氧化锌相对于氰尿酸的摩尔比为2~3,且上述混合粉末的水分量为9~18质量%的混合粉末优选具有下述粒度分布:筛孔1000μm的筛余物小于1质量%,且筛孔400μm的筛余物小于10质量%。因此,作为原料使用的氧化锌及氰尿酸优选分别具有下述粒度分布:筛孔1000μm的筛余物小于1质量%,且筛孔400μm的筛余物小于10质量%。本发明中,筛余物可以如下求出:将试料放入规定的筛孔内径为200mm的不锈钢制筛上,将该筛放在Ro-tap震荡机上,由残留在筛上的试料的质量求出。
本发明中,氧化锌相对于氰尿酸的摩尔比为2~3,优选为2.5。混合粉末中的氧化锌相对于氰尿酸的摩尔比大于3时,反应后氧化锌残存多,故不优选。另外,上述摩尔比小于2时,反应后氰尿酸残存多,故不优选。另外,为了得到碱式氰尿酸锌的单一相,混合粉末中的氧化锌相对于氰尿酸的摩尔比优选为2.5。
本发明中,包含氧化锌、氰尿酸及水的混合粉末的水分量为9~18质量%。该水分量少于8%时,未反应的氧化锌和氰尿酸残存,故不优选。另外,上述混合粉末的水分量大于18质量%且至80质量%时,不是粉末,而变成湿的饼或糊剂,不能通过筛孔1000μm的筛,其筛余物为100质量%。进而,水分量大于80质量%时,成为高粘度浆料,在30~300℃的范围进行加热处理后,成为硬的饼或高粘度的糊剂。为了将反应物粉末化,进一步需要干燥和干式粉碎,所以生产效率差,不是廉价的制造方法。该水分量较优选为11~15质量%。
另外,本发明中,上述混合粉末的制备也可以选择下述任一方法:将氧化锌、氰尿酸和水同时添加并混合的方法;将氧化锌和氰尿酸预先混合,接着添加水并混合的方法;将氧化锌和水预先混合,接着添加氰尿酸并混合的方法;或者将氰尿酸和水预先混合,接着添加氧化锌并混合的方法。
本发明中,加热处理在密闭或开放的条件下在30~300℃进行。若在低于30℃温度进行加热处理,则反应变得非常缓慢,未反应的氧化锌和氰尿酸粉大量残存,故不优选。另外,加热处理虽然能够在300℃以下进行,但若超过100℃,则需要昂贵的高压装置。因此,为了以更低成本进行制造,较优选在不使用高压装置的50~100℃进行加热处理。
本发明中,对于加热处理而言,在工业上大量生产的情况下,优选使用具有混合机构和加热机构的粉体混合机进行。作为具体的例子,可举出亨舍尔混合机、lodige混合机、nauta混合机、回转炉等开放型或密闭型且能够进行搅拌混合的加热式反应槽。
对于通过本发明得到的碱式氰尿酸锌而言,筛孔1000μm的筛余物小于1质量%。另外,对于通过本发明得到的碱式氰尿酸锌而言,优选筛孔400μm的筛余物小于10质量%。
另外,对于通过本发明得到的碱式氰尿酸锌而言,优选利用透射型电子显微镜观察所观察到的一次粒子的长轴为50~2000nm,且短轴为20~300nm。而且,更优选上述长轴与短轴之比为2~10。
通过本发明得到的碱式氰尿酸锌是与国际公开第2011/162353号公报的实施例1中得到的碱式氰尿酸锌显示相同的粉末X射线衍射图案、以化学式Zn5(C3N3O3)2(OH)3·3H2O表示的化合物。
另外,通过本发明得到的碱式氰尿酸锌粉末的表面电荷在水系中在pH3~pH10的范围具有负电荷。因此,在酸性至碱性区域在水中的分散性良好,不仅如此,在制备水系防锈涂料时,与合成树脂、乳液等的相容性良好,能够得到稳定的水系防锈涂料。
本发明的防锈颜料组合物的制造方法具有下述(a)工序及(b)工序。
(a)工序:将混合粉末在密闭或开放的条件下在50~300℃的范围进行加热处理从而得到筛孔1000μm的筛余物小于1质量%的碱式氰尿酸锌粉末的工序,所述混合粉末是含有氧化锌、氰尿酸及水的混合粉末,氧化锌相对于氰尿酸的摩尔比为2~3,且上述混合粉末的水分量为9~18质量%;
(b)工序:将(a)工序中得到的筛孔1000μm的筛余物小于1质量%的氰尿酸锌粉末、粘合剂成分和稀释剂混合的工序。
在上述(b)工序中,作为粘合剂,可举出油性系粘合剂或合成树脂粘合剂。作为油性系粘合剂,可举出熟油(boiled oil)、亚麻籽油、大豆油、红花油、蓖麻油等。另外,作为合成树脂粘合剂,可举出苯二甲酸树脂、丙烯酸树脂、氨基树脂、环氧树脂、硅树脂、聚氨酯树脂、氟系树脂、氯乙烯树脂等。
另外,作为上述稀释剂,可举出水或松节油、矿物油精、甲苯、二甲苯、甲基乙基酮、甲基异丁基酮、乙酸溶纤剂、乙醇、丁醇、异丙醇、环己烷、乙酸乙酯、乙酸丁酯等有机溶剂。
