一种遗传性Usher综合征的致病突变及其检测试剂的制作方法
本发明属于生物医药领域,涉及一种遗传性Usher综合征的致病突变及其检测试剂。
背景技术:
Usher综合征,又称遗传性耳聋色素性视网膜炎综合征,视网膜色素变性感音神经性耳聋综合征,是一种常染色体隐性遗传性疾病。Usher综合征以先天性感音神经性聋、渐进性视网膜色素变性而致视野缩小、视力障碍为主要表现。临床上将Usher综合征主要分为以下三型:Ⅰ型为先天性重深度感音神经性聋,患者前庭反应消失,10岁左右出现视网膜色素变性;Ⅱ型为先天性中重度感音神经性聋,患者前庭反应正常,18~20岁出现视网膜色素变性;Ⅲ型为进行性感音神经性聋,前庭反应不确定,视网膜色素变性发生时间不确定。Usher综合征是临床上常见且危害较为严重的一类遗传性致聋、致盲性疾病,针对Usher综合征的遗传诊断能够辅助Usher综合征的临床诊断,也能为基因治疗提供必要的支持。
Usher综合征为单基因遗传病,具有显著的遗传异质性。目前已知的遗传模式为常染色体隐性遗传,已知的致病基因有13个(www.RetNet.org),且随着研究深入,该数目正逐年递增。然而,这些基因的突变并不能够解释全部Usher综合征患者的遗传病因,仍有40%到50%的Usher综合征患者的致病基因及突变尚未找到,提示存在未知的新致病基因有待挖掘。针对Usher综合征的分子遗传学研究必须建立在一定的分子生物学技术的基础上。研究Usher综合征致病基因的一个重要目的是进行分子诊断,鉴于其遗传异质性,寻求一种全新的Usher综合征致病基因的研究方法显得尤为迫切。高通量二代测序技术能够高效的针对大量基因的遗传信息进行捕获,具有传统技术所不可及的优势。
Centrosomal protein of 78 kDa(CEP78)基因位于9号染色体,该基因含有16个外显子,其编码的Cep78蛋白是一种含有722个氨基酸的中心体蛋白,目前关于Cep78蛋白功能的研究较少,CEP78基因突变与疾病,尤其与Usher综合征间的关系国内外均未见任何报道。
技术实现要素:
本发明的目的是针对上述缺陷,提供一种遗传性Usher综合征的致病突变。
本发明的另一目的是提供该致病突变的应用。
本发明的又一目的是提供该致病突变的检测试剂。
本发明的目的可通过如下技术方案实现:
一种用于检测遗传性Usher综合征的CEP78基因,突变的CEP78基因为剪切位点纯合突变CEP78c.1629-2A>G。
野生型CEP78基因在Ensemble数据库中的基因编号为:ENSG00000148019,所述的突变的CEP78基因在物理位置为80880286(human genome reference 19;hg19)的碱基由A突变为G其他部分与野生型相同。
本发明所述的突变的CEP78基因在制备遗传性Usher综合征检测试剂或检测设备中的应用。
其中所述的检测试剂优选自:引物或引物对、探针、抗体、或核酸芯片中的一种或多种。
所述的检测设备优选包括含有检测突变的CEP78基因的基因芯片的检测平台。
一种检测遗传性Usher综合征的试剂盒,所述的试剂盒包括:
(a)检测CEP78基因物理位置为80880286核苷酸位点的试剂;
(b)说明书;其中记载:发现CEP78基因在物理位置为80880286的碱基由A变为G的突变为遗传性Usher综合征患者的致病突变。
其中,所述的试剂优选自:引物或引物对、探针、抗体、或核酸芯片中的一种或多种。
作为本发明的一种优选,所述的试剂为基于深度测序为平台的基因芯片杂交探针。
所述的试剂进一步优选包含如序列表中SEQ ID NO.28所示的检测80880286核苷酸位点的杂交探针chr9|80880268-80880387。
作为本发明的另一种优选,所述的试剂为PCR检测80880286核苷酸位点的试剂。
所述的PCR检测80880286核苷酸位点的试剂进一步优选包含PCR检测80880286位点的引物对,其正向引物序列为SEQ ID NO.36,反向引物序列为SEQ ID NO.37。
一种以深度测序为平台筛查Usher综合征患者中CEP78基因突变的方法,包括以下步骤:
(1)建立Usher综合征患者家系临床与遗传资源库,收集Usher综合征家系的临床资料及血液标本,提取基因组DNA;
(2)设计SEQ ID NO.1~SEQ ID NO.35所示的用于检测CEP78基因突变的杂交探针,并将其整合到基因芯片上;
