树脂膜以及树脂膜的制造方法与流程
本发明涉及树脂膜以及树脂膜的制造方法。
背景技术:
已知有以下的技术:在包含具有结晶性的含有脂环式结构的聚合物的树脂的膜中,通过加热该膜从而将含有脂环式结构的聚合物晶化的技术(参考专利文献1及2)。由包含像这样晶化了的含有脂环式结构的聚合物的树脂形成的膜通常耐热性优异。
另一方面,对于树脂膜,根据用途存在有要求耐折性、低吸水性以及耐热性这些特性的情况。因此,一直以来都在进行着对这些特性优异的树脂膜的开发。
现有技术文献
专利文献
专利文献1:日本特开2002-194067号公报;
专利文献2:日本特开2013-10309号公报;
专利文献3:日本特开2006-309266号公报。
技术实现要素:
发明要解决的问题
根据本发明人的研究明确,包含含有脂环式结构的聚合物的树脂通常低吸水性优异。此外,在包含含有脂环式结构的聚合物的树脂中,特别是包含具有结晶性的含有脂环式结构的聚合物的树脂如上所述那样通常耐热性优异。然而,利用包含具有结晶性的含有脂环式结构的聚合物的树脂所制造的现有技术的膜的耐折性差。
本发明是鉴于上述课题而创立的,因此其目的在于提供耐折性、低吸水性及耐热性均优异的树脂膜以及该树脂膜的制造方法。
用于解决问题的方案
本发明人为了解决上述课题进行了深入研究,结果发现通过在对由包含具有结晶性的含有脂环式结构的聚合物的树脂形成的膜进行了拉伸后对其进行加热,从而能够实现耐折性、低吸水性以及耐热性均优异的树脂膜,进而完成了本发明。
即,本发明如下所述。
[1]一种树脂膜,是由树脂形成的,上述树脂包含具有结晶性的含有脂环式结构的聚合物,
上述树脂膜的耐折度为2000次以上,吸水率为0.1%以下,且耐热温度为180℃以上。
[2]一种树脂膜,是由树脂形成的,上述树脂包含具有结晶性的含有脂环式结构的聚合物,
上述含有脂环式结构的聚合物的晶化度为10%以上,
上述树脂膜的耐折度为2000次以上,吸水率为0.1%以下,且平滑。
[3]根据[1]或[2]所述的树脂膜,其中,面取向系数的绝对值为0.01以上。
[4]一种树脂膜的制造方法,为[1]~[3]中任一项所述的树脂膜的制造方法,包含:
第一工序,将由树脂形成的拉伸前膜拉伸而得到拉伸膜,上述树脂包含具有结晶性的含有脂环式结构的聚合物,及
第二工序,在上述第一工序后将上述拉伸膜加热。
[5]根据[4]所述的树脂膜的制造方法,其中,在上述第一工序中,在(tg-30℃)以上、(tg+60℃)以下的温度范围将上述拉伸前膜拉伸,在此,tg为上述树脂的玻璃化转变温度。
[6]根据[4]或[5]所述的树脂膜的制造方法,其中,在上述第二工序中,将上述拉伸膜在上述含有脂环式结构的聚合物的玻璃化转变温度以上、上述含有脂环式结构的聚合物的熔点以下的温度加热。
[7]根据[4]~[6]中任一项所述的树脂膜的制造方法,其中,在上述第二工序中,在将上述拉伸膜的至少两边保持而使拉伸膜绷紧的状态下加热上述拉伸膜。
发明效果
根据本发明能够提供耐折性、低吸水性及耐热性均优异的树脂膜以及该树脂膜的制造方法。特别是本发明的树脂膜即使长时间地放置在高温及高湿度的使用环境下也不易产生断裂。特别是即使在本发明的树脂膜上设置了硬层(导电层、硬涂层等)的情况下,也能够抑制由于树脂膜与硬层的热膨胀率的差导致的变性而在树脂膜上产生裂缝、断裂的情况。这样的特征在将树脂膜或包含树脂膜的叠层体弯折而使用时特别有用。
附图说明
图1为示意性地表示判断树脂膜是否平滑的判断方法中的试验片的情况的剖面图。
图2为示意性地表示判断树脂膜是否平滑的判断方法中的试验片的情况的平面图。
具体实施方式
以下,对本发明示出实施方式及示例物进行详细地说明。但是,本发明并不限定于示于以下的实施方式及示例物,在不脱离本发明的权利要求的范围及其同等的范围的范围内可以任选地变更实施。
在以下的说明中,“长条”的膜是指相对于宽具有5倍以上的长度的膜,优选具有10倍或其以上的长度,具体而言是指具有可卷成辊状而保管或搬运的程度的长度的膜。
在以下的说明中,要素的方向“平行”、“垂直”以及“正交”只要没有其它特别说明,也可以包含在不损害本发明的效果的范围内例如±5°的范围内的误差。
[1.树脂膜]
[1.1.树脂膜的概要]
本发明的树脂膜为由包含具有结晶性的含有脂环式结构的聚合物的树脂形成的膜,满足下述的要件(a)以及(b)的任一个。
要件(a):耐折度为2000次以上、吸水率为0.1%以下、且耐热温度为180℃以上。
要件(b):耐折度为2000次以上、吸水率为0.1%以下、含有脂环式结构的聚合物的晶化度为10%以上、且平滑。
进而,本发明的树脂膜优选满足要件(a)以及要件(b)的两者。
在以下的说明中,有时将包含具有结晶性的含有脂环式结构的聚合物的树脂称为“结晶性树脂”。
[1.2.结晶性树脂]
结晶性树脂包含具有结晶性的含有脂环式结构的聚合物。在此,含有脂环式结构的聚合物指的是在分子内具有脂环式结构的聚合物,是可以通过将环状烯烃作为单体使用的聚合反应从而得到的聚合物或其加氢物。含有脂环式结构的聚合物通常吸水性低。因此,能够使由包含这样的含有脂环式结构的聚合物的结晶性树脂形成的本发明的树脂膜的吸水性小。
含有脂环式结构的聚合物可以单独使用1种,也可以以任选的比率组合2种以上使用。
作为含有脂环式结构的聚合物所具有的脂环式结构,可举出例如环烷烃结构以及环烯烃结构。在这些中,从容易得到热稳定性等特性优异的树脂膜的观点出发,优选环烷烃结构。1个脂环式结构所包含的碳原子数优选为4个以上,更优选为5个以上,优选为30个以下,更优选为20个以下,特别优选为15个以下。通过使1个脂环式结构所包含的碳原子数处在上述范围内,从而使机械强度、耐热性以及成型性高度地平衡。
在含有脂环式结构的聚合物中,相对于全部的结构单元的具有脂环式结构的结构单元的比例优选为30重量%以上,更优选为50重量%以上,特别优选为70重量%以上。通过使在含有脂环式结构的聚合物中的具有脂环式结构的结构单元的比例像上述那样地多,从而能够提高耐热性。
此外,在含有脂环式结构的聚合物中,具有脂环式结构的结构单元以外的剩余部分没有特别限定,可以根据使用目的适宜选择。
结晶性树脂所包含的含有脂环式结构的聚合物具有结晶性。在此,“具有结晶性的含有脂环式结构的聚合物”是指具有熔点[即,能够用差示扫描量热计(dsc)观测熔点]的含有脂环式结构的聚合物。含有脂环式结构的聚合物的熔点优选为200℃以上,更优选为230℃以上,优选为290℃以下。通过使用这样的具有熔点的含有脂环式结构的聚合物,从而能够得到成型性与耐热性的平衡更加优异的树脂膜。
含有脂环式结构的聚合物的重均分子量(mw)优选为1000以上,更优选为2000以上,优选为1000000以下,更优选为500000以下。具有这样的重均分子量的含有脂环式结构的聚合物的成型加工性与耐热性的平衡优异。
含有脂环式结构的聚合物的分子量分布(mw/mn)优选为1.0以上,更优选为1.5以上,优选为4.0以下,更优选为3.5以下。在此,mn表示数均分子量。具有这样的分子量分布的含有脂环式结构的聚合物的成型加工性优异。
含有脂环式结构的聚合物的重均分子量(mw)以及分子量分布(mw/mn)可以利用将四氢呋喃作为洗脱液的凝胶渗透色谱法(gpc),作为聚苯乙烯换算值测定。
含有脂环式结构的聚合物的玻璃化转变温度tg没有特别限定通常为85℃以上、170℃以下的范围。
作为上述的含有脂环式结构的聚合物,可举出例如下述的聚合物(α)~聚合物(δ)。在这些中,从容易得到耐热性优异的树脂膜的观点出发,作为结晶性的含有脂环式结构的聚合物,优选聚合物(β)。