本发明的防锈颜料组合物的制造方法中,筛孔1000μm的筛余物小于1质量%的氰尿酸锌粉末与粘合剂与稀释剂的配合比例是任意的,但例如通过配合30~80质量份的筛孔1000μm的筛余物小于1质量%的氰尿酸锌粉末、5~20质量份的粘合剂、2~50质量份的稀释剂,能够制造防锈性能优异的防锈颜料组合物。所得的防锈颜料组合物是具有粘性的浆料。
利用本发明得到的防锈颜料组合物是发挥与含有铅丹、氰氨化铅等有害物质的防锈颜料组合物同等或为其以上的防锈效果的无公害的防锈颜料组合物。该防锈颜料组合物由于不含铅、铬有害物质,因此可以作为无公害防锈颜料组合物应用于在人易于接触的车站建筑、道路的护栏等中使用的防锈涂料。
另外,由于利用本发明得到的碱式氰尿酸锌粉末为白色,因此利用本发明制造的防锈颜料组合物也可以为与着色颜料混合而着色为任意色的防锈颜料组合物。
利用本发明得到的防锈颜料组合物可以使用刷子、辊、喷枪等通常已知的涂布方法进行涂布。
另外,利用本发明得到的防锈颜料组合物与富锌涂料等长期耐久性的重防腐防锈组合物不同,是短期耐久性用途的防锈颜料组合物。因此,适于前述的车站建筑、道路的护栏等污垢显眼,以5~10年周期重新涂布的用途。
实施例
以下,基于合成例、实施例、比较例及参考例进行更详细地叙述,但本发明不受该实施例的任何限定。
(测定装置)
实施例及比较例中的分析以下述的装置、条件进行。
1)透射型电子显微镜观察:JEM-1010型(日本电子(株)制、加速电压100kV)
2)激光衍射法粒径测定:SALD-7000型((株)岛津制作所制),利用试料浓度为0.04质量%的水分散液进行测定。
3)粉末的粒度分布:从上开始组合设置筛孔1000μm、筛孔400μm、筛孔160μm、筛孔75μm、筛孔45μm的不锈钢制筛,使用Ro-tap震荡机筛分试料并计量各筛的筛余物。
4)比表面积测定:氮吸附法表面积测定装置Monosorb(Quantachrome社制),将试料于200℃干燥后进行测定。
5)粉体的水分量:将约2g试料加入瓷制坩埚并精确称量,然后由在110℃干燥后的质量算出水分量(质量%)。
6)粉末X射线衍射分析:使用粉末X射线衍射装置RINT Ultima型((株)リガク制)。
7)粘度:使用B型粘度计((株)东京计器制)。
8)Zeta电位:使用激光Zeta电位计ELS-6000(大塚电子(株)制)。
(实施例1)
将具有筛孔45μm的筛余物为0.01质量%以下的粒度的氧化锌粉末(堺化学(株)制2种氧化锌)480g和具有筛孔400μm的筛余物为0质量%、筛孔160μm的筛余物为1.1质量%、筛孔45μm的筛余物为82.6质量%的粒度的氰尿酸粉末(日产化学工业(株)制、水分量5.1质量%)320g装入容积为3升的聚乙烯制容器,进而添加101g纯水,将该容器置于震荡机上混合10分钟。通过该操作,制备了氧化锌/氰尿酸的摩尔比为2.5、水分量为13质量%、筛孔1000μm的筛余物为0质量%、筛孔400μm的筛余物为7.6质量%、筛孔160μm的筛余物为47.7质量%、筛孔45μm的筛余物为97.1质量%的原料粉末901g。将装有901g该原料粉末的聚乙烯制容器密闭,然后放入70℃的干燥机中,加热处理210分钟。在210分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:从干燥机取出聚乙烯制容器,置于震荡机上,将原料粉末充分混合1分钟,送回至干燥机。加热处理后所得的物质是水分量为13质量%的干爽的白色粉末。进行该白色粉末的粉末X射线衍射分析,结果是,如图1所示,仅检测到碱式氰尿酸锌的衍射峰,未检测到归属于原料氰尿酸及氧化锌的衍射峰。利用透射型电子显微镜对所得的白色粉末进行观察的结果是,长轴为60~1000nm、短轴为40~140nm、长轴与短轴的轴比为5~8的微粒被确认。另外,所得的白色粉末的110℃干燥后的比表面积为18m2/g。另外,对于白色粉末的粒度而言,筛孔1000μm的筛余物为0质量%,筛孔400μm的筛余物为1.0质量%,筛孔160μm的筛余物为41.8质量%,筛孔45μm的筛余物为90.5质量%。将该白色粉末850g放入110℃的干燥机中,干燥至水分量为0.6%。使该白色粉末分散于纯水中制作5质量%的浆料,结果,pH为6.5。