(3)利用制备的基因芯片捕获目标区域并进行深度测序;
(4)对测序结果进行优化的生物信息学分析,筛选到一个新的Usher综合征的致病突变为纯合突变CEP78 c.1629-2A>G。突变CEP78 c.1629-2A>G位于9号染色体,物理位置为80880286(hg19)的碱基由A突变为G。
其中,步骤(2)所述的基因芯片优选Illumina公司生产的基因芯片。
步骤(3)所述的利用制备的基因芯片捕获目标区域并进行深度测序优选利用美国Illumina公司的HiSeq 2000仪器完成。
步骤(3)所述的利用制备的基因芯片捕获目标区域并进行深度测序优选流程为:将基因组DNA片段化,在DNA末端标记“A”并与Illumina PE接头-寡核苷酸混合物相连接;连接产物经PCR富集,获得DNA文库,并将DNA文库与已知致病基因捕获芯片杂交、洗脱、纯化,获得编码序列;创建配对末端,在Illumina HiSeq 2000平台上对目标序列进行测序。
有益效果
1.Usher综合征是常见的、严重的遗传性致聋、致盲疾病,在我国发病率较高,严重危害了国民健康。挖掘Usher综合征的新致病突变和新致病基因有利于进一步探索Usher综合征的分子遗传学病因,是充分利用我国的眼科遗传病资源,造福Usher综合征患者的现实需要,是后基因组时代最重要的研究方向之一。本专利旨在探索Usher综合征的遗传学病因,从而帮助了解发病机制、辅助临床诊断、产前诊断和转基因治疗。
2.目前并没有研究指出CEP78基因突变与Usher综合征间的关系,因此,针对CEP78基因与Usher综合征的相关研究显得尤为必要。本专利选择CEP78基因作为Usher综合征的候选基因,是由申请人在多年从事临床医疗、遗传学研究的基础上所筛选出的,通过本发明方法进一步明确了CEP78基因与Usher综合征的关系。
3.一个基因可以对应多个不同的转录本,且不同转录本所编码的RNA及蛋白有所不同。本专利中申请人根据长期从事遗传学研究的经验,筛选出CEP78基因的最优转录本,并根据不同的转录本设计相应的探针,从而使筛查效益达到最高,最终获得Usher综合征的致病突变CEP78 c.1629-2A>G。
4.Usher综合征具有一定的遗传异质性,目前已知的致病基因有13个,但仍存在未知的致病基因。本发明提供了新的致病基因的致病位点,为该疾病的诊断提供了新的分子生物学基础。
附图说明
图1 家系图
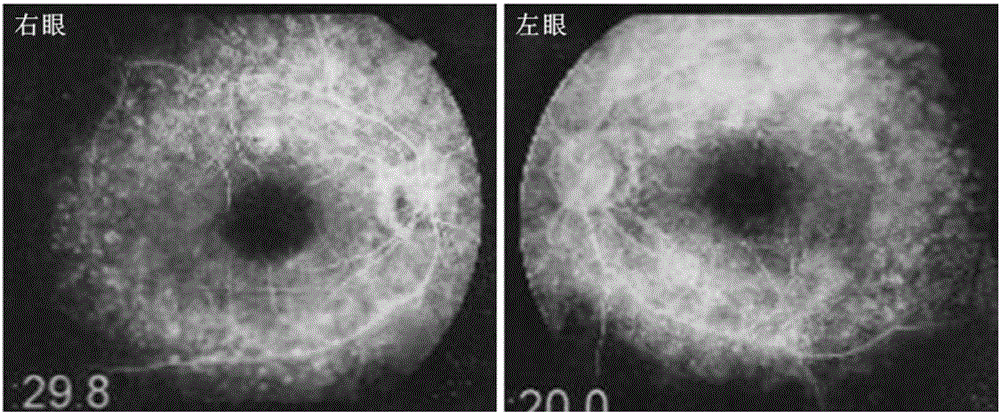
图2 患者双眼眼底荧光血管造影检查结果
图3 患者双眼光学相干断层成像检查结果
图4 CEP78基因突变序列测序图谱
具体实施方式
实施例1
对一个近亲结婚Usher综合征家系的CEP78基因突变进行检测。
实验方法:
1.该家系临床资源的采集及遗传资源库的建立:
收集该家系患者的临床资料及血液样本,家系图见图1。临床资料主要包括个人病史、家族史、最佳矫正视力(best corrected visual acuities;BCVAs)、裂隙灯检查、眼底照、色觉检查、视野检查(Humphrey视野计)、全视野电生理检查(electroretinography;ERG)、眼底荧光血管造影检查(fundus fluorescein angiography;FFA)及光学相干断层成像检查(optical coherence tomography;OCT)等。并用血液基因组DNA提取试剂盒(Qiagen,Hilden,Germany)对家系各成员的血液基因组DNA进行提取。
2.借助于高通量二代测序挖掘该患者的致病突变:
2.1设计并订制捕获芯片:
2.1.1 CEP78基因及转录本序列信息:
该基因捕获芯片为全外显子组捕获芯片,可对目前所有已知基因进行检测,其中包括全部目前已知的Usher综合征相关的致病基因。我们所参照的CEP78基因在Ensemble数据库中的基因编号为:ENSG00000148019(注:该编号来自Ensemble数据库,www.ensembl.org,可输入基因编码检索基因详细信息及基因序列)。
2.1.2转录本的选择:
针对不同基因选择特定的转录本,每个基因均含有多个转录本,在选择转录本时,我们的原则是:首先考虑拥有CCDS编码蛋白的转录本,若一个基因有多个转录本均编码蛋白,则首选含氨基酸数最多的蛋白相对应的转录本,若多个转录本氨基酸含量相同,则进一步选择含碱基数最多的转录本。据以上原则,我们所筛选出的CEP78基因转录本编号为:ENST00000376597(注:该编号来自Ensemble数据库,www.ensembl.org,可输入转录本编码检索转录本详细信息及转录本序列)。
2.1.2杂交探针的设计:
杂交探针的设计标准为:(1)探针覆盖所有候选基因的目标区域,即外显子区域以及外显子和内含子拼接处(外显子上下游各100个bp);(2)去除重复序列:对于在基因组出现的高度重复序列以及在人类基因组中出现2-5倍的较低频率的重复片段,我们予以去除,避免捕获其他同源基因,增加假阳性,从而降低检测效率;(3)在探针设计过程中,我们对临近的外显子进行了特定的整合,其相邻探针整合标准为:当相邻外显子的综合目标区域(即前个外显子的上游100bp起至后一个外显子的下游100bp止)总和小于600bp,即将其整合为一个探针,以求通过一对探针完成多对外显子区域的捕获;(4)当所设计探针序列小于250bp时,在其两端各包含上下游100bp的内含子的基础上,各继续增加相同bp数的内含子,使探针大小达到250bp。根据以上设计原则,我们针对CEP78基因所设计的探针序列如下:
用于筛选Usher综合征致病基因CEP78的杂交探针序列共35条,序列如SEQ ID
NO.1~SEQ ID NO.35所示;
2.2目标区域捕获及深度测序:
首先将基因组DNA片段化,并在DNA末端标记“A”,与Illumina PE接头-寡核苷酸混合物相连接,连接产物经PCR富集,获得DNA文库。然后将DNA文库与已知致病基因捕获芯片杂交、洗脱、纯化,获得编码序列。最后创建配对末端,在HiSeq 2000(Illumina)平台上对目标序列进行测序。
2.3对测序数据进行生物信息学分析,筛选出候选致病基因:
2.3.1采用Mosaik软件(http://bioinformatics.bc.edu/marthlab/Mosaik)处理Illumina原始测序数据(配对末端数据),产生.bam类型文件。将.bam文件输入GATK,利用GATK检测单核苷酸变异体(single nucleotide variant)和小的插入或缺失(insertion/deletions),同时进行质量评估,便于下游的生物信息学分析,最后产生.vcf类型文件。
2.3.2将患者的测序结果在包括dbSNP144
(http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/snp144.txt.gz.),HapMap计划
(ftp://ftp.ncbi.nlm.nih.gov/hapmap),1000 Genome Project
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp),炎黄数据库(http://yh.genomics.org.cn/),Exome Variant Server(http://evs.gs.washington.edu/EVS/)及Exome Aggregation Consortium
(http://exac.broadinstitute.org/)在内的6个单核苷酸多态性(single nucleotide polymorphism,SNP)数据库中筛查,滤过所有已知的SNP及Indel位点;
2.3.3将患者的测序结果所对应的基因序列进行比较和分析,优先分析纯合的插入/缺失突变、无义突变、错义突变和剪切位点的突变。
2.4经Sanger测序验证,鉴定致病基因:
PCR法分别针对筛选出的突变位点及邻近DNA序列在相应家系中进行扩增,所用引物序列采用Primer 3(http://frodo.wi.mit.edu/)引物设计软件设计。所用PCR的反应体系(20μL体系)为:5*buffer 4μL,25mM MgCl2 2μL,DNA 1μL,正向引物F 1μL,反向引物R 1μL,10mM dNTP 0.4μL,Taq酶0.1μL,ddH2O 10.5μL。PCR反应程序:98℃5min,35个循环(98℃10s,60℃15s,72℃1min),72℃7min,4℃5min。1%琼脂糖凝胶电泳检测,与紫外线切胶仪下切取PCR产物凝胶并纯化。对所有的PCR产物分别以正、反向引物测序,并对测序结果进行进一步分析,使用NCBI在线对比工具BLAST(http://blast.ncbi.nlm.nih.gov/),排除假阳性结果。其中检测80880286核苷酸位点的正向引物序列为SEQ ID NO.36,反向引物序列为SEQ ID NO.37。