聚合物(α):环状烯烃单体的开环聚合物,具有结晶性。
聚合物(β):聚合物(α)的加氢物,具有结晶性。
聚合物(γ):为环状烯烃单体的加成聚合物,具有结晶性。
聚合物(δ):聚合物(γ)的加氢物等,具有结晶性。
具体而言,作为含有脂环式结构的聚合物,更优选为二环戊二烯的开环聚合物且具有结晶性的聚合物、以及二环戊二烯的开环聚合物的加氢物且具有结晶性的聚合物,特别优选为二环戊二烯的开环聚合物的加氢物且具有结晶性的聚合物。在此,二环戊二烯的开环聚合物是指相对于全部结构单元的来自二环戊二烯的结构单元的比例通常为50重量%以上,优选为70重量%以上,更优选为90重量%以上,进一步优选为100重量%的聚合物。
以下,对聚合物(α)以及聚合物(β)的制造方法进行说明。
可以用于制造聚合物(α)以及聚合物(β)的环状烯烃单体为具有用碳原子形成的环结构、在该环中具有碳-碳双键的化合物。作为环状烯烃单体的例子,可举出降冰片烯系单体等。此外,在聚合物(α)为共聚物的情况下,作为环状烯烃单体,也可以使用单环的环状烯烃。
降冰片烯系单体为包含降冰片烯环的单体。作为降冰片烯系单体,可举出例如:双环[2.2.1]庚-2-烯(常用名:降冰片烯)、5-乙叉基-双环[2.2.1]庚-2-烯(常用名:乙叉基降冰片烯)及其衍生物(例如在环上具有取代基)等2环式单体;三环[4.3.0.12.5]癸-3,7-二烯(常用名:二环戊二烯)及其衍生物等3环式单体;7,8-苯并三环[4.3.0.12.5]癸-3-烯(常用名:桥亚甲基四氢芴:也称为1,4-桥亚甲基-1,4,4a,9a-四氢芴)及其衍生物、四环[4.4.0.12,5.17,10]十二碳-3-烯(常用名:四环十二碳烯)、8-乙叉基四环[4.4.0.12,5.17,10]-3-十二碳烯及其衍生物等4环式单体等。
在上述的单体中作为取代基,可举出例如:甲基、乙基等烷基;乙烯基等烯基;丙烷-2-叉基等烷叉基;苯基等芳基;羟基;酸酐基;羧基;甲氧基羰基等烷氧基羰基等。此外,上述的取代基可以单独具有1种,也可以以任选的比率具有2种以上。
作为单环的环状烯烃,可举出例如:环丁烯、环戊烯、甲基环戊烯、环己烯、甲基环己烯、环庚烯、环辛烯等环状单烯烃;环己二烯、甲基环己二烯、环辛二烯、甲基环辛二烯、苯基环辛二烯等环状二烯烃等。
环状烯烃单体可以单独使用1种,也可以以任选的比率组合2种以上使用。在使用2种以上的环状烯烃单体的情况下,聚合物(α)可以是嵌段共聚物,也可以是无规共聚物。
对于环状烯烃单体,能够有内型及外型的立体异构体存在的环状烯烃单体。作为环状烯烃单体,也可以使用内型及外型的任一种。此外,可以仅单独地使用内型及外型中的一种的异构体,也可以使用以任选的比例包含内型及外型的异构体混合物。其中,从容易得到含有脂环式结构的聚合物的结晶性增高、耐热性更优异的树脂膜的观点出发,优选提高一种的立体异构体的比例。例如,内型或外型的比例优选为80%以上,更优选为90%以上,进一步优选为95%以上。此外,从容易合成的观点出发,优选提高内型的比例。
聚合物(α)及聚合物(β)通常通过提高其间规立构规整性的程度(间同二单元组(ラセモ·ダイアッド、racemo·diad)的比例),从而能够提高结晶性。从提高聚合物(α)及聚合物(β)的立构规整性的程度的观点出发,对于聚合物(α)及聚合物(β)的结构单元的间同二单元组的比例优选为51%以上,更优选为60%以上,特别优选为70%以上。
间同二单元组的比例可以利用13c-nmr光谱分析进行测定。具体地可以按照下述方法测定。
将邻二氯苯-d4作为溶剂,在200℃应用inverse-gateddecoupling法进行聚合物试样的13c-nmr测定。根据该13c-nmr测定的结果,将邻二氯苯-d4的127.5ppm的峰值作为基准偏移,基于来自全同二单元组的43.35ppm的信号与来自间同二单元组的43.43ppm的信号的强度比,可以求得聚合物试样的间同二单元组的比例。
对于聚合物(α)的合成通常使用开环聚合催化剂。开环聚合催化剂可以单独使用1种,也可以以任选的比率组合2种以上使用。作为这样的聚合物(α)的合成用的开环聚合催化剂,优选可以使环状烯烃单体开环聚合、使具有间规立构规整性的开环聚合物生成的开环聚合催化剂。作为优选的开环聚合催化剂,可举出包含用下述式(1)表示的金属化合物的开环聚合催化剂。
m(nr1)x4-a(or2)a·lb(1)
(在式(1)中,
m表示选自元素周期表第6族的过渡金属原子中的金属原子,
r1表示用在3位、4位及5位的至少1个位置可以具有取代基的苯基、或-ch2r3(r3表示选自氢原子、可以具有取代基的烷基及可以具有取代基的芳基中的基团。)所表示的基团,
r2表示选自可以具有取代基的烷基及可以具有取代基的芳基中的基团,
x表示选自卤原子、可以具有取代基的烷基、可以具有取代基的芳基及烷基甲硅烷基中的基团,
l表示供电子性的中性配体,
a表示0或1的数,
b表示0~2的整数。)
在式(1)中,m表示选自元素周期表第6族的过渡金属原子中的金属原子。作为该m,优选铬、钼及钨,更优选钼及钨,特别优选钨。
在式(1)中,r1表示用在3位、4位及5位的至少1个位置可以具有取代基的苯基、或-ch2r3所表示的基团。
r1的在3位、4位及5位的至少1个位置可以具有取代基的苯基的碳原子数优选为6~20,更优选为6~15。此外,作为上述取代基,可举出例如:甲基、乙基等烷基;氟原子、氯原子、溴原子等卤原子;甲氧基、乙氧基、异丙氧基等烷氧基等。这些取代基可以单独具有1种,也可以以任选的比率具有2种以上。进而,在r1中,在3位、4位及5位的至少2个位置存在的取代基也可以互相键合,形成环结构。
作为在3位、4位及5位的至少1个位置可以具有取代基的苯基,可举出例如:无取代苯基;4-甲基苯基、4-氯苯基、3-甲氧基苯基、4-环己基苯基、4-甲氧基苯基等一取代苯基;3,5-二甲基苯基、3,5-二氯苯基、3,4-二甲基苯基、3,5-二甲氧基苯基等二取代苯基;3,4,5-三甲基苯基、3,4,5-三氯苯基等三取代苯基;2-萘基、3-甲基-2-萘基、4-甲基-2-萘基等可以具有取代基的2-萘基等。
在r1的用-ch2r3所表示的基团中,r3表示选自氢原子、可以具有取代基的烷基及可以具有取代基的芳基中的基团。
r3的可以具有取代基的烷基的碳原子数优选为1~20,更优选为1~10。该烷基可以是直链状也可以是支链状。进而,作为上述取代基,可举出例如:苯基,4-甲基苯基等可以具有取代基的苯基;甲氧基、乙氧基等烷氧基等。这些取代基可以单独使用1种,也可以以任选的比率组合2种以上使用。
作为r3的可以具有取代基的烷基,可举出例如甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、新戊基、苄基、新苯基等。
r3的可以具有取代基的芳基的碳原子数优选为6~20,更优选为6~15。进而,作为上述取代基,可举出例如:甲基、乙基等烷基;氟原子、氯原子、溴原子等卤原子;甲氧基、乙氧基、异丙氧基等烷氧基等。这些取代基可以单独使用1种,也可以以任选的比率组合2种以上使用。
作为r3的可以具有取代基的芳基,可举出例如苯基、1-萘基、2-萘基、4-甲基苯基、2,6-二甲基苯基等。
在这些中,作为用r3所表示的基团,优选碳原子数为1~20的烷基。
在式(1)中,r2表示选自可以具有取代基的烷基及可以具有取代基的芳基中的基团。作为r2的可以具有取代基的烷基及可以具有取代基的芳基,可以各自任选地使用选自作为r3的可以具有取代基的烷基及可以具有取代基的芳基所示的范围中的基团。