将该5质量%的浆料用1质量%的氢氧化钠水溶液调节为pH10,然后一边用1质量%的盐酸水溶液降低pH一边测定Zeta电位,结果,pH9.5时Zeta电位为-19mV,pH8.9时Zeta电位为-21mV,pH6.9时Zeta电位为-26mV,pH4.8时Zeta电位为-24mV,pH4.0时Zeta电位为-16mV。在容积为50毫升的聚乙烯制容器中投入所得的白色粉末的110℃干燥品2g和纯水38g后,进行密闭,然后用40KHz的超声波清洗机(W-222本田电子(株)制)分散处理5分钟,制作5质量%的浆料。对于该浆料而言,未检测到沉降物,另外用激光衍射法对粒径进行测定的结果是,平均粒径为1.0μm,最大粒径为45.6μm,在水中显示优异的分散性。
(实施例2)
分取28.8g利用与实施例1相同的操作制作的氧化锌/氰尿酸的摩尔比为2.5、水分量为13质量%的原料粉末。将该原料粉末装入容积250毫升的聚乙烯制容器中并进行密闭后,装入80℃的干燥机中,加热处理150分钟。在150分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯制容器从干燥机取出,置于震荡机上,将原料粉末充分混合1分钟,送回至干燥机。加热处理后所得的物质是水分量为13质量%的干爽的白色粉末。进行该白色粉末的粉末X射线衍射分析,结果是,与实施例1同样的,仅检测到碱式氰尿酸锌的衍射峰,未检测到归属于原料氰尿酸及氧化锌的衍射峰。利用透射型电子显微镜对所得的白色粉末进行观察的结果是,长轴为60~1000nm、短轴为40~150nm、长轴与短轴之比为5~8的微粒被确认。另外,110℃干燥后的比表面积为17m2/g。另外,关于白色粉末的粒度,筛孔1000μm的筛余物为0质量%,筛孔400μm的筛余物为5.9质量%,筛孔160μm的筛余物为87.7质量%,筛孔45μm的筛余物为93.1质量%。在容积为50毫升的聚乙烯制容器中投入所得的白色粉末的110℃干燥品2g和纯水38g后,进行密闭,然后用40KHz的超声波清洗机(W-222本田电子(株)制)分散处理5分钟,制作5质量%的浆料。对于该浆料,用激光衍射法测定粒径的结果是,平均粒径为3.4μm,最大粒径为195.8μm。
(实施例3)
分取28.8g利用与实施例1相同的操作制作的氧化锌/氰尿酸的摩尔比为2.5、水分量为13质量%的原料粉末。将该原料粉末装入容积为250毫升的聚乙烯制容器中并进行密闭后,装入90℃的干燥机中,加热处理150分钟。在150分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯制容器从干燥机取出,置于震荡机上,将原料粉末充分混合1分钟,送回至干燥机。加热处理后所得的物质是水分量为13质量%的干爽的白色粉末。进行该白色粉末的粉末X射线衍射分析,结果是,与实施例1同样的,仅检测到碱式氰尿酸锌的衍射峰,未检测到归属于原料氰尿酸及氧化锌的衍射峰。利用透射型电子显微镜对所得的白色粉末进行观察的结果是,长轴为100~1000nm、短轴为40~200nm、长轴与短轴之比为5~8的微粒被确认。另外,110℃干燥后的比表面积为14m2/g。另外,关于白色粉末的粒度,筛孔1000μm的筛余物为0质量%,筛孔400μm的筛余物为6.4质量%,筛孔160μm的筛余物为67.9质量%,筛孔45μm的筛余物为98.5质量%。在容积为50毫升的聚乙烯制容器中投入所得的白色粉末的110℃干燥品2g和纯水38g后,进行密闭,然后用40KHz的超声波清洗机(W-222本田电子(株)制)分散处理5分钟,制作5质量%的浆料。对于该浆料,用激光衍射法测定粒径的结果是,平均粒径为6.9μm,最大粒径为297.0μm。
(实施例4)
将实施例1的氧化锌粉末(堺化学(株)制2种氧化锌)15.0g和实施例1的氰尿酸粉末(日产化学工业(株)制、水分量5.1质量%)10.0g装入容积为250毫升的聚乙烯制容器中,进而添加纯水2.2g,将该容器置于震荡机上混合10分钟。通过该操作,制备氧化锌/氰尿酸的摩尔比为2.5、水分量为10质量%、筛孔1000μm的筛余物为0质量%、筛孔400μm的筛余物为4.6质量%、筛孔160μm的筛余物为40.4质量%、筛孔45μm的筛余物为99.3质量%的原料粉末27.2g。将装有该原料粉末的聚乙烯制容器密闭,装入80℃的干燥机中,加热处理150分钟。