用于Chip‑seq全基因组结合谱的内源蛋白标记的方法与流程
本发明属于蛋白标记领域,涉及一种应用于大规模筛选癌症转录因子转录调控机制的方法,尤其是涉及一种用于Chip-seq全基因组结合谱的内源蛋白标记的方法。
背景技术:
CRISPR/Cas9是一种细菌和古细菌在长期演化过程中形成的适应性免疫防御系统,新兴基因编辑技术,被认为是改变世界的革命性技术。2013年初开始,CRISPR/Cas9作为基因编辑技术在哺乳动物细胞中使用。CRISPR/Cas9介导的基因编辑具有效率高、准确性好的特点,受到众多研究者的青睐,并被用于癌症中特定基因的功能研究、癌症模型的建立及靶基因的大规模筛选。2015年本研究团队开发了CRISPR/Cas9大规模基因敲除数据的分析算法MAGeCK。针对sgRNA可能存在的脱靶问题,国内外研究者开发出一系列基于CRISPR/Cas9的改良技术和sgRNA设计及算法。目前常用的sgRNA在线设计网站有:张锋实验室建立的http://crispr.mit.edu/和Michael Boutros实验室建立的http://e-crisp.org/E-CRISPR/designcrispr/。
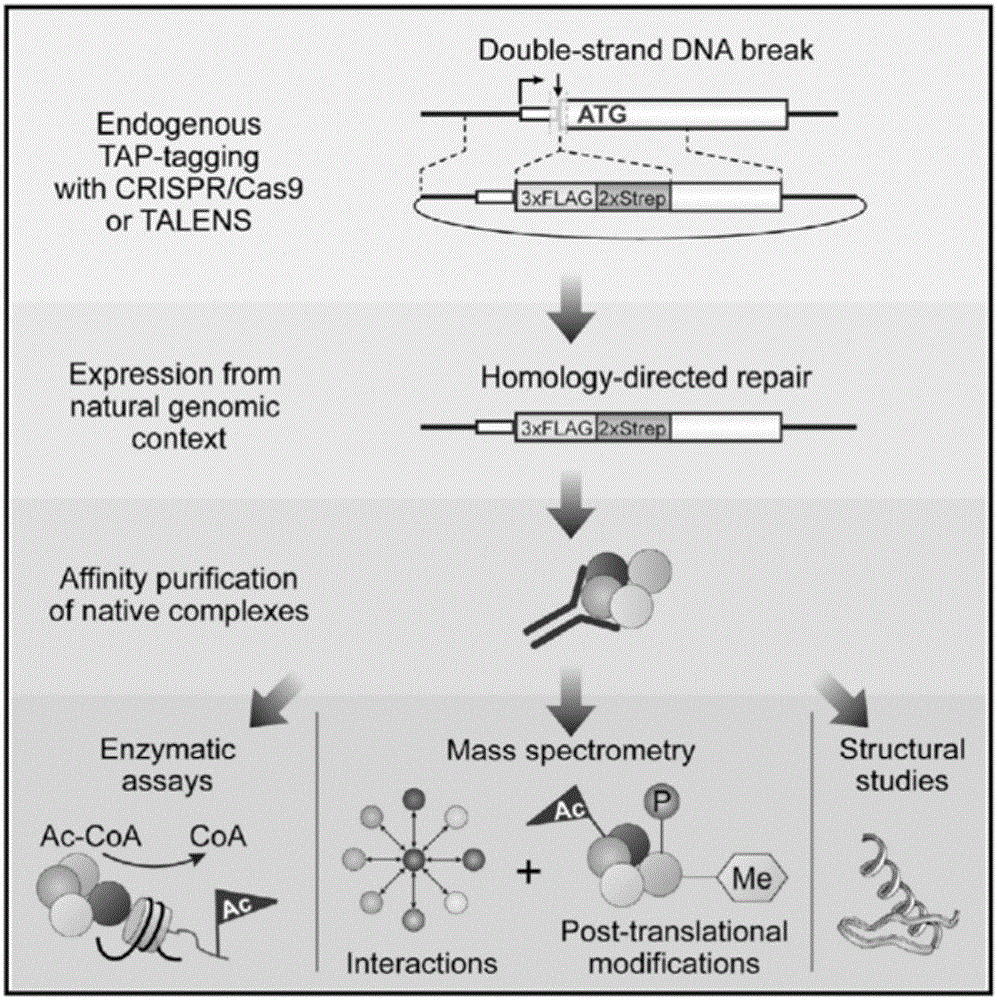
自2014年起,CRISPR/Cas9的应用变得多元化。研究者们开始将CRISPR/Cas9用于控制基因转录。2015年10月,Dalvai等人用CRISPR/Cas9技术,在基因组上对标签插入靶点进行双链切割(Double-strand DNA break);并通过同源重组修复(homology-directed repair,HR)在内源基因上标记3xFLAG和2xSTREP标签;结合亲和纯化和质谱技术(AP-MS),成功分离了NuA4蛋白复合体的组成亚单位。其结果与前的研究相符,研究中还首次分离了复合体MCM8/9的亚单位(见图1)。同年,Dellaire研究团队将绿色荧光蛋白(GFP)的变体Clover标记在内源基因上的第一外显子,用于筛选能够增强CRISPR/Cas9介导的同源重组修复途径的小分子,并成功筛选出小分子RS-1。以上CRISPR/Cas9内源蛋白标记技术为转录因子的转录调控功能研究提供了新的思路。Chip-seq技术已经广泛用于转录因子结合高通量分析,但是目前由于转录因子抗体特异性的限制,相当一部分转录因子的转录调控机制尚不清楚。
技术实现要素:
本发明的目的就是为了克服上述现有技术存在的缺陷而提供一种用于Chip-seq全基因组结合谱的基于CRISPR/Cas9技术的内源蛋白标记的方法。
本发明的目的可以通过以下技术方案来实现:
一种用于Chip-seq全基因组结合谱的内源蛋白标记的方法,该方法基于CRISPR/Cas9技术,具体包括以下步骤:
(1)设计和筛选具有切割效率的sgRNA:
在线设计sgRNA,针对三种ERα共转录因子基因的终止编码位点(stop codon site)上下游50bp区域,设计sgRNA,同时引入DNA开放性参数,进一步筛选位于开放程度高的基因组DNA上的sgRNA;
(2)构建含有sgRNA的慢病毒载体:
sgRNA序列的5’端加入序列5’-GACCA-3’,sgRNA的反向互补序列的5’端加入序列5’-GACCA-3’。合成修饰过的sgRNA序列。慢病毒质粒选用带有Cas9和Puro元件的lentiCRISPRv2质粒。用BsmBI酶对lentiCRISPRv2质粒进行酶切和去磷酸化,将线性化的质粒进行胶回收纯化,并用双蒸水溶解。用退火的方法将sgRNA的两条单链以互补配对的方式结合,并用T4PNK将sgRNA磷酸化。最后用连接酶将线性化质粒,并与sgRNA连接。用Stbl3感受态细胞进行转化,从而得到含有sgRNA的lentiCRISPRv2载体。用菌液测序鉴定sgRNA插入的正确性。
(3)制备表达sgRNA的慢病毒并确定病毒感染效率:
病毒包装质粒选用psPAX2和pMD2.G。将含有sgRNA的lentiCRISPRv2、psPAX2和pMD2.G转染293FT细胞,转染后培养48h,吸取细胞培养基,收获病毒,继续培养24h后第二次收获病毒。将两次收取的病毒合并,用0.45μm的滤器过滤,去除细胞碎片。在MCF-7细胞中鉴定病毒感染效率。在6孔板中培养MCF-7,18h后,每个孔分别用0μL、50μL、100μL、200μL、400μL、600μL的病毒,同时加入终浓度为10μg/ml的polybrene加强病毒感染效率。感染48后,用1.5μg/ml的puromycin筛选72h,此时对照组(0μL病毒组)的细胞全部死亡,选用能够使细胞100%存活的最小病毒量作进行后续实验。
(4)鉴定sgRNA效率:
采用两种方法鉴定sgRNA效率。
一种为T7E1核酸酶酶切实验。具体方案为:PCR扩增含有预期切割位点的DNA片段,使DNA片段长度为700bp左右,PCR产物进行切胶纯化。检测纯化后的DNA浓度,实验组和对照组取相同DNA量,用T7E1对DNA片段进行酶切。其中实验组为感染含有待检sgRNA的病毒的MCF-7,对照组为野生型MCF-7。酶切后用琼脂糖凝胶电泳检测,若实验组DNA产生小于700bp的两条条带,则说明sgRNA有效。
T7E1核酸酶酶切实验中,T7E1内切酶的用量为0.5μL/10μL体系。
PCR体系为:
另一种为PCR结合一代测序。具体方案为:用PCR扩增含有预期切割位点的DNA片段,送公司测序,以分析结果PAM序列(NGG)前3个碱基附近起出现明显杂峰为sgRNA有效的标准。
(5)建立NCOA1带有Flag标签的MCF-7细胞模型
以NCOA1的Flag标记为例。用CRISPR/Cas9技术在NCOA1的转录终止位点(stop codon site)处插入Flag标签(Flag-tag),插入位置和donor的设计如图2所示。用lonza电转仪将含有靶向NCOA1的sgRNA的lentiCRISPRv2,转染到含有dox诱导ERα537位氨基酸突变的MCF-7细胞,培养24h后,用puromycin筛选24h,用PCR结合一代测序检测NCOA1基因3’端是否有Flag-tag的插入。若有Flag-tag的插入,则用细胞稀释的方法分离和培养单个细胞,建立单克隆细胞株。并再次用PCR结合一代测序鉴定Flag-tag插入的正确性。使用Flag-tag抗体,采用Western blot检测Flag蛋白的表达,确认内源标记成功。