在式(1)中,x表示选自卤原子、可以具有取代基的烷基、可以具有取代基的芳基及烷基甲硅烷基中的基团。
作为x的卤原子,可举出例如氯原子、溴原子、碘原子。
作为x的可以具有取代基的烷基及可以具有取代基的芳基,可以各自任选地使用选自作为r3的可以具有取代基的烷基及可以具有取代基的芳基所示的范围中的基团。
作为x的烷基甲硅烷基,可举出例如三甲基甲硅烷基、三乙基甲硅烷基、叔丁基二甲基甲硅烷基等。
用式(1)所表示的金属化合物在1个分子中具有2个以上的x的情况下,这些x可以互相相同,也可以不同。进而,2个以上的x也可以互相键合,形成环结构。
在式(1)中,l表示供电子性的中性配体。
作为l的供电子性的中性配体,可举出例如含有元素周期表第14族或第15族的原子的供电子性化合物。作为其具体例子,可举出:三甲基膦、三异丙基膦、三环己基膦、三苯基膦等膦类;二乙醚、二丁醚、1,2-二甲氧基乙烷、四氢呋喃等醚类;三甲胺、三乙胺、吡啶、二甲基吡啶等胺类等。在这些中,优选醚类。此外,用式(1)所表示的金属化合物在1个分子中具有2个以上的l的情况下,这些l可以互相相同,也可以不同。
作为用式(1)所表示的金属化合物,优选具有苯基酰亚胺基的钨化合物。即,在式(1)中,优选m为钨原子、且r1为苯基的化合物。进而,在其中更优选四氯化钨苯基酰亚胺(四氢呋喃)配位化合物。
用式(1)所表示的金属化合物的制造方法没有特别限定。例如,如日本特开平5-345817号公报所记载的那样,能够通过将第6族过渡金属的卤氧化物;在3位、4位及5位的至少1个位置可以具有取代基的苯基异氰酸酯类或一取代甲基异氰酸酯类;供电子性的中性配体(l)进行混合以及根据需要混合醇类、金属烷氧化物、金属芳氧化物,从而制造用式(1)所表示的金属化合物。
在上述的制造方法中,用式(1)所表示的金属化合物通常以包含于反应液的状态而得到。金属化合物的制造后,也可以将上述的反应液直接作为开环聚合反应的催化剂液使用。此外,也可以通过晶化等纯化处理将金属化合物从反应液中分离及纯化后,将得到的金属化合物供给到开环聚合反应。
开环聚合催化剂可以单独地使用用式(1)所表示的金属化合物,也可以将用式(1)所表示的金属化合物与其它成分组合使用。例如,通过将用式(1)所表示的金属化合物与有机金属还原剂组合使用,从而能够使聚合活性提高。
作为有机金属还原剂,可举出例如具有碳原子数为1~20的烃基的元素周期表第1族、第2族、第12族、第13族或第14族的有机金属化合物。作为这样的有机金属化合物,可举出例如:甲基锂、正丁基锂、苯基锂等有机锂;丁基乙基镁、丁基辛基镁、二己基镁、乙基氯化镁、正丁基氯化镁、烯丙基溴化镁等有机镁;二甲基锌、二乙基锌、二苯基锌等有机锌;三甲基铝、三乙基铝、三异丁基铝、二乙基氯化铝、乙基三氯化二铝、乙基二氯化铝、二乙基乙氧基铝、二异丁基异丁氧基铝、乙基二乙氧基铝、异丁基二异丁氧基铝等有机铝;四甲基锡、四正丁基锡、四苯基锡等有机锡等。在这些中,优选有机铝或有机锡。此外,有机金属还原剂可以单独使用1种,也可以以任选的比率组合2种以上使用。
开环聚合反应通常在有机溶剂中进行。有机溶剂可使用能够使开环聚合物及其加氢物在规定的条件溶解或分散、且不妨碍开环聚合反应及其氢化反应的有机溶剂。作为这样的有机溶剂,可举出例如:戊烷、己烷、庚烷等脂肪族烃类;环戊烷、环己烷、甲基环己烷、二甲基环己烷、三甲基环己烷、乙基环己烷、二乙基环己烷、十氢化萘、双环庚烷、三环癸烷、六氢茚、环辛烷等脂环族烃类;苯、甲苯、二甲苯等芳香族烃类;二氯甲烷、氯仿、1,2-二氯乙烷等卤素系脂肪族烃类;氯苯、二氯苯等卤素系芳香族烃类;硝基甲烷、硝基苯、乙腈等含氮烃类;二乙醚、四氢呋喃等醚类;将它们组合的混合溶剂等。在这些中,作为有机溶剂,优选芳香族烃类、脂肪族烃类、脂环族烃类、醚类。此外,有机溶剂可以单独使用1种,也可以以任选的比率组合2种以上使用。
开环聚合反应例如能够通过将环状烯烃单体、用式(1)所表示的金属化合物和根据需要的有机金属还原剂混合,从而使开环聚合反应开始。混合这些成分的顺序没有特别限定。例如,可以在包含环状烯烃单体的溶液中混合包含用式(1)所表示的金属化合物及有机金属还原剂的溶液。此外,也可以在包含有机金属还原剂的溶液中混合包含环状烯烃单体及用式(1)所表示的金属化合物的溶液。进而,还可以在包含环状烯烃单体及有机金属还原剂的溶液中混合用式(1)所表示的金属化合物的溶液。在将各成分混合时,可以一次性混合各个成分的全部的量,也可以分成多次进行混合。此外,也可以历经比较长的时间(例如1分钟以上)连续地混合。
在开环聚合反应开始时的反应液中的环状烯烃单体的浓度优选为1重量%以上,更优选为2重量%以上,特别优选为3重量%以上,优选为50重量%以下,更优选为45重量%以下,特别优选为40重量%以下。通过将环状烯烃单体的浓度设为上述范围的下限值以上,从而能够提高生产率。此外,通过设为上限值以下,从而能够使开环聚合反应后的反应液的粘度低,因此能够易于进行其后的氢化反应。
开环聚合反应所使用的用式(1)所表示的金属化合物的量期望以“金属化合物∶环状烯烃单体”的摩尔比计处于规定的范围的方式设定。具体而言,上述的摩尔比优选为1∶100~1∶2000000,更优选为1∶500~1000000,特别优选为1∶1000~1∶500000。通过将金属化合物的量设为上述范围的下限值以上,从而能够得到充分的聚合活性。此外,通过设为上限值以下,从而能够在反应后容易地去除金属化合物。
有机金属还原剂的量相对于1摩尔的用式(1)所表示的金属化合物优选为0.1摩尔以上,更优选为0.2摩尔以上,特别优选为0.5摩尔以上,优选为100摩尔以下,更优选为50摩尔以下,特别优选为20摩尔以下。通过将有机金属还原剂的量设为上述范围的下限值以上,从而能够充分地提高聚合活性。此外,通过设为上限值以下,从而能够抑制副反应的产生。
聚合物(α)的聚合反应体系也可以包含活性调节剂。通过使用活性调节剂,从而能够稳定开环聚合催化剂、调整开环聚合反应的反应速度、调整聚合物的分子量分布。
作为活性调节剂,可以使用具有官能团的有机化合物。作为这样的活性调节剂,可举出例如:含氧化合物、含氮化合物、含磷有机化合物等。
作为含氧化合物,可举出例如:二乙醚、二异丙醚、二丁醚、苯甲醚、呋喃、四氢呋喃等醚类;丙酮、二苯甲酮、环己酮等酮类;乙酸乙酯等酯类等。
作为含氮化合物,可举出例如:乙腈、苯甲腈等腈类;三乙基胺、三异丙基胺、奎宁环、n,n-二乙基苯胺等胺类;吡啶、2,4-二甲基吡啶、2,6-二甲基吡啶、2-叔丁基吡啶等吡啶类等。
作为含磷化合物,可举出例如:三苯基膦、三环己基膦、磷酸三苯酯、磷酸三甲酯等膦类;氧化三苯基膦等氧化膦类等。
活性调节剂可以单独使用1种,也可以以任选的比率组合2种以上使用。
聚合物(α)的聚合反应体系中的活性调节剂的量相对于100摩尔%的用式(1)所表示的金属化合物优选为0.01摩尔%~100摩尔%。
为了调节聚合物(α)的分子量,聚合物(α)的聚合反应体系也可以包含分子量调节剂。作为分子量调节剂,可举出例如:1-丁烯、1-戊烯、1-己烯、1-辛烯等α-烯烃类;苯乙烯、乙烯基甲苯等芳香族乙烯基化合物;乙基乙烯基醚、异丁基乙烯基醚、烯丙基缩水甘油醚、乙酸烯丙酯、烯丙醇、甲基丙烯酸缩水甘油酯等含氧乙烯基化合物;烯丙基氯等含卤乙烯基化合物;丙烯酰胺等含氮乙烯基化合物;1,4-戊二烯、1,4-己二烯、1,5-己二烯、1,6-庚二烯、2-甲基-1,4-戊二烯、2,5-二甲基-1,5-己二烯等非共轭二烯;1,3-丁二烯、2-甲基-1,3-丁二烯、2,3-二甲基-1,3-丁二烯、1,3-戊二烯、1,3-己二烯等共轭二烯等。