在150分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯制容器从干燥机取出,置于震荡机上,将原料粉末充分混合1分钟,送回至干燥机。加热处理后所得的物质是水分量为10%的干爽的白色粉末。进行该白色粉末的粉末X射线衍射分析,结果是,与实施例1同样的,仅检测到碱式氰尿酸锌的衍射峰,未检测到归属于原料氰尿酸及氧化锌的衍射峰。利用透射型电子显微镜对所得的白色粉末进行观察的结果是,长轴为60~1000nm、短轴为40~140nm、长轴与短轴之比为5~8的微粒被确认。另外,110℃干燥后的比表面积为17m2/g。另外,关于白色粉末的粒度,筛孔1000μm的筛余物为0质量%,筛孔400μm的筛余物为5.2质量%,筛孔160μm的筛余物为54.1质量%,筛孔45μm的筛余物为98.2质量%。在容积为50毫升的聚乙烯制容器中投入所得的白色粉末的110℃干燥品2g和纯水38g后,进行密闭,然后用40KHz的超声波清洗机(W-222本田电子(株)制)分散处理5分钟,制作5质量%的浆料。对于该浆料,用激光衍射法测定粒径的结果是,平均粒径为6.7μm,最大粒径为195.8μm。
(实施例5)
分取28.8g利用与实施例1相同的操作制作的氧化锌/氰尿酸的摩尔比为2.5、水分量为13质量%的原料粉末。将该原料粉末装入容积为120毫升的氟系树脂制容器中并密闭后,装入150℃的干燥机中,加热处理50分钟。加热处理后,冷却至室温,取出该聚乙烯制容器并置于震荡机上,混合原料粉末1分钟。将该氟系树脂制容器再次放入150℃的干燥机中,加热处理50分钟。反复进行3次该加热处理的操作,合计进行150分钟150℃的加热处理。加热处理后所得的物质是水分量为13质量%的干爽的白色粉末。进行该白色粉末的粉末X射线衍射分析,结果是,与实施例1同样的,仅检测到碱式氰尿酸锌的衍射峰,未检测到归属于原料氰尿酸及氧化锌的衍射峰。利用透射型电子显微镜对所得的白色粉末进行观察的结果是,长轴为100~800nm、短轴为100~400nm、长轴与短轴之比为2~5的微粒被确认。另外,110℃干燥后的比表面积为10m2/g。另外,关于白色粉末的粒度,筛孔1000μm的筛余物为0质量%,筛孔400μm的筛余物为9.0质量%,筛孔160μm的筛余物为43.7质量%,筛孔45μm的筛余物为99.1质量%。在容积为50毫升的聚乙烯制容器中投入所得的白色粉末的110℃干燥品2g和纯水38g后,进行密闭,然后用40KHz的超声波清洗机(W-222本田电子(株)制)分散处理5分钟,制作5质量%的浆料。对于该浆料,用激光衍射法测定粒径的结果是,平均粒径为11.7μm、最大粒径为129.1μm。
(实施例6)
向实施例1的氰尿酸粉末(日产化学工业(株)制、水分量5.1质量%)320g添加纯水123g。此时的氰尿酸粉末为干爽的粉。在该氰尿酸粉末中加入实施例1的氧化锌粉末(堺化学(株)制2种氧化锌)480g并进行混合,制备氧化锌/氰尿酸的摩尔比为2.5、水分量为15质量%、筛孔400μm的筛余物为7.3质量%、筛孔160μm的筛余物为84.8质量%、筛孔45μm的筛余物为98.6质量%的原料粉末923g。将该原料粉装入容积为3升的聚乙烯制容器并密闭后,装入80℃的干燥机中,加热处理270分钟。在270分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯制容器从干燥机取出,置于震荡机上,将原料粉末充分混合1分钟,送回至干燥机。加热处理后所得的物质是水分量为15质量%的干爽的白色粉末。进行该白色粉末的粉末X射线衍射分析,结果是,与实施例1同样的,仅检测到碱式氰尿酸锌的衍射峰,未检测到归属于原料氰尿酸及氧化锌的衍射峰。利用透射型电子显微镜对所得的白色粉末进行观察的结果是,长轴为60~1000nm、短轴为40~150nm、长轴与短轴之比为5~8的微粒被确认。另外,110℃干燥后的比表面积为16m2/g。另外,关于白色粉末的粒度,筛孔1000μm的筛余物为0质量%,筛孔400μm的筛余物为3.3质量%,筛孔160μm的筛余物为43.6质量%,筛孔45μm的筛余物为82.4质量%。在容积为50毫升的聚乙烯制容器中投入所得的白色粉末的110℃干燥品2g和纯水38g后,进行密闭,然后用40KHz超声波清洗机(W-222本田电子(株)制)分散处理5分钟,制作5质量%的浆料。对于该浆料,用激光衍射法测定粒径的结果是,平均粒径为10.8μm,最大粒径为159.0μm。
