基因mRNA的m6A单碱基位点鉴定方法与流程
本发明属生物
技术领域:
,涉及一种基因mRNA的m6A单碱基位点鉴定方法。
背景技术:
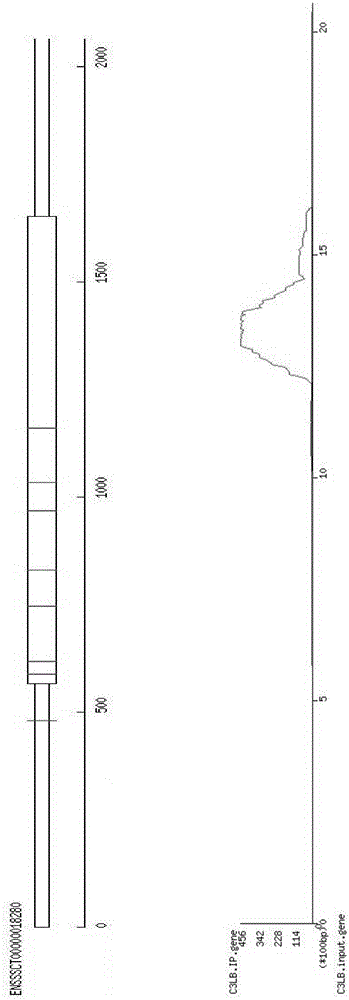
:m6A是指发生在碱基A第6位N原子上的甲基化修饰是一种最常见的转录后水平修饰,大约占全部RNA修饰的三分之二。在真核生物中,m6A大约占细胞mRNA全部腺苷含量的0.1%-0.4%。随着二代测序技术的高速发展,各生物基因组库逐步建立完整,利用测序来检测基因的甲基化分布并筛选出甲基化差异基因已成为较好的研究基因甲基化的重要方式。本实验室建立了mRNA甲基化高通量检测方法,实现了多个样本全转录组甲基化程度的比较,对mRNAm6A多样性的生物学研究开辟了新的技术。但是m6A并不能影响其配对碱基的能力,且没有化学试剂来改变或特异性标记这一修饰,现有技术无法精确确定m6A位点。本发明在高通量测序的基础上,利用位点预测并结合分子生物学技术可以实现单个碱基的m6A位点准确鉴定,这为进一步研究m6A的功能提供了技术保障。技术实现要素:为了解决上述问题,本发明提供一个基因mRNA的m6A单碱基位点鉴定方法,包括以下步骤:1)、根据《mRNA甲基化高通量检测方法》建立的方法对样品m6A高通量测序,根据测序结果找到靶基因,并确定其m6A峰大致位置;2)、利用在线工具SRAMP(http://www.cuilab.cn/sramp/)得到该基因的m6A峰区域序列中所有可能存在m6A位点;3)、将所有可能的m6A位点进行点突变,并将该基因的CDS序列和突变序列分别连入哺乳动物表达载体pSilencer4.1,构建该基因的超表达载体和突变载体;4)、将步骤3)中得到的两种载体分别转染细胞,收集细胞提取RNA;将提取的RNA片段化,利用免疫沉淀反应,得到富集的含有m6A的RNA片段;5)、将步骤4)得到的富集的含有m6A的RNA反转录为cDNA,设计各个突变位点区域的荧光定量引物;将超表达载体组和突变载体组的荧光定量结果比较,若某位点突变载体组表达量小于超表达载体组且P<0.05(t-test),则判定该位点是m6A位点。作为本发明的基因mRNA的m6A单碱基位点鉴定方法的改进,靶基因为FAM134B基因,其突变(同义突变)为:位点1:GGACA→GGCCA位点2:TGACC→TGCCC位点3:GGACA→GGCCA;荧光定量PCR引物为:位点1:F5’-ACGGGACCTTCAACCTTTCA-3’R5’-AGTGCCTCTCTTTGCTTGGT-3’位点2:F5’-CCAAGCAAAGAGAGGCACTCA-3’R5’-CTAACTGGTCTTTGATGGCGG-3’位点3:F5’-CCGCCATCAAAGACCAGTTAG-3’R5’-TGGCCTCCGAGTAGATTTGA-3’。作为本发明的基因mRNA的m6A单碱基位点鉴定方法的进一步改进:所述步骤1):按照2016103629813的发明《mRNA甲基化高通量检测方法》进行高通量测序;根据测序结果挑选出靶基因,并确定其m6A峰大致位置。本发明提供了一种快速简便的方法,检测出单个基因所有可能的甲基化特征序列(RRACH)中m6A位点所在的具体位置。本发明首次利用简单快速可靠的生物学技术方法,确定m6A单碱基位点;本发明可以为实现m6A单碱基位点的生物学功能研究提供重要技术支撑。附图说明下面结合附图对本发明的具体实施方式作进一步详细说明。图1:长白猪背部脂肪(C-LB)组织m6A测序所得FAM134B基因甲基化分布(m6A峰区域1250-1490bp);图2:FAM134B测序结果m6A峰区域中的m6A位点;图3:为FAM134B的m6A峰区域序列(下划线序列为可能含有m6A的密码子);图4:FAM134B的m6A峰区域突变后序列(下划线序列为突变后的密码子)图5:荧光定量结果显示位点3为FAM134B基因的m6A位点。具体实施方式下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:实施例1、1、按照2016103629813的发明《mRNA甲基化高通量检测方法》进行高通量测序;根据测序结果挑选猪FAM134B为靶基因(如图1所示),并确定其m6A峰大致位置为区域1250-1490bp处。