步骤(1)中在线设计sgRNA时,可以利用本实验室开发的在线设计网站http://cistrome.org/crispr/tool进行设计。
步骤(2)中,Lenti CRISPRv2、psPAX2、pMD2.G按照质量比4:3:1的比例转染293FT细胞,Lenti CRISPRv2与X-tremeGENE HP DNA Transfection Reagent的比例关系为6μg:30μL。
步骤(2)中,细胞转染后15小时换液,并于之后的24小时和48小时分别收取病毒液,并将两次收取的病毒液合并待用。
与现有技术相比,本发明具有以下优点及有益效果:
1.目前常用的sgRNA在线设计工具有张锋实验室建立的http://crispr.mit.edu/和Michael Boutros实验室建立的http://e-crisp.org/E-CRISPR/designcrispr/。而本发明开发的sgRNA设计方法,设计参数中加入了染色质开放性,能显著提高提高设计的sgRNA的效率(图3)。如图3所示,针对NCOA1共设计5种sgRNA。其中根据加入染色质开放性参数的方法得到sgRNA1和sgRNA2,应用在线设计工具http://crispr.mit.edu/设计sgRNA3和sgRNA4,应用在线设计工具http://e-crisp.org/E-CRISPR/designcrispr/设计sgRNA4,PCR结合一代测序结果显示加入染色质开放性参数的方法得到sgRNA1和sgRNA2的Cas9切割效率明显高于其他两种方法。
2.本发明在慢病毒包装中,选用Roche的转染试剂X-tremeGENE HP DNA Transfection Reagent将Lenti CRISPRv2及另外两种病毒包装质粒psPAX2和pMD2.G共转染至293FT细胞中,培养细胞,收获病毒液;并且通过大量实验确定了CRISPRv2、psPAX2、pMD2.G按照质量比4:3:1的比例转染293FT细胞,并且确定Lenti CRISPRv2与X-tremeGENE HP DNA Transfection Reagent的比例关系为6μg:30μL。之前的相关方法中常用的CRISPRv2、psPAX2、pMD2.G按照质量比为3:2:1。本发明在此基础上做了一系列质量比,确定4:3:1的比例包装的病毒的感染效率最高,并且此质量比例更接近质粒摩尔比1:1:1,更符合慢病毒在自然状态下的包装比例。
3、本发明采用内源基因标记与高通量描绘结合谱方法相结合。由于目前对于相当多的一部分转录因子,因其抗体的特异性不好或效率不高,绘制起全基因组结合谱存在技术瓶颈。运用的CRISPR/Cas9标记技术对内源基因进行标记,规避了转录因子抗体质量的难题。
4、本发明运用的CRISPR/Cas9标记技术是最新发展的同源重组标记技术,具有技术创新性。与其他的同源重组标记技术(TALEN、ZFN)相比,具有准确性好、效率高的特点。PCR结合一代测序的实验结果显示TALEN、ZFN同源重组技术的阳性率一般在3%-6%,而CRISPR/Cas9标记技术则为20%。