(实施例7)
将实施例1的氧化锌粉末(堺化学(株)制2种氧化锌)5.3kg和实施例1的氰尿酸粉末(日产化学工业(株)制、水分量0%)3.4kg装入开放型的lodige混合机装置(M-20:中央机工(株)制)后,一边使混合机的主轴以230rpm旋转一边进行3分钟预混合。接着,将lodige混合机装置的夹套部的冷却水更换为110℃的蒸汽,使原料粉末的温度升温至80℃后,以26g/分钟经58分钟在装置内喷雾纯水1.5kg。该时刻装置内的原料粉末的水分量为13质量%,关于原料粉末的粒度,筛孔1000μm的筛余物为0质量%,筛孔400μm的筛余物为2.1质量%,筛孔160μm的筛余物为64.8质量%,筛孔45μm的筛余物为95.0质量%。进而一边以230rpm使混合机的主轴旋转,一边于80℃对原料粉末进行60分钟加热处理,然后将经加热处理的粉末从装置排出。被取出的粉末是水分量为9质量%的干爽的白色粉末。进行该白色粉末的粉末X射线衍射分析,结果是,与实施例1同样的,仅检测到碱式氰尿酸锌的衍射峰,未检测到归属于原料氰尿酸及氧化锌的衍射峰。利用透射型电子显微镜对所得的白色粉末进行观察的结果是,长轴为200~600nm、短轴为30~100nm、长轴与短轴之比为5~7的微粒被确认。另外,110℃干燥后的比表面积为15m2/g。另外,关于白色粉末的粒度,筛孔1000μm的筛余物为0质量%,筛孔400μm的筛余物为4.6质量%,筛孔160μm的筛余物为39.3质量%,筛孔45μm的筛余物为95.7质量%。在容积为50毫升的聚乙烯制容器中投入所得的白色粉末的110℃干燥品2g和纯水38g后,进行密闭,然后用40KHz超声波清洗机(W-222本田电子(株)制)分散处理5分钟,制作5质量%的浆料。对于该浆料,用激光衍射法测定粒径的结果是,平均粒径为8.4μm,最大粒径为159.0μm。
(实施例8)
将实施例1的氧化锌粉末(堺化学(株)制2种氧化锌)15.0g和实施例1的氰尿酸粉末(日产化学工业(株)制、水分量5.1质量%)10.0g装入容积为250毫升的聚乙烯制容器,进而添加1.3g纯水,将该容器置于震荡机上混合10分钟。通过该操作,制备氧化锌/氰尿酸的摩尔比为2.5、水分量为7质量%、筛孔1000μm的筛余物为0质量%、筛孔400μm的筛余物为3.0质量%、筛孔160μm的筛余物为45.6质量%、筛孔45μm的筛余物为98.0质量%的原料粉末26.3g。将装有该原料粉末并进行了密闭的聚乙烯制容器装入90℃的干燥机中,加热处理150分钟。在150分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯容器从干燥机取出,置于震荡机上,将原料粉末充分混合1分钟,送回至干燥机。加热处理后所得的物质是水分量为7%的干爽的白色粉末。进行该白色粉末的粉末X射线衍射分析,结果是,观察到碱式氰尿酸锌的衍射峰,除此之外,检测到归属于原料氰尿酸及氧化锌的弱的衍射峰。另外,关于白色粉末的粒度,筛孔400μm的筛余物为2.0质量%,筛孔160μm的筛余物为31.8质量%,筛孔45μm的筛余物为90.0质量%。
(实施例9)
分取28.8g利用与实施例1相同的操作制作的氧化锌/氰尿酸的摩尔比为2.5、水分量为13质量%的原料粉末。将该原料粉末装入容积为250毫升的聚乙烯制容器中并进行密闭后,放入40℃的干燥机中,加热处理300分钟。在300分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯制容器从干燥机取出,置于震荡机上,将原料粉末充分混合1分钟,送回至干燥机。加热处理后所得的物质是水分量为13质量%的干爽的白色粉末。进行该白色粉末的粉末X射线衍射分析,结果是,观察到碱式氰尿酸锌的衍射峰,除此之外,检测到归属于原料氰尿酸及氧化锌的弱的衍射峰。用透射型电子显微镜对白色粉末进行观察的结果是,长轴为60~1000nm、短轴为40~150nm、长轴与短轴之比为5~8的微粒被确认。另外,110℃干燥后的比表面积为17m2/g。另外,关于白色粉末的粒度,筛孔1000μm的筛余物为0质量%,筛孔400μm的筛余物为5.1质量%,筛孔160μm的筛余物为46.6质量%,筛孔45μm的筛余物为95.5质量%。
(实施例10)