2、在NCBI上搜索并得到该区域的序列。进入SRAMP网站(http://www.cuilab.cn/sramp/),选择prediction,在右侧的MaturemRNAmode框中将序列输入,选择submit,得到猪FAM134B基因1250-1490bp处所有的m6A预测位点。3、将3处可能的m6A位点进行点突变,分别为:位点1:GGACA→GGCCA位点2:TGACC→TGCCC位点3:GGACA→GGCCA将3处突变的CDS序列合成并连接到哺乳动物表达载体pSilencer4.1上,构建FAM134B突变载体。另外,将FAM134B的原始CDS序列并连接到哺乳动物表达载体pSilencer4.1上,构建成FAM134B超表达载体作为对照。4、培养人源胚胎肾细胞系(293T),待6个10cm平皿的细胞均长到80-90%密度时,用脂质体转染试剂lipofectamine3000进行载体的转染,超表达载体和突变载体各转染3个平皿,具体步骤如下:1)、按照转染时密度为80-90%左右的要求,转染前一天将培养基替换为无抗培养基。备注说明:培养293T细胞所用培养基为含有10%胎牛血清和1%青链霉素的DMEMBasic培养基。所替换的无抗培养基为含10%胎牛血清的DMEMBasic培养基。胎牛血清购自杭州四季青生物有限公司,青链霉素与DMEMBasic培养基购自Gibco公司。2)、每个皿细胞均进行如下操作:使用DMEMBasic或OPTI-MEM培养基0.5ml分别稀释10ugFAM134B超表达载体/突变载体和25uLlipofectamine3000转染试剂,37℃放置5min混匀,室温保存15min后,将复合物逐滴加入皿中。lipofectamine3000转染试剂可购自life生物有限公司。3)、轻轻摇晃细胞后,放于CO2培养箱中培养6h(37摄氏度恒温,5%CO2);更换无抗完全培养基。转染24h后,收集细胞。5、细胞甲基化mRNA提取将步骤4所得的细胞用Trizol法提取样品的总RNA,具体方法为:①将细胞置于RNase-free的1.5mleppendorf管中;②加入1mlTrizol剧烈摇晃至均一溶液;③加入200ul氯仿后,剧烈摇晃至均一溶液,4C,12000g离心15min;④取上清到另一个RNase-free的1.5mleppendorf管中,加入相同体积异丙醇,轻轻混匀,室温孵育10min,4C,12000g离心10min;⑤去掉上清,用1ml75%乙醇洗涤沉淀,4C,7500g离心5min⑥用20-50ulRNase-freeH2O溶解。将上步所得的总RNA用GenelutemRNAminiprepKits(Sigma)提取mRNA,具体方法为:①将totalRNA体积扩大到250ul,加入250ulbindingsolution,混匀;②加入15ulbeads,剧烈摇晃至均一溶液,置于70C孵育3min后,置于室温10min;③最大速度离心2min,弃掉上清;④用500ulwashbuffer重悬beads,并转移至spinfilter;⑤离心1-2min,弃掉flowthrough;⑥重复步骤④、⑤;⑦加入预先加热至70C的ElutionBuffer50ul,70C孵育2-5min;⑧离心产物即是mRNA;⑨将所得mRNA利用真空浓缩至9ul。6、将总mRNA经过超声断裂或者金属离子(如Zn2+、Ca2+、Fe2+等)断裂成100~200nt的mRNA片段,具体方法入下:RNA片段化体系温度、孵育时间为:70℃、15min,加入EDTA终止反应。片段化缓冲液(10X)为:800μLRNase-free水,100μL1MTris-HCl(pH=7.0),100μL1MZnCl2溶液。7、免疫沉淀1)、将片段化的mRNA取出50ng左右作为Input(对照),其余部分定义为IP(免疫沉淀),首先利用免疫沉淀反应,与m6A抗体在4℃低温下孵育2小时,具体步骤如下:在4℃孵育2h。2)、封闭beads(ProteinA,Lifetechnologies,10002D)取40μlbeads置于磁力架上,弃去上清,用1ml的1*IPbuffer洗三次,重悬于1*IP封闭buffer1ml中,于4℃孵育2h。