附图说明
图1为内源基因3xFLAG和2xSTREP标记流程及亲和纯化分析图;
图2为CRISPR/Cas9介导的同源重组示意图;
图3为5种sgRNA的切割效率对比图;
图4为lentiCRISPRv2质粒结构示意图。
具体实施方式
下面结合附图和具体实施例对本发明进行详细说明。
实施例1
对乳腺癌细胞MCF7中NCOA1的基因的标记及其基因组转录谱绘制。基于本发明的具体方法如下:
(1)sgRNA文库建立:
利用本实验室开发的在线设计网站http://cistrome.org/crispr/tool,针对三种ERα共转录因子基因的终止编码位点(stop codon site)上下游50bp区域,设计sgRNA,引入DNA开放性参数,进一步筛选位于开放程度高的基因组DNA上的sgRNA。
(2)构建含有sgRNA的慢病毒载体:
sgRNA序列的5’端加入序列5’-GACCA-3’,sgRNA的反向互补序列的5’端加入序列5’-GACCA-3’。合成修饰过的sgRNA序列。慢病毒质粒选用带有Cas9和Puro元件的lentiCRISPRv2质粒,如图4所示。用BsmBI酶对lentiCRISPRv2质粒进行酶切和去磷酸化,将线性化的质粒进行胶回收纯化,并用双蒸水溶解。用退火的方法将sgRNA的两条单链以互补配对的方式结合,并用T4PNK将sgRNA磷酸化。最后用连接酶将线性化质粒,并与sgRNA连接。用Stbl3感受态细胞进行转化,从而得到含有sgRNA的lentiCRISPRv2载体。用菌液测序鉴定sgRNA插入的正确性。
(3)制备表达sgRNA的慢病毒并确定病毒感染效率:
病毒包装质粒选用psPAX2和pMD2.G。将含有sgRNA的lentiCRISPRv2、psPAX2和pMD2.G转染293FT细胞,转染后培养48h,吸取细胞培养基,收获病毒,继续培养24h后第二次收获病毒。将两次收取的病毒合并,用0.45μm的滤器过滤,去除细胞碎片。在MCF-7细胞中鉴定病毒感染效率。在6孔板中培养MCF-7,18h后,每个孔分别用0μL、50μL、100μL、200μL、400μL、600μL的病毒,同时加入终浓度为10μg/ml的polybrene加强病毒感染效率。感染48后,用1.5μg/ml的puromycin筛选72h,此时对照组(0μL病毒组)的细胞全部死亡,选用能够使细胞100%存活的最小病毒量作进行后续实验。
(4)鉴定sgRNA效率:
采用两种方法鉴定sgRNA效率。
一种为T7E1核酸酶酶切实验。具体方案为:PCR扩增含有预期切割位点的DNA片段,使DNA片段长度为700bp左右,PCR产物进行切胶纯化。检测纯化后的DNA浓度,实验组和对照组取相同DNA量,用T7E1对DNA片段进行酶切。其中实验组为感染含有待检sgRNA的病毒的MCF-7,对照组为野生型MCF-7。酶切后用琼脂糖凝胶电泳检测,若实验组DNA产生小于700bp的两条条带,则说明sgRNA有效。