将实施例1的氧化锌粉末(堺化学(株)制2种氧化锌)12.8g和实施例1的氰尿酸粉末(日产化学工业(株)制、水分量5.1质量%)10.5g装入容积为250毫升的聚乙烯制容器,进而添加纯水3.0g,将该容器置于震荡机上混合10分钟。通过该操作,制备氧化锌/氰尿酸的摩尔比为2.0、水分量为13质量%、筛孔1000μm的筛余物为0质量%、筛孔400μm的筛余物为9.8质量%、筛孔160μm的筛余物为75.0质量%、筛孔45μm的筛余物为99.2质量%的原料粉末26.3g。将装有该原料粉末的聚乙烯制容器密闭后,装入80℃的干燥机中,加热处理150分钟。在150分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯容器从干燥机取出,置于震荡机上,将原料粉末充分混合1分钟,送回至干燥机。加热处理后所得的物质是水分量为13质量%的干爽的白色粉末。进行该白色粉末的粉末X射线衍射分析,结果是,观察到碱式氰尿酸锌的衍射峰,除此之外,检测到归属于原料氰尿酸的弱的衍射峰。另外,关于白色粉末的粒度,筛孔1000μm的筛余物为0质量%,筛孔400μm的筛余物为5.8质量%,筛孔160μm的筛余物为27.5质量%,筛孔45μm的筛余物为96.0质量%。
(实施例11)
将实施例1的氧化锌粉末(堺化学(株)制2种氧化锌)15.0g和实施例1的氰尿酸粉末(日产化学工业(株)制、水分量5.1质量%)8.3g装入容积为250毫升的聚乙烯制容器,进而添加纯水3.1g,将该容器置于震荡机上混合10分钟。通过该操作,制备氧化锌/氰尿酸的摩尔比为3.0、水分量为13质量%、筛孔1000μm的筛余物为0质量%、筛孔400μm的筛余物为7.1质量%、筛孔160μm的筛余物为80.7质量%、筛孔45μm的筛余物为99.8质量%的原料粉末26.4g。将装有该原料粉末的聚乙烯制容器密闭后,装入80℃的干燥机中,加热处理150分钟。在150分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯容器从干燥机取出,置于震荡机上,将原料粉末充分混合1分钟,送回至干燥机。加热处理后所得的物质是水分量为13质量%的干爽的白色粉末。进行该白色粉末的粉末X射线衍射分析,结果是,观察到碱式氰尿酸锌的衍射峰,除此之外,检测到归属于原料氧化锌的衍射峰。另外,关于白色粉末的粒度,筛孔1000μm的筛余物为0质量%,筛孔400μm的筛余物为5.6质量%,筛孔160μm的筛余物为32.2质量%,筛孔45μm的筛余物为95.8质量%。
(实施例12)
在容积为3升的聚乙烯制容器中向实施例1的氰尿酸粉末(日产化学工业(株)制、水分量5.1质量%)401g添加纯水130g并进行密闭后,置于震荡机上,混合5分钟。此时的氰尿酸粉末为干爽的粉。在装有该氰尿酸粉末的容积为3升的聚乙烯制容器中加入实施例1的氧化锌粉末(堺化学(株)制2种氧化锌)600g并进行混合、密闭,然后置于震荡机上,混合30分钟,制备氧化锌/氰尿酸的摩尔比为2.5、水分量为13质量%、筛孔400μm的筛余物为5.0质量%、筛孔160μm的筛余物为86.8质量%、筛孔45μm的筛余物为99.0质量%的原料粉末1131。将装有该原料粉的容积为3升的聚乙烯制容器密闭后,装入90℃的干燥机中,加热处理150分钟。在150分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯制容器从干燥机取出,置于震荡机上,将原料粉末充分混合1分钟,送回至干燥机。加热处理后所得的物质是水分量为11质量%的干爽的白色粉末。进行该白色粉末的粉末X射线衍射分析,结果是,与实施例1同样的,仅检测到碱式氰尿酸锌的衍射峰,未检测到归属于原料氰尿酸及氧化锌的衍射峰。利用透射型电子显微镜对所得的白色粉末进行观察的结果是,长轴为80~1200nm、短轴为30~200nm、长轴与短轴之比为5~8的微粒被确认。另外,110℃干燥后的比表面积为14m2/g。另外,关于白色粉末的粒度,筛孔1000μm的筛余物为0质量%,筛孔400μm的筛余物为9.1质量%,筛孔160μm的筛余物为41.5质量%,筛孔45μm的筛余物为99.5质量%。在容积为50毫升的聚乙烯制容器中投入所得的白色粉末的110℃干燥品2g和纯水38g后,进行密闭,然后用40KHz超声波清洗机(W-222本田电子(株)制)分散处理5分钟,制作5质量%的浆料。对于该浆料,未检测到沉降物,另外用激光衍射法对粒径进行测定的结果是,平均粒径为2.0μm,最大粒径为45.6μm,在水中显示优异的分散性。