分子量调节剂可以单独使用1种,也可以以任选的比率组合2种以上使用。
在用于聚合聚合物(α)的聚合反应体系中的分子量调节剂的量可以根据作为目标的分子量适宜决定。分子量调节剂的具体的量相对于环状烯烃单体优选为0.1摩尔%~50摩尔%的范围。
聚合温度优选为-78℃以上,更优选为-30℃以上,优选为+200℃以下,更优选为+180℃以下。
聚合时间可以依赖于反应规模。具体的聚合时间优选为从1分钟到1000小时的范围。
通过上述的制造方法从而可得到聚合物(α)。通过将该聚合物(α)氢化,从而能够制造聚合物(β)。
聚合物(α)的氢化能够通过例如按照常规方法在氢化催化剂的存在下,将氢供给到包含聚合物(α)的反应体系内而进行。在该氢化反应中,只要适宜的设定反应条件,则通常基于氢化反应的加氢物的立构规整度不会变化。
作为氢化催化剂,可以使用作为烯烃化合物的氢化催化剂公知的均相催化剂及非均相催化剂。
作为均相催化剂,可举出例如:乙酸钴/三乙基铝、乙酰丙酮镍/三异丁基铝、二氯二茂钛/正丁基锂、二氯二茂锆/仲丁基锂、四丁氧基钛酸酯/二甲基镁等由过渡金属化合物和碱金属化合物组合形成的催化剂;双(三苯基膦)二氯化钯、氯氢羰基三(三苯基膦)钌、氯氢羰基双(三环己基膦)钌、双(三环己基膦)亚苄基二氯化钌(ⅳ)、三(三苯基膦)氯化铑等贵金属配位化合物催化剂等。
作为非均相催化剂,可举出例如:镍、钯、铂、铑、钌等金属催化剂;镍/二氧化硅、镍/硅藻土、镍/氧化铝、钯/碳、钯/二氧化硅、钯/硅藻土、钯/氧化铝等使上述金属负载在碳、二氧化硅、硅藻土、氧化铝、氧化钛等载体上而成的固体催化剂。
氢化催化剂可以单独使用1种,也可以以任选的比率组合2种以上使用。
氢化反应通常在非活性有机溶剂中进行。作为非活性有机溶剂,可举出:苯、甲苯等芳香族烃类;戊烷、己烷等脂肪族烃类;环己烷、十氢化萘等脂环族烃类;四氢呋喃、乙二醇二甲基醚等醚类等。非活性有机溶剂可以单独使用1种,也可以以任选的比率组合2种以上使用。此外,非活性有机溶剂可以是与用于开环聚合反应的有机溶剂相同的有机溶剂,也可以是与其不同的有机溶剂。进而,也可以在开环聚合反应的反应液中混合氢化催化剂而进行氢化反应。
氢化反应的反应条件通常根据使用的氢化催化剂而不同。
氢化反应的反应温度优选为-20℃以上,更优选为-10℃以上,特别优选为0℃以上,优选为+250℃以下,更优选为+220℃以下,特别优选为+200℃以下。通过将反应温度设为上述范围的下限值以上,从而能够加快反应速度。此外,通过设为上限值以下,从而能够抑制副反应的产生。
氢压优选为0.01mpa以上,更优选为0.05mpa以上,特别优选为0.1mpa以上,优选为20mpa以下,更优选为15mpa以下,特别优选为10mpa以下。通过将氢压设为上述范围的下限值以上,从而能够加快反应速度。此外,通过设为上限值以下,从而能够不需要高耐压反应装置等特别的装置、抑制设备成本。
氢化反应的反应时间可以设定为期望的加氢率可达成的任选的时间,优选为0.1小时~10小时。
氢化反应后通常按照常规方法回收聚合物(α)的加氢物即聚合物(β)。
氢化反应中的加氢率(被氢化的主链双键的比例)优选为98%以上,更优选为99%以上。加氢率越增高,含有脂环式结构的聚合物的耐热性能够越良好。
在此,聚合物的加氢率可以以邻二氯苯-d4作为溶剂在145℃利用1h-nmr测定。
接下来,对聚合物(γ)以及聚合物(δ)的制造方法进行说明。
作为用于制造聚合物(γ)以及(δ)的环状烯烃单体,可以任选地使用选自作为可以用于制造聚合物(α)以及聚合物(β)的环状烯烃单体所示的范围中的环状烯烃单体。此外,环状烯烃单体可以单独使用1种,也可以以任选的比率组合2种以上使用。
在聚合物(γ)的制造中,作为单体,可以使用与环状烯烃单体组合能够与环状烯烃单体共聚的任选的单体。作为任选的单体,可举出例如:乙烯、丙烯、1-丁烯、1-戊烯、1-己烯等碳原子数为2~20的α-烯烃;苯乙烯、α-甲基苯乙烯等芳香环乙烯基化合物;1,4-己二烯、4-甲基-1,4-己二烯、5-甲基-1,4-己二烯、1,7-辛二烯等非共轭二烯等。在它们之中,优选α-烯烃,更优选乙烯。此外,任选的单体可以单独使用1种,也可以以任选的比率组合2种以上使用。
环状烯烃单体与任选的单体的量的比例以重量比(环状烯烃单体∶任选的单体)计优选为30∶70~99∶1,更优选为50∶50~97∶3,特别优选为70∶30~95∶5。
在使用2种以上的环状烯烃单体的情况、以及将环状烯烃单体与任选的单体组合使用的情况下,聚合物(γ)可以是嵌段共聚物,也可以是无规共聚物。
对于聚合物(γ)的合成通常使用加成聚合催化剂。作为这样的加成聚合催化剂,可举出例如由钒化合物及有机铝化合物所形成的钒系催化剂、由钛化合物及有机铝化合物所形成的钛系催化剂、由锆配位化合物及铝氧烷所形成的锆系催化剂等。此外,加成聚合催化剂可以单独使用1种,也可以以任选的比率组合2种以上使用。
加成聚合催化剂的量相对于1摩尔的单体优选为0.000001摩尔以上,更优选为0.00001摩尔以上,优选为0.1摩尔以下,更优选为0.01摩尔以下。
环状烯烃单体的加成聚合通常在有机溶剂中进行。作为有机溶剂,可任选地使用选自作为可用于环状烯烃单体的开环聚合的有机溶剂所示的范围中的有机溶剂。此外,有机溶剂可以单独使用1种,也可以以任选的比率组合2种以上使用。
在用于制造聚合物(γ)的聚合时的聚合温度优选为-50℃以上,更优选为-30℃以上,特别优选为-20℃以上,优选为250℃以下,更优选为200℃以下,特别优选为150℃以下。此外,聚合时间优选为30分钟以上,更优选为1小时以上,优选为20小时以下,更优选为10小时以下。
通过上述的制造方法从而可得到聚合物(γ)。通过将该聚合物(γ)氢化,从而能够制造聚合物(δ)。
聚合物(γ)的氢化可通过与作为将聚合物(α)氢化的方法而先前所示的方法同样的方法而进行。
在结晶性树脂中,具有结晶性的含有脂环式结构的聚合物的比例优选为50重量%以上,更优选为70重量%以上,特别优选为90重量%以上。通过将具有结晶性的含有脂环式结构的聚合物的比例设为上述范围的下限值以上,从而能够提高本发明的树脂膜的耐热性。
结晶性树脂除了包含具有结晶性的含有脂环式结构的聚合物之外,还可以包含任选的成分。作为任选的成分,可举出例如:酚系抗氧化剂、磷系抗氧化剂、硫系抗氧化剂等抗氧化剂;受阻胺系光稳定剂等光稳定剂;石油系蜡、费托蜡(fischer-tropsch蜡)、聚烯烃蜡等蜡;山梨糖醇系化合物、有机磷酸的金属盐、有机羧酸的金属盐、高岭土及滑石等成核剂;二氨基茋衍生物、香豆素衍生物、唑系衍生物(例如,苯并恶唑衍生物、苯并三恶唑衍生物、苯并咪唑衍生物及苯并噻唑衍生物)、咔唑衍生物、吡啶衍生物、萘二甲酸衍生物及咪唑啉酮衍生物等荧光增白剂;二苯甲酮系紫外线吸收剂、水杨酸系紫外线吸收剂、苯并三唑系紫外线吸收剂等紫外线吸收剂;滑石、二氧化硅、碳酸钙、玻璃纤维等无机填充材料;着色剂;阻燃剂;阻燃助剂;防静电剂;增塑剂;近红外线吸收剂;滑剂;填充剂及软质聚合物等具有结晶性的含有脂环式结构的聚合物以外的任选的聚合物等。此外,任选的成分可以单独使用1种,也可以以任选的比率组合2种以上使用。
[1.3.树脂膜的物性]
本发明的树脂膜的耐折性优异。本发明的树脂膜的耐折性具体而言可以用耐折度表示。本发明的树脂膜的耐折度通常为2000次以上,优选为2200次以上,更优选为2400次以上。因为耐折度越高越好,所以对耐折度的上限没有限制,耐折度通常为100000次以下。