1*IP封闭buffer配制如下:5×IPbuffer200μlRNAaseinhibitor10μlBSA25μlRNAasefreewater765μlTotal1000μl孵育2h后,用1ml的1*IPbuffer洗三次,用100μl1*IPbuffer重悬。3)、beads与mRNA-抗体免疫沉淀:将上述1)和2)得到的样品混合,4℃孵育2h。4)、洗脱I将步骤3)所得样品置于磁力架上,弃上清,用500μl1*IPbuffer洗三次,重悬于洗脱buffer450ul,置于4℃孵育1h。洗脱buffer配制如下:5×IPbuffer90μlRNAaseinhibitor7μlM6A150μlRNAasefreewater203μlTotal450μl5)、洗脱II将4)所得样品置于磁力架上,收集上清;然后在beads中再加入50ul洗脱buffer,置于4℃孵育30min,再次置于磁力架上,再次收集上清;6)、将步骤5)的2份上清进行合并,得到200ul洗脱液(RNA洗脱液)。7)、乙醇沉淀:将200ulRNA洗脱液+20ulNaAc(浓度为3M)+500ul乙醇(100%)+4ulglycogen(5mg/ml),-80℃,过夜沉淀。8)、第二天在4℃,14000rpm离心30min,弃上清,加入1ml75%乙醇在4℃,14000rpm离心30min,弃上清,风干后,加入9ulRNase-free水,并测定浓度。从而获得富集的含有m6A的RNA片段。8、引物设计将步骤7得到的富集的含有m6A的RNA反转录为cDNA,设计突变位点区域的荧光定量引物:根据NCBI上提供的突变位点区域序列,在涵盖甲基化高度保守的RRACH(R=G,A;H=A,C,T)序列区域设计荧光定量PCR引物(为多对荧光定量PCR引物),用于分析基因的m6A分布。各位点的引物:位点1:F5’-ACGGGACCTTCAACCTTTCA-3’R5’-AGTGCCTCTCTTTGCTTGGT-3’位点2:F5’-CCAAGCAAAGAGAGGCACTCA-3’R5’-CTAACTGGTCTTTGATGGCGG-3’位点3:F5’-CCGCCATCAAAGACCAGTTAG-3’R5’-TGGCCTCCGAGTAGATTTGA-3’9、实时荧光相对定量PCR将超表达载体组(FAM)和突变试验组(mF)的RNA反转录成cDNA,按常规荧光定量PCR步骤操作。以m6A-IP和Input的靶基因Ct差值作为参照,结果分析采用2-△△Ct的方法,其中△△Ct的计算公式如下:△△Ct=(CtmF-CtCon)X1-(CtmF-CtCon)X2X1和X2表示待比较的任意两个样本,通过上述公式计算出X1相对于X2的靶基因甲基化相对水平。结果如图5所示,得到以超表达载体组(FAM)的FAM134B表达量为“1”,突变试验组(mF)的FAM134B表达量可变的柱状图。结果表明,位点3的FAM134b表达量突变组显著低于超表达组,其他位点无显著差异,由于突变试验组(mF)转染进入细胞的载体不会产生含有m6A的FAM134BRNA片段,因此步骤7中得到的突变试验组(mF)富集m6A的RNA中不含FAM134B,导致荧光定量结果中该组的理论值会下降。结合m6A测序所得FAM134B基因甲基化分布图,证明位点3(1356-1361)是长白猪FAM134B基因的甲基化位点。对比例1、将实施例1步骤8中的引物改成如下:位点1:F5’-ACCGCTCGATGAGTGATCCTTACC-3’R5’-AAACGGTAAGGATCACTCATCGAG-3’位点2:F5’-CCAGGCCTATGACGATGGAG-3’R5’-ACCGTCGCACGATATCACTC-3’位点3:F5’-AATCAAGGCTGTTGAACCCG-3’R5’-TCCTGTGAACACCTGCTGAT-3’其余内容等同于实施例1。荧光定量结果中溶解曲线不正常,表明引物特异性不佳。此结果不可用。最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1