(比较例1)
将实施例1的氧化锌粉末(堺化学(株)制2种氧化锌)15.0g和实施例1的氰尿酸粉末(日产化学工业(株)制、水分量5.1质量%)10.0g装入容积250毫升的聚乙烯制容器,进而添加纯水6.2g,将该容器置于震荡机上混合10分钟。通过该操作,制备氧化锌/氰尿酸的摩尔比为2.5、水分量为20质量%的混合物。所得的混合物为数mm~1cm的凝集块,筛孔1000μm的筛余物为100质量%。将装有该凝集块的容积250毫升的聚乙烯制容器密闭后,装入80℃的干燥机中,加热处理150分钟。在150分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯制容器从干燥机取出,置于震荡机上,充分震荡凝集块1分钟,送回至干燥机。加热处理后所得的物质是数mm~1cm的白色的硬的凝集块,筛孔1000μm的筛余物为100质量%。将该白色的硬的凝集块用乳钵粉碎后,进行粉末X射线衍射分析,结果,观察到碱式氰尿酸锌的衍射峰,除此之外,检测到归属于原料氰尿酸及氧化锌的衍射峰。
(比较例2)
将实施例1的氧化锌粉末(堺化学(株)制2种氧化锌)15.0g和实施例1的氰尿酸粉末(日产化学工业(株)制、水分量5.1质量%)10.0g装入容积为250毫升的聚乙烯制容器中,进而添加纯水16.0g,将该容器置于震荡机上混合10分钟。通过该操作,制备氧化锌/氰尿酸的摩尔比为2.5、水分量为40质量%的混合物。所得的混合物为直径2~3cm的湿的饼,筛孔1000μm的筛余物为100质量%。将装有该湿的饼的容积250毫升的聚乙烯制容器密闭后,装入80℃的干燥机中,加热处理150分钟。在150分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯制容器从干燥机取出,置于震荡机上,充分震荡饼1分钟,送回至干燥机。加热处理后所得的物质是直径2~3cm的白色的硬且湿的饼,筛孔1000μm的筛余物为100质量%。将该白色的湿的饼于50℃进行干燥,用乳钵粉碎后,进行粉末X射线衍射分析,结果,观察到碱式氰尿酸锌的衍射峰,除此之外,检测到归属于原料氰尿酸及氧化锌的衍射峰。
(比较例3)
将实施例1的氧化锌粉末(堺化学(株)制2种氧化锌)15.0g和实施例1的氰尿酸粉末(日产化学工业(株)制、水分量5.1质量%)10.0g装入容积250毫升的聚乙烯制容器中,进而添加纯水47.0g,将该容器置于震荡机上混合10分钟。通过该操作,制备氧化锌/氰尿酸的摩尔比为2.5、水分量为66质量%的混合物。所得的混合物为直径约为5cm的圆盘状的湿的白色饼,筛孔1000μm的筛余物为100质量%。将装有该白色的湿的饼的容积250毫升的聚乙烯制容器密闭后,装入80℃的干燥机中,加热处理150分钟。在150分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯制容器从干燥机取出,置于震荡机上,充分震荡饼1分钟,送回至干燥机。加热处理后所得的物质是直径约为5cm的圆盘状的白色的硬且湿的饼,筛孔1000μm的筛余物为100质量%。将该白色的硬且湿的饼于50℃干燥,用乳钵粉碎后,进行粉末X射线衍射分析,结果,观察到碱式氰尿酸锌的衍射峰,除此之外,检测到归属于原料氰尿酸及氧化锌的衍射峰。
(比较例4)
将实施例1的氧化锌粉末(堺化学(株)制2种氧化锌)15.0g和实施例1的氰尿酸粉末(日产化学工业(株)制、水分量5.1质量%)10.0g装入容积250毫升的聚乙烯制容器中,进而添加纯水57.0g,将该容器置于震荡机上混合10分钟。通过该操作,制备氧化锌/氰尿酸的摩尔比为2.5、水分量为70质量%的混合物。所得的混合物是粘度为90,000mPa·s的糊剂,筛孔1000μm的筛余物为100质量%。将装有该糊剂的容积为250毫升的聚乙烯制容器密闭后,装入80℃的干燥机中,加热处理150分钟。在150分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯制容器从干燥机取出,置于震荡机上,将糊剂充分震荡1分钟,送回至干燥机。加热处理后所得的物质是直径约为5cm的圆盘状的白色的硬且湿的饼,筛孔1000μm的筛余物为100质量%。将该白色的硬且湿的饼于50℃干燥,用乳钵粉碎后,进行粉末X射线衍射分析,结果,观察到碱式氰尿酸锌的衍射峰,除此之外,检测到归属于原料氰尿酸及氧化锌的衍射峰。在容积为50毫升的聚乙烯制容器中投入所得的白色粉末的110℃干燥品2g和纯水38g后,进行密闭,然后用40KHz超声波清洗机(W-222本田电子(株)制)分散处理5分钟,制作5质量%的浆料。该浆料中存在多个沉降物形式的约5mm的块。对于该浆料,用激光衍射法测定粒径的结果是,平均粒径为325.6μm,最大粒径为500μm以上,为测定范围以上的尺寸。