树脂膜的耐折度可以通过按照jisp8115“纸及纸板-耐折强度试验方法-mit试验机法”的mit耐折试验,用下述的方法进行测定。
从作为试样的树脂膜中切取宽15mm±0.1mm、长约110mm的试验片。这时,以树脂膜被更强拉伸的方向成为与试验片的约110mm的边平行的方式制作试验片。然后,使用mit耐折度试验机(安田精机制作所制造“no.307”),在负荷9.8n、弯曲部的曲率0.38±0.02mm、弯折角度135°±2°、弯折速度175次/分钟的条件,以在试验片的宽度方向上显现折痕的方式弯折上述的试验片。继续该弯折,测定直到试验片断裂为止的往复弯折次数。
制作10张试验片,按照上述的方法测定10次直到试验片断裂为止的往复弯折次数。将这样所测定的10次的测定值的平均作为该树脂膜的耐折度(mit耐折次数)。
此外,本发明的树脂膜的低吸水性优异。本发明的树脂膜的低吸水性具体而言可以用吸水率表示。本发明的树脂膜的吸水率通常为0.1%以下,优选为0.08%以下,更优选为0.05%以下。
树脂膜的吸水率可以用下述的方法测定。
从作为试样的树脂膜中切取试验片,测定试验片的质量。其后,将该试验片在23℃的水中浸渍24小时,测定浸渍后的试验片的质量。然后,将由于浸渍而增加的试验片的质量相对于浸渍前的试验片的质量的比例作为吸水率(%)而算出。
进而,本发明的树脂膜的耐热温度为180℃以上或含有脂环式结构的聚合物的晶化度为10%以上。这样的树脂膜的耐热性优异。对于本发明的树脂膜而言,耐热温度可以为180℃以上;含有脂环式结构的聚合物的晶化度可以为10%以上;也可以耐热温度为180℃以上且含有脂环式结构的聚合物的晶化度为10%以上。
本发明的树脂膜的耐热温度通常为180℃以上,优选为200℃以上,更优选为220℃以上。因为耐热温度越高越好,所以对耐热温度的上限没有限制,但耐热温度通常为含有脂环式结构的聚合物的熔点tm以下。
树脂膜的耐热温度在上述范围的情况可以用下述的方法进行确认。
以不对作为试样的树脂膜施加张力的状态,将该树脂膜在某温度tx的环境下放置10分钟。之后,用目视来确认树脂膜的表面状况。在树脂膜的表面的形状上不能确认凹凸的情况下,可以知道该树脂膜的耐热温度为上述的温度tx以上。
本发明的树脂膜所包含的含有脂环式结构的聚合物的晶化度优选为10%以上,更优选为15%以上,特别优选为20%以上,优选为70%以下,更优选为60%以下,特别优选为50%以下。通过树脂膜所包含的含有脂环式结构的聚合物的晶化度为上述范围的下限值以上,从而能够提高树脂膜的耐热性,此外,通过为上限值以下,从而能够使膜的透明性良好。
树脂膜所包含的含有脂环式结构的聚合物的晶化度可以利用x射线衍射法测定。
如上述的那样,本发明的树脂膜的低吸水性、耐热性及耐折性均优异。可推测上述的优异的低吸水性通过使用吸水性低的聚合物即含有脂环式结构的聚合物而得到。此外,可推测上述的优异的耐热性通过除了使用耐热性优异的聚合物即含有脂环式结构的聚合物之外,进一步使该含有脂环式结构的聚合物晶化而提高对热的耐性从而得到。进而可推测上述的优异的耐折性通过以下的方式得到,即,经拉伸而使含有脂环式结构的聚合物的分子取向,从而即使含有脂环式结构的聚合物晶化也能够抑制脆化。但是,本发明并不限定于上述的推测。
进而本发明的树脂膜通常是平滑的。通过树脂膜平滑,从而能够使树脂膜的处理良好、树脂膜的光学特性等特性良好。
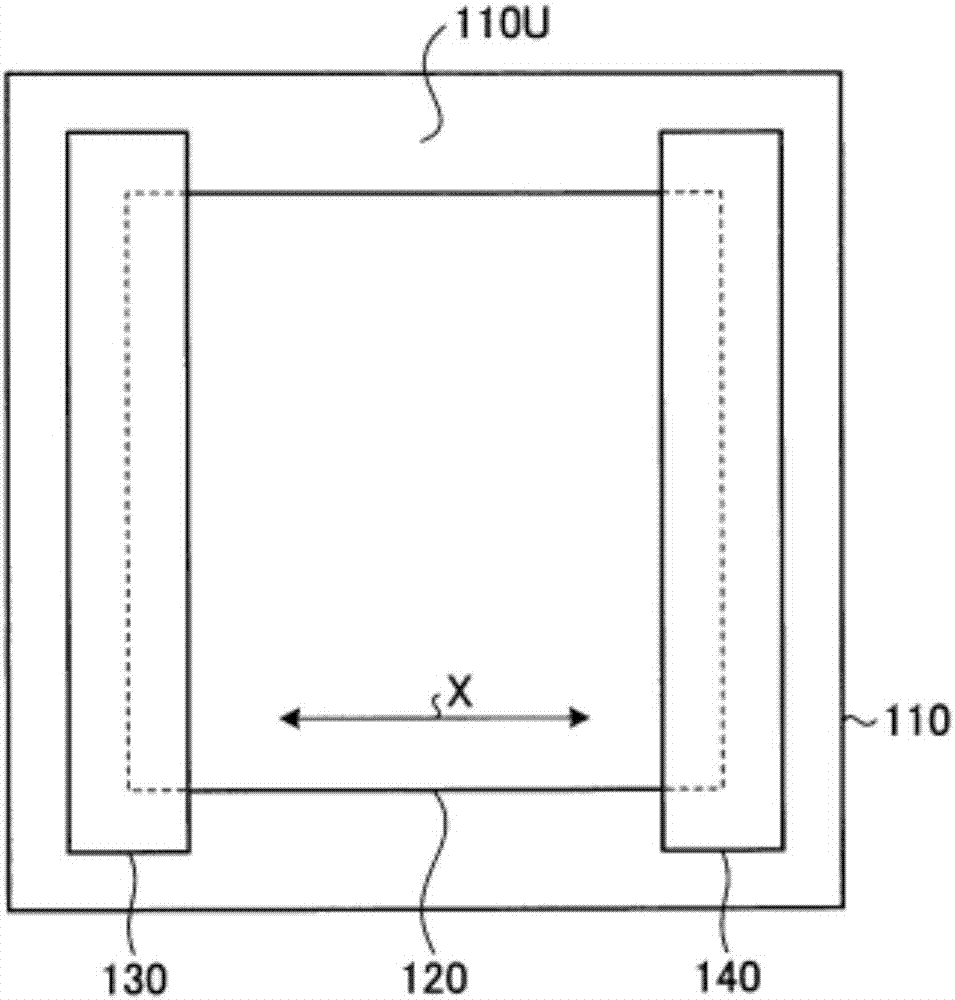
在此,树脂膜是否平滑可按照包含下述的(i)~(viii)的步骤的判断方法进行判断。图1为示意性地表示判断树脂膜是否平滑的判断方法中的试验片的情况的剖面图。此外,图2为示意性地表示判断树脂膜是否平滑的判断方法中的试验片的情况的平面图。
(i)从作为试样的树脂膜中切取5张150mm×150mm的正方形的试验片。这时,使试验片的正方形的边与树脂膜被最强拉伸的方向平行或垂直。
(ii)如图1及图2所示的那样,在具有水平的平面状的支承面110u的平台110的上述支承面110u上,载置试验片120。
(iii)为了防止试验片120的卷曲,在树脂膜被最强拉伸的方向x的试验片120的两端部10mm处载置秤锤130及140。
(iv)在该状态下,将试验片120使用三坐标形状测定机(nikon公司制造“多关节式三坐标测定机mcax20”)扫描,由此测定试验片120的立体形状。
(v)根据所测定的立体形状,求得从平台110的支承面110u到最远离该支承面110u的试验片120上的点p120的距离l。
(vi)把试验片反过来,进行上述(ii)~(v)的步骤,求得距离l。
(vii)对于剩余的4张试验片,也分别进行上述(ii)~(vi)的步骤,求得距离l。
(viii)在用5张的试验片测定的距离l均小于2mm的情况下,判断该树脂膜“平滑”。此外,用5张的试验片测定的距离l中,即使一张超过2mm的情况下,也判断该树脂膜不平滑。
本发明的树脂膜所包含的含有脂环式结构的聚合物优选取向。因此,树脂膜优选具有规定范围的绝对值的面取向系数δne。
本发明的树脂膜可以具有的上述的规定范围的面取向系数的绝对值|δne|优选为0.010以上,更优选为0.012以上,特别优选为0.014以上,优选为0.100以下,更优选为0.090以下,特别优选为0.080以下。
在此,树脂膜的面取向系数的绝对值|δne|为“δne=(nx+ny)/2-nz”所表示的值δne的绝对值。nx表示与树脂膜的厚度方向垂直的方向(面内方向)即提供最大的折射率的方向的折射率。ny表示树脂膜的上述面内方向即与nx的方向垂直的方向的折射率。nz表示树脂膜的厚度方向的折射率。只要没有特别限定,上述的折射率nx、ny及nz的测定波长为550nm。
本发明的树脂膜优选透明性优异。具体而言,本发明的树脂膜的全光线透过率优选为70%以上,更优选为80%以上,特别优选为90%以上。
树脂膜的全光线透过率可以使用紫外·可见光分光光度计在波长400nm~700nm的范围测定。
本发明的树脂膜优选雾度小。具体而言,本发明的树脂膜的雾度优选为10%以下,更优选为5%以下,特别优选为3%以下。
对于树脂膜的雾度,将该树脂膜在选择的任意的部位切取50mm×50mm的正方形的薄膜样品,之后,对于薄膜样品使用雾度计进行测定。
本发明的树脂膜也可以根据用途具有延迟(retardation)。例如,在将本发明的树脂膜作为相位差膜、光学补偿膜等光学膜使用的情况下,优选树脂膜具有延迟。
[1.4.树脂膜的厚度]
本发明的树脂膜的厚度优选为1μm以上,更优选为3μm以上,特别优选为5μm以上,优选为400μm以下,更优选为200μm以下,特别优选为100μm以下。
[1.5.树脂膜的用途]
本发明的树脂膜可以用于任意的用途。其中,本发明的树脂膜例如适合作为光学各向同性膜和相位差膜等光学膜、电气电子用膜、阻挡膜(barrierfilm)用的基材膜、以及导电性膜用的基材膜。作为上述的光学膜,可举出例如液晶显示装置用的相位差膜、偏振片保护膜、有机el显示装置的圆偏振片用的相位差膜等。作为电气电子用膜,可举出例如柔性布线基板、膜电容器用绝缘材料等。作为阻挡膜,可举出例如有机el元件用的基板、密封膜、太阳电池的密封膜等。作为导电性膜,可举出例如有机el元件或太阳电池的柔性电极、触控式面板构件等。
[2.树脂膜的制造方法]
本发明的树脂膜例如可以通过包含将由包含具有结晶性的含有脂环式结构的聚合物的树脂形成的拉伸前膜拉伸而得到拉伸膜的第一工序和在上述第一工序后将上述拉伸膜加热的第二工序的本发明的树脂膜的制造方法而制造。以下,对该制造方法进行说明。