实验结果:
1.家系情况:
该家系为近亲结婚家系,家系内除患者V:1外,并无其他成员有类似的疾病史,家系图见图1。
2.临床资料:
临床专家对患者V:1进行了详细全面的临床检查后,对该患者作出了Usher综合征的临床诊断。该患者,女,自幼夜盲,8岁时开始出现轻度色觉障碍及听力障碍。最近一次眼科临床检查时间为2013年,患者当时35岁。患者存在轻度的色觉异常。眼底照相检查提示双眼底呈典型的视网膜色素变性改变,包括视乳头苍白,视网膜血管迂曲、变细,周边部视网膜萎缩,可见骨细胞样色素沉着。眼底FFA检查结果与眼底照相一致,可见视网膜血管形态迂曲、变形,后极部视网膜出现散在点状高荧光区域,提示存在视网膜色素上皮的萎缩,见图2。OCT检查结果进一步提示该患者双眼视网膜色素上皮层及外核层均明显变薄,周边部视网膜内、外节盘层消失,见图3。ERG检查结果提示患者双眼视锥、视杆细胞反应均呈熄灭状。综上,患者的临床表现符合Usher综合征的诊断。
3.该家系遗传检测结果:
通过对患者V:1进行了全外显子组测序及生物信息学分析后,我们发现该患者的可疑致病突变为CEP78 c.1629-2A>G,该突变为剪切位点突变,未发现其他可疑的致病基因突变位点。Sanger测序进一步证实了该突变,排除了假阳性,测序结果见图4。突变CEP78c.1629-2A>G位于9号染色体,物理位置为80880286(Ensemble数据库)的碱基由A突变为G,该基因相关突变从未在Usher综合征患者中被发现。
根据我们所设计的筛选流程,借助我们所设计的基因芯片及深度测序技术,我们成功证实所检测到的CEP78基因为Usher综合征新致病基因,c.1629-2A>G为该疾病的新致病位点。
实施例2:
针对实施例1中所检测出的致病突变CEP78 c.1629-2A>G可能引起的剪切异常进行预测。
实验方法:
1.剪切位点改变预测:
我们所找到的突变位于第14号外显子与第13、14号外显子间内含子的受体剪切位点附近,突变与正常剪切位点的位置关系见图4。我们借助一系列剪切位点预测网站对可能产生的剪切位点改变进行了预测,包括:
(1)Human Splicing Finder,网址:http://www.umd.be/HSF3/HSF.html;
(2)NetGene2 Server,网址:http://www.cbs.dtu.dk/services/NetGene2/;
(3)Splicing Regulation Online Graphical Engine,网址:http://sroogle.tau.ac.il/;
(4)Berkeley Drosophila Genome Project,网址:http://www.fruitfly.org/seq_tools/splice.html;
(5)SplicePort,网址:http://spliceport.cbcb.umd.edu/。
实验结果:
1.剪切位点改变预测结果:
(1)Human Splicing Finder软件预测结果提示该突变能够引起第14号外显子与第13、14号外显子间内含子的正常受体剪切位点的消失,并产生新的异常受体剪切位点,从而引起CEP78基因所编码的Cep78蛋白结构的改变;
(2)NetGene2 Server软件预测结果提示该突变能够引起正常剪切位点的消失,引起CEP78基因所编码的Cep78蛋白结构的改变;
(3)Splicing Regulation Online Graphical Engine软件预测结果提示该突变能够引起正常剪切位点的消失,引起CEP78基因所编码的Cep78蛋白结构的改变;
(4)Berkeley Drosophila Genome Project软件预测结果提示该突变能够引起正常剪切位点的消失,引起CEP78基因所编码的Cep78蛋白结构的改变;
(5)SplicePort软件预测结果提示该突变能够引起正常剪切位点的消失,引起CEP78基因所编码的Cep78蛋白结构的改变。
- 还没有人留言评论。精彩留言会获得点赞!