(比较例5)
将实施例1的氧化锌粉末(堺化学(株)制2种氧化锌)15.0g和实施例1的氰尿酸粉末(日产化学工业(株)制、水分量5.1质量%)10.0g装入容积为250毫升的聚乙烯制容器,进而添加纯水98.5g,将该容器置于震荡机上混合10分钟。通过该操作,制备氧化锌/氰尿酸的摩尔比为2.5、水分量为80质量%的混合物。所得的混合物是粘度为500mPa·s的高粘度浆料。关于该浆料,筛孔1000μm的筛余物为97.1质量%,筛孔400μm的筛余物为97.9质量%,筛孔160μm的筛余物为99.2质量%,筛孔175μm的筛余物为100质量%。将装有该高粘度浆料的容积250毫升的聚乙烯制容器密闭后,装入80℃的干燥机中,加热处理150分钟。在150分钟的加热处理期间,以30分钟一次的比例反复进行下述操作:将聚乙烯制容器从干燥机取出,置于震荡机上,将浆料充分震荡1分钟,送回至干燥机。加热处理后所得的物质是粘度为100,000mPa·s以上的白色糊剂,筛孔1000μm的筛余物为100质量%。将该白色糊剂在50℃干燥,用乳钵粉碎后,进行粉末X射线衍射分析,结果,仅检测到碱式氰尿酸锌的衍射峰,未检测到归属于原料氰尿酸及氧化锌的衍射峰。在容积为50毫升的聚乙烯制容器中投入所得的白色粉末的110℃干燥品2g和纯水38g后,进行密闭,然后用40KHz超声波清洗机(W-222本田电子(株)制)分散处理5分钟,制作5质量%的浆料,但浆料中存在多个沉降物形式的约5mm的块。对于该浆料,用激光衍射法测定粒径的结果是,平均粒径为300.8μm、最大粒径为500μm以上,是测定范围以上的尺寸。
[表1]
(防锈评价)
基于JIS-K5674,在500毫升的广口玻璃瓶中装入フタリット(注册商标)清漆(透明涂料:関西ペイント(株)制)96g、作为分散剂的DISPERBYK-103(ビックケミー·ジャパン(株)制)4g、作为体质颜料的试剂硫酸钡80g及实施例7中得到的碱式氰尿酸锌粉末的110℃干燥品20g,用高速搅拌机(分散机)进行混合后,使用均化器以转速20,000rpm分散5分钟,制作防锈性能评价用涂料200g。另外,作为比较,基于JIS-K5674,在广口玻璃瓶中装入フタリット(注册商标)清漆(透明涂料:関西ペイント(株)制)96g、作为分散剂的DISPERBYK-103(ビックケミー·ジャパン(株)制)4g、作为体质颜料的试剂硫酸钡80g,用高速搅拌机分散并混合后,用均化器以转速20000rpm分散5分钟,制作比较用涂料180g。
在利用耐水研磨纸制备的钢板SPCC-SS(纵150mm×横70mm×厚0.8mm)上刷涂所制备的上述防锈性能评价用涂料1.0g后,于40℃干燥2小时,然后在最初的涂膜上刷涂相同的涂料1.0g,进一步于40℃使其干燥2小时,制作3枚形成有涂膜的钢板。用刀将该钢板的涂膜面划伤后,用保护封条将里面覆盖,制作试验片。将3枚试验片分别设置于复合循环试验机(スガ试验机(株)制)内,将下述操作作为1个循环(需要时间为6小时):于30℃喷雾5质量%的食盐水5分钟后,于30℃、湿度95%进行1.5小时湿润试验,接着于50℃热风干燥2小时,然后于30℃温风干燥2小时。进行36次该循环后,取出试验片,用自来水清洗后,于25℃进行干燥。然后,测定试验片的突起宽度。其结果是,涂布了防锈性能评价用涂料的试验片的最大突起宽度为2mm,比JIS标准的规定的3mm小,因此判断为没有突起,确认到具有优异的防锈性能。另一方面,同样地涂布有比较用涂料的试验片的最大突起宽度为10mm。
产业上的可利用性
根据本发明,能够高效且廉价地制造防锈性能高、不需要粉碎工序的粉末状的碱式氰尿酸锌,使用所得的碱式氰尿酸锌粉末能够制造防锈性能优异的无铅、铬的无公害防锈涂料。
- 还没有人留言评论。精彩留言会获得点赞!