[2.1.拉伸前膜的准备]
在本发明的树脂膜的制造方法中,进行准备拉伸前膜的工序。拉伸前膜为由结晶性树脂形成的膜。
作为制造由结晶性树脂形成的拉伸前膜的方法,可举出例如注射成型法、挤出成型法、压制成型法、吹胀成型法、吹塑成型法、压延成型法、注塑成型法、压缩成型法等树脂成型法。在这些中,从容易控制厚度的观点出发,优选挤出成型法。
在利用挤出成型法制造拉伸前膜的情况下,该挤出成型法的制造条件优选为如下所述。料筒温度(熔融树脂温度)优选为tm以上,更优选为(tm+20℃)以上,优选为(tm+100℃)以下,更优选为(tm+50℃)以下。此外,铸辊温度优选为(tg-50℃)以上,优选为(tg+70℃)以下,更优选为(tg+40℃)以下。通过在这样的条件制造拉伸前膜,从而能够容易地制造期望厚度的拉伸前膜。在此,“tm”表示含有脂环式结构的聚合物的熔点,“tg”表示含有脂环式结构的聚合物的玻璃化转变温度。
拉伸前膜的厚度可以根据想要制造的树脂膜的厚度而任选地设定,优选为5μm以上,更优选为20μm以上,特别优选为40μm以上,优选为400μm以下,更优选为300μm以下,特别优选为200μm以下。
[2.2.第一工序:拉伸工序]
准备拉伸前膜后,进行将该拉伸前膜进行拉伸而得到拉伸膜的第一工序。
对于拉伸前膜的拉伸方法没有特别限定,可以使用任选的拉伸方法。可举出例如:将拉伸前膜沿长度方向单轴拉伸的方法(纵向单轴拉伸法)、将拉伸前膜沿宽度方向单轴拉伸的方法(横向单轴拉伸法)等单轴拉伸法;将拉伸前膜沿长度方向拉伸的同时沿宽度方向拉伸的同时双轴拉伸法、将拉伸前膜沿长度方向及宽度方向的一方拉伸后再沿另一方向拉伸的依次双轴拉伸法等双轴拉伸法;将拉伸前膜沿即不平行也不垂直于宽度方向的斜方向拉伸的方法(斜向拉伸法)等。
作为上述的纵向单轴拉伸法,可举出例如利用辊间的周速的差的拉伸方法等。
此外,作为上述的横向单轴拉伸法,可举出例如使用拉幅拉伸机的拉伸方法。
进而,作为上述的同时双轴拉伸法,可举出例如:使用具有设置成能够沿导轨移动且可以保持拉伸前膜的多个夹子的拉幅拉伸机,分开夹子的间隔来将拉伸前膜沿长度方向拉伸,同时利用导轨的扩展角度将拉伸前膜沿宽度方向拉伸的拉伸方法等。
此外,作为上述的依次双轴拉伸法,可举出例如利用辊间的周速的差而将拉伸前膜沿长度方向拉伸后,将该拉伸前膜的两端部用夹子保持,利用拉幅拉伸机沿宽度方向拉伸的拉伸方法等。
进而,作为上述的斜向拉伸法,可举出例如使用可对于拉伸前膜沿长度方向或宽度方向施加左右不同速度的推力、拉力或牵引力的拉幅拉伸机而将拉伸前膜沿斜方向连续地拉伸的拉伸方法等。
在第一工序中,将拉伸前膜拉伸的情况下的拉伸温度优选为(tg-30℃)以上,更优选为(tg-10℃)以上,优选为(tg+60℃)以下,更优选为(tg+50℃)以下。在此,“tg”表示结晶性树脂的玻璃化转变温度。通过在这样的温度范围进行拉伸,从而能够使拉伸前膜所包含的聚合物分子适当地取向,因此能够使树脂膜的耐折性高效地提高。
将拉伸前膜拉伸的情况下的拉伸倍率优选为1.2倍以上,更优选为1.5倍以上,通常为20倍以下,优选为15倍以下,更优选为10倍以下。在此,在第一工序中,例如在像双轴拉伸那样沿不同的多个方向进行拉伸的情况下,用各个拉伸方向的拉伸倍率的积所表示的总拉伸倍率优选处于上述的范围。通过使拉伸倍率处于上述的范围,从而能够使拉伸前膜所包含的聚合物分子适当地取向,因此能够使树脂膜的耐折性高效地提高。
通过将上述的这样的拉伸施加于拉伸前膜,从而能够得到拉伸膜。在该拉伸膜中,该拉伸膜所包含的含有脂环式结构的聚合物的分子取向。因此,能够抑制在通过第二工序的加热而晶化含有脂环式结构的聚合物的情况下的树脂膜的脆化,能够提高树脂膜的耐折性。进而,通过将拉伸前膜拉伸,从而能够抑制第二工序的加热而导致的大的晶粒的产生。因此,能够抑制起因于晶粒的白化,因此能够提高树脂膜的透明性。
拉伸膜的厚度可以根据想要制造的树脂膜的厚度任选地设定,优选为1μm以上,更优选为3μm以上,优选为500μm以下,更优选为200μm以下。
[2.3.第二工序:加热工序]
在上述第一工序得到拉伸膜后,进行将该拉伸膜加热的第二工序。在第二工序所加热的拉伸膜中,通常在含有脂环式结构的聚合物维持了其取向状态的情况下进行含有脂环式结构的聚合物的晶化。因此,通过上述的第二工序,可得到包含维持了取向状态的、晶化的含有脂环式结构的聚合物的树脂膜。
第二工序中的拉伸膜的加热温度优选设为拉伸膜所包含的含有脂环式结构的聚合物的玻璃化转变温度tg以上、含有脂环式结构的聚合物的熔点tm以下这样的特定的温度范围。由此,能够使含有脂环式结构的聚合物的晶化高效地进行。进而,在上述特定的温度范围中,优选设定为晶化的速度增大的这样的温度。例如,在使用二环戊二烯的开环聚合物的氢化物作为含有脂环式结构的聚合物的情况下,第二工序中的拉伸膜的加热温度优选为110℃以上,更优选为120℃以上,优选为240℃以下,更优选为220℃以下。
作为用于将拉伸膜加热的加热装置,从不需要将加热装置与拉伸膜接触的观点出发,优选使拉伸膜的环境温度上升的加热装置。如果举出合适的加热装置的具体例子的话,可举出烘箱及加热炉。
进而,在第二工序中,拉伸膜的加热优选在将拉伸膜的至少两边保持而使拉伸膜绷紧的状态下进行。在此,使拉伸膜绷紧的状态是指对拉伸膜施加张力的状态。但是,在使该拉伸膜绷紧的状态下不包含拉伸膜实质上被拉伸的状态。此外,实质上被拉伸是指拉伸膜的向任一方向的拉伸倍率为通常1.1倍以上。
通过在将拉伸膜的至少两边保持而绷紧的状态下进行加热,从而能够在被保持的边之间的区域中阻止拉伸膜的热收缩导致的变形。这时,为了在拉伸膜的广泛的面积中阻止变形,优选保持包含相向的两边的边,使该被保持的边之间的区域成为绷紧的状态。例如,对于矩形的单张的拉伸膜,优选通过保持相向的两边(例如,两个长边或两个短边)而使上述两边之间的区域成为绷紧的状态从而在该单张的拉伸膜的整面中防止变形。此外,对于长条的拉伸膜,优选通过保持位于宽度方向的端部的两边(即,长边)而使上述两边之间的区域成为绷紧的状态从而在该长条的拉伸膜的整面中防止变形。这样的防止了变形的拉伸膜即使由于热收缩而在膜内产生应力也能抑制褶皱等变形的产生。因此,因为能够抑制由于加热而损害树脂膜的平滑性的情况,所以能够得到波纹及褶皱少的平滑的树脂膜。
进而,为了更加可靠地抑制加热时的变形,优选保持更多的边。因此,例如对于单张的拉伸膜,优选保持其全部的边。如果举出具体例子的话,对于矩形的单张的拉伸膜优选保持四边。
在将拉伸膜保持的情况下,可以利用合适的保持件保持拉伸膜的边。保持件可以是连续地保持拉伸膜的边的全长的保持件,也可以是空出间隔断续地保持的保持件。例如,也可以利用以规定的间隔排列的保持件将拉伸膜的边断续地保持。
此外,作为保持件,优选在拉伸膜的边以外的部分与拉伸膜不接触。通过使用这样的保持件,从而能够得到平滑性更优异的树脂膜。
进而,作为保持件,优选可以将保持件之间的相对的位置在第二工序中保持。这样的保持件在第二工序中保持件之间的位置不会相对移动,因此容易抑制加热时的拉伸膜的实质上的拉伸。
作为合适的保持件,例如作为矩形的拉伸膜用的保持件,可举出以规定间隔设置于模具的、可以夹持拉伸膜的边的夹子等夹具。此外,例如作为用于保持位于长条的拉伸膜的宽度方向的端部的两边的保持件,可举出设置于拉幅拉伸机、可以夹持拉伸膜的边的夹具。
在使用长条的拉伸膜的情况下,可以保持位于该拉伸膜的长度方向的端部的边(即,短边),也可以代替保持上述的边而保持拉伸膜的加热到特定的温度范围的区域的长度方向的两侧。例如,也可以在拉伸膜的加热到特定的温度范围的区域的长度方向的两侧设置能够以拉伸膜不会热收缩的方式将其保持而使其成为绷紧的状态的保持装置。作为这样的保持装置,可举出例如2个辊的组合等。通过利用这些的组合对拉伸膜施加输送张力等张力,从而能够在加热到特定的温度范围的区域中抑制该拉伸膜的热收缩。因此,只要使用上述的组合作为保持装置,就能够一边将拉伸膜沿长度方向输送一边保持该拉伸膜,因此能够高效地制造树脂膜。
在第二工序中,将拉伸膜维持在上述的特定的温度范围的处理时间优选为5秒以上,更优选为10秒以上,优选为1小时以下。由此,因为能够使含有脂环式结构的聚合物的晶化充分地进行,所以能够特别提高树脂膜的耐热性。
[2.4.任选的工序]
本发明的树脂膜的制造方法可以与上述的工序组合包含任选的工序。
例如,本发明的树脂膜的制造方法也可以进行对树脂膜实施任选的表面处理的工序。
实施例
以下,出示实施例对本发明具体地说明。但是,本发明并不限定于以下所示的实施例中,在不脱离本发明的权利要求范围及其同等的范围的范围中可以任选地变更实施。
在以下的说明中,表示量的“%”和“份”,只要没有特别说明则为重量基准。此外,以下所说明的操作只要没有特别说明均是在常温常压的条件进行。
[评价方法]
[重均分子量及数均分子量的测定方法]
聚合物的重均分子量及数均分子量使用凝胶渗透色谱法(gpc)系统(tosoh公司制造“hlc-8320”)作为聚苯乙烯换算值测定。测定时,使用h型柱(tosoh公司制造)作为色谱柱,使用四氢呋喃作为溶剂。此外,测定时的温度为40℃。
[熔点tm的测定方法]
在氮环境下将加热到300℃的试样用液体氮急剧冷却,使用差示扫描量热计(dsc)以10℃/分钟升温求得试样的熔点。
[聚合物的加氢率的测定方法]
聚合物的加氢率以邻二氯苯-d4作为溶剂,在145℃利用1h-nmr测定来进行测定。
[聚合物的间同二单元组的比例的测定方法]
以邻二氯苯-d4为溶剂,在150℃应用inverse-gateddecoupling法进行聚合物的13c-nmr测定。根据该13c-nmr测定的结果,将邻二氯苯-d4的127.5ppm的峰值作为基准偏移,基于来自全同二单元组的43.35ppm的信号与来自间同二单元组的43.43ppm的信号的强度比,求得聚合物的间同二单元组的比例。
[聚合物的晶化度的测定方法]
膜所包含的聚合物的晶化度利用x射线衍射法进行测定。
[耐折度的评价方法]
膜的耐折度通过按照jisp8115“纸及纸板-耐折强度试验方法-mit试验机法”的mit耐折试验,按照下述的顺序进行测定。
从作为试样的膜中切取宽15mm±0.1mm、长约110mm的试验片。这时,在上述膜为经拉伸处理而被制造的膜的情况下,以被更强拉伸的方向成为与试验片的约110mm的边平行的方式制作试验片。
使用mit耐折度试验机(安田精机制作所制造“no.307”,在负荷9.8n、弯曲部的曲率0.38±0.02mm、弯折角度135°±2°、弯折速度175次/分钟的条件,以在试验片的宽度方向上显现折痕的方式弯折上述的试验片。继续该弯折,测定直到试验片断裂为止的往复弯折次数。
制作10张试验片,按照上述的方法测定10次直到试验片断裂为止的往复弯折次数。将这样所测定的10次的测定值的平均作为该膜的耐折度(mit耐折次数)。
如果耐折度为2000次以上则判断为“良”,如果耐折度小于2000次则判断为“不良”。
[吸水率的评价方法]
从作为试样的膜中以宽100mm、长100mm切取试验片,测定试验片的质量。其后,将该试验片在23℃的水中浸渍24小时,测定浸渍后的试验片的质量。然后,将由于浸渍而增加的试验片的质量相对于浸渍前的试验片的质量的比例作为吸水率(%)而算出。
如果吸水率的值为0.1%以下则判断为“良”,如果吸水率的值比0.1%大则判断为“不良”。
[耐热温度的评价方法]
在不对作为试样的膜施加张力的状态下,将该树脂膜在180℃的环境下放置10分钟。之后,用目视来确认膜的表面状况。
在膜的表面的形状能够确认凹凸的情况下,作为耐热温度小于180℃而判断为“不良”。此外,在膜的表面的形状不能够确认凹凸的情况下,作为耐热温度为180℃以上而判断为“良”。
[平滑性的评价方法]
将下述的步骤(i)~(viii)以该顺序进行来评价树脂膜的平滑性。
(i)将作为试样的树脂膜沿宽度方向5等分而得到5张的分割膜。从这样得到的分割膜各自的中央部分切取150mm×150mm的正方形的试验片,得到5张试验片。这时,使试验片的正方形的边与树脂膜被最强拉伸的方向平行或垂直。
(ii)如图1及图2所示的那样,在具有水平的平面状的支承面110u的平台110的上述支承面110u上,载置试验片120。
(iii)为了防止试验片120的卷曲,在树脂膜被最强拉伸的方向x的试验片120的两端部10mm处载置秤锤130及140。
(iv)在该状态下,将试验片120使用三坐标形状测定机(nikon公司制造“多关节式三坐标测定机mcax20”)扫描,由此测定试验片120的立体形状。
(v)根据所测定的立体形状,求得从平台110的支承面110u到最远离该支承面110u的试验片120上的点p120的距离l。
(vi)把试验片反过来,进行上述(ii)~(v)的步骤,求得距离l。
(vii)对于剩余的4张试验片,也分别进行上述(ii)~(vi)的步骤,求得距离l。
(viii)在用5张的试验片测定的距离l均小于2mm的情况下,判断该树脂膜“平滑”,将平滑性设为“良”。此外,用5张的试验片测定的距离l中,即使一张超过2mm的情况下,也判断该树脂膜不平滑,将平滑性设为“不良”。
[制造例1.二环戊二烯的开环聚合物的加氢物的制造]
将金属制的耐压反应器充分地干燥后,进行氮置换。在该金属制耐压反应器中加入154.5份的环己烷、42.8份的二环戊二烯(内型含有率为99%以上)的浓度70%的环己烷溶液(作为二环戊二烯的量为30份)以及1.9份的1-己烯,加热到53℃。
在将0.014份的四氯化钨苯基酰亚胺(四氢呋喃)配位化合物溶解于0.70份的甲苯的溶液中,加入0.061份的浓度19%的二乙基乙氧基铝/正己烷溶液,搅拌10分钟制备催化剂溶液。
将该催化剂溶液加入耐压反应器中,开始开环聚合反应。之后,一边保持53℃一边使反应进行4小时,得到二环戊二烯的开环聚合物的溶液。
得到的二环戊二烯的开环聚合物的数均分子量(mn)及重均分子量(mw)分别为8750和28100,由此求得的分子量分布(mw/mn)为3.21。
在200份的得到的二环戊二烯的开环聚合物的溶液中加入0.037份的作为终止剂的1,2-乙二醇,加热到60℃,搅拌1小时,使聚合反应终止。在其中加入1份的类水滑石化合物(协和化学工业公司制造“kyoward(注册商标)2000”),加热到60℃,搅拌1小时。其后,加入0.4份的过滤助剂(昭和化学工业公司制造“radiolight(注册商标)#1500”),使用聚丙烯褶筒式过滤器(pppleatscartridgefilter、advantec东洋公司制造“tcp-hx”),过滤吸附剂和溶液。
在200份的过滤后的二环戊二烯的开环聚合物的溶液(聚合物量为30份)中加入100份的环己烷,添加0.0043份的氯氢羰基三(三苯基膦)钌,在氢压6mpa、180℃进行4小时的氢化反应。由此,得到包含二环戊二烯的开环聚合物的加氢物的反应液。对于该反应液,加氢物析出而成为浆料液。
将上述的反应液中所包含的加氢物和溶液使用离心分离器分离,在60℃减压干燥24小时,得到28.5份的具有结晶性的二环戊二烯的开环聚合物的加氢物。该加氢物的加氢率为99%以上,玻璃化转变温度(tg)为95℃,熔点(tm)为262℃,间同二单元组的比例为89%。
[实施例1]
(1-1.拉伸前膜的制造)
在100份的制造例1中得到的二环戊二烯的开环聚合物的加氢物中,混合1.1份的抗氧化剂(四[亚甲基-3-(3’,5’-二叔丁基-4’-羟基苯基)丙酸酯]甲烷;basf日本公司制造“irganox(注册商标)1010”),得到成为膜的材料的树脂。
将上述的树脂投入到具有4个内径φ3mm的模孔的双轴挤出机(东芝机械公司制造“tem-37b”)中。利用上述的双轴挤出机将树脂通过热熔融挤出成型成型为股状的成型体。将该成型体用线料切粒机切碎,得到树脂的颗粒。上述的双轴挤出机的运行条件如下所示。
筒设定温度:270℃~280℃;
模具设定温度:250℃;
螺杆转速:145rpm;
进料器转速:50rpm。
接着,将得到的颗粒提供给具有t模的热熔融挤出膜成型机。使用该膜成型机,将由上述的树脂形成的长条的拉伸前膜(厚度100μm)采用以2m/分钟的牵引速度卷成辊的方法制造。上述的膜成型机的运行条件如下所示。
筒温度设定:280℃~290℃;
模温度:270℃;
螺杆转速:30rpm。
(1-2.拉伸膜的制造)
准备具有可以夹持长条的拉伸前膜的宽度方向的端部的两边的夹子的拉幅拉伸机。将长条的拉伸前膜提供给上述的拉幅拉伸机,用夹子将拉伸前膜的宽度方向的端部的两边保持,沿宽度方向拉伸,由此对拉伸前膜实施单轴拉伸处理。这时的拉伸条件为拉伸温度100℃、拉伸倍率2.0倍。由此,得到拉伸膜。
(1-3.加热处理)
一边在用拉幅装置的夹子将拉伸膜的宽度方向的端部的两边保持而使其绷紧的状态下输送拉伸膜,一边对拉伸膜实施加热处理。这时的加热条件为处理温度200℃、处理时间20分钟。由此,拉伸膜所包含的含有脂环式结构的聚合物的晶化进行,得到厚度50μm的长条的树脂膜。
从这样得到的树脂膜中切除被夹子夹持的端部,对剩余的部分用上述方法评价聚合物的晶化度、面取向系数δne、耐折度、吸水率、耐热温度及平滑性。
[实施例2]
在上述工序(1-1)中,以得到厚度50μm的树脂膜的方式调整拉伸前膜的厚度。
此外,在上述工序(1-2)中,对拉伸前膜不仅沿宽度方向实施拉伸而且沿长度方向也实施拉伸的同时双轴拉伸处理,由此制造拉伸膜。这时的拉伸条件为拉伸温度100℃、宽度方向的拉伸倍率2.0倍、长度方向的拉伸倍率2.0倍。
除了以上的事项以外,与实施例1同样地进行树脂膜的制造及评价。
[比较例1]
在上述工序(1-1)中,以得到厚度50μm的拉伸膜的方式调整拉伸前膜的厚度。
此外,不进行上述工序(1-3)。
除了以上的事项以外,与实施例1同样地进行,将未实施加热处理的拉伸膜作为树脂膜制造及进行评价。
[比较例2]
在上述工序(1-1)中,以得到厚度50μm的拉伸膜的方式调整拉伸前膜的厚度。
此外,在上述工序(1-2)中,对拉伸前膜不仅沿宽度方向实施拉伸而且沿长度方向也实施拉伸的同时双轴拉伸处理,由此制造拉伸膜。这时的拉伸条件为拉伸温度100℃、宽度方向的拉伸倍率2.0倍、长度方向的拉伸倍率2.0倍。
进而,不进行上述工序(1-3)。
除了以上的事项以外,与实施例1同样地进行,将未实施加热处理的拉伸膜作为树脂膜制造及进行评价。
[比较例3]
在上述工序(1-1)中,将拉伸前膜的厚度调整成50μm。
此外,不进行上述工序(1-2)及工序(1-3)。
除了以上的事项以外,与实施例1同样地进行,将未实施拉伸处理及加热处理的拉伸前膜作为树脂膜制造及进行评价。
[比较例4]
在上述工序(1-1)中,将挤出条件保持原样,将牵引速度设为2倍。此外,不进行上述工序(1-2)及工序(1-3)。
除了以上的事项以外,与实施例1同样地进行,将未实施拉伸处理及加热处理的拉伸前膜作为树脂膜制造及进行评价。
[比较例5]
在上述工序(1-1)中,作为成为膜的材料的树脂,使用聚对苯二甲酸乙二酯树脂。
此外,在上述工序(1-1)中,以得到厚度50μm的树脂膜的方式调整拉伸前膜的厚度。
进而,在上述工序(1-2)中,对拉伸前膜不仅沿宽度方向实施拉伸而且沿长度方向也实施拉伸的同时双轴拉伸处理,由此制造拉伸膜。这时的拉伸条件为拉伸温度120℃、宽度方向的拉伸倍率2.0倍、长度方向的拉伸倍率2.0倍。
除了以上的事项以外,与实施例1同样地进行树脂膜的制造及评价。
[比较例6]
在上述工序(1-1)中,作为成为膜的材料的树脂,使用不具有结晶性的环状烯烃树脂(日本瑞翁公司制造“zeonor”、玻璃化转变温度120℃)。
此外,在上述工序(1-1)中,将拉伸前膜的厚度调整成50μm。
进而,不进行上述工序(1-2)及工序(1-3)。
除了以上的事项以外,与实施例1同样地进行,将未实施拉伸处理及加热处理的拉伸前膜作为树脂膜制造及进行评价。
[比较例7]
在上述工序(1-1)中,作为成为膜的材料的树脂,使用不具有结晶性的环状烯烃树脂(日本瑞翁公司制造“zeonor”、玻璃化转变温度120℃)。
此外,在上述工序(1-1)中,以得到厚度50μm的拉伸膜的方式调整拉伸前膜的厚度。
进而,在上述工序(1-2)中,对拉伸前膜不仅沿宽度方向实施拉伸而且沿长度方向也实施拉伸的同时双轴拉伸处理,由此制造拉伸膜。这时的拉伸条件为拉伸温度120℃、宽度方向的拉伸倍率2.0倍、长度方向的拉伸倍率2.0倍。
此外,不进行上述工序(1-3)。
除了以上的事项以外,与实施例1同样地进行,将未实施加热处理的拉伸膜作为树脂膜制造及进行评价。
[比较例8]
在上述工序(1-1)中,作为成为膜的材料的树脂,使用不具有结晶性的乙烯-降冰片烯加成共聚物树脂。
此外,在上述工序(1-1)中,将拉伸前膜的厚度调整成50μm。
进而,不进行上述工序(1-2)及工序(1-3)。
除了以上的事项以外,与实施例1同样地进行,将未实施拉伸处理及加热处理的拉伸前膜作为树脂膜制造及进行评价。
[比较例9]
在上述工序(1-1)中,作为成为膜的材料的树脂,使用聚碳酸酯树脂(旭化成公司制造“wonderlightpc-115”、玻璃化转变温度145℃)。
此外,在上述工序(1-1)中,以得到厚度50μm的拉伸膜的方式调整拉伸前膜的厚度。
进而,在上述工序(1-2)中,对拉伸前膜不仅沿宽度方向实施拉伸而且沿长度方向也实施拉伸的同时双轴拉伸处理,由此制造拉伸膜。这时的拉伸条件为拉伸温度150℃、宽度方向的拉伸倍率2.0倍、长度方向的拉伸倍率2.0倍。
此外,不进行上述工序(1-3)。
除了以上的事项以外,与实施例1同样地进行,将未实施加热处理的拉伸膜作为树脂膜制造及进行评价。
[比较例10]
在上述工序(1-1)中,以得到厚度50μm的树脂膜的方式调整拉伸前膜的厚度。
此外,不进行上述工序(1-2)。因此,在上述工序(1-3)中,代替拉伸膜使用拉伸前膜。
除了以上的事项以外,与实施例1同样地进行未实施拉伸处理的树脂膜的制造及评价。
[结果]
将上述的实施例及比较例的操作概要示于表1,将结果示于表2。此外,在下述的表中,简称的意思为如下:
pdcpd:二环戊二烯的开环聚合物的加氢物
pet:聚对苯二甲酸乙二酯
cop:环状烯烃聚合物
coc:环状烯烃共聚物
pc:聚碳酸酯
δne:面取向系数
[表1]
[表1.实施例及比较例的操作概要]
[表2]
[表2.实施例及比较例的结果]
[讨论]
由表1及表2可知,在实施例1及实施例2中,得到耐折度、吸水率及耐热温度均良好的结果。由此可确认,根据本发明能够实现耐折性、低吸水性及耐热性均优异的树脂膜。
附图标记说明
110:平台;
110u:支承面;
120:试验片;
130:秤锤;
140:秤锤。
- 还没有人留言评论。精彩留言会获得点赞!