另一种为PCR结合一代测序。具体方案为:用PCR扩增含有预期切割位点的DNA片段,送公司测序,以分析结果PAM序列(NGG)前3个碱基附近起出现明显杂峰为sgRNA有效的标准。
建立NCOA1带有Flag标签的MCF-7细胞模型
以NCOA1的Flag标记为例。用CRISPR/Cas9技术在NCOA1的转录终止位点(stop codon site)处插入Flag标签(Flag-tag)。用lonza电转仪将含有靶向NCOA1的sgRNA的lentiCRISPRv2,转染到含有dox诱导ERα537位氨基酸突变的MCF-7细胞,培养24h后,用puromycin筛选24h,用PCR结合一代测序检测NCOA1基因3’端是否有Flag-tag的插入。若有Flag-tag的插入,则用细胞稀释的方法分离和培养单个细胞,建立单克隆细胞株。并再次用PCR结合一代测序鉴定Flag-tag插入的正确性。使用Flag-tag抗体,采用Western blot检测Flag蛋白的表达,确认内源标记成功。
(5)Chip-seq文库构建与测序:
文库按照illumina公司的建库试剂盒说明书构建,将建好文库进行Hiseq2000的50SE进行高通量测序,结果进行生物信息分析。
目前对乳腺癌细胞抗体质量差的转录因子还没有成熟的常规方法。
本实施例结合现有的CRISPR/Cas9同源重组方法,结合Chip-seq得到了一个新的方法。与常规sgRNA设计方法相比,步骤(1)的使用染色质开放程度作为设计参数的方法能够更加有效的设计出sgRNA,其sgRNAu有效率为80%以上,而常规方法设计的sgRNA有效率小于30%。本发明步骤(4)中运用两种方法鉴定sgRNA效率,保证了实验的可信性,其中另一种为PCR结合一代测序。具体方案为:用PCR扩增含有预期切割位点的DNA片段,送公司测序,以分析结果PAM序列(NGG)前3个碱基附近起出现明显杂峰为sgRNA有效的标准。此方法操作简单用时短(PCR仅需2个小时),测序结果第二天可以得到。
实施例2
应用于肝癌细胞系HepG2的CRISPR/Cas9应用于大规模筛选癌症基因的CRISPR/Cas9富集测序方法,包括以下步骤:
(1)设计sgRNA:
利用本实验室开发的在线设计网站http://cistrome.org/crispr/tool,针对三种ERα共转录因子基因的终止编码位点(stop codon site)上下游50bp区域,设计sgRNA,引入DNA开放性参数,进一步筛选位于开放程度高的基因组DNA上的sgRNA。
(2)构建含有sgRNA的慢病毒载体:
sgRNA序列的5’端加入序列5’-GACCA-3’,sgRNA的反向互补序列的5’端加入序列5’-GACCA-3’。合成修饰过的sgRNA序列。慢病毒质粒选用带有Cas9和Puro元件的lentiCRISPRv2质粒。用BsmBI酶对lentiCRISPRv2质粒进行酶切和去磷酸化,将线性化的质粒进行胶回收纯化,并用双蒸水溶解。用退火的方法将sgRNA的两条单链以互补配对的方式结合,并用T4PNK将sgRNA磷酸化。最后用连接酶将线性化质粒,并与sgRNA连接。用Stbl3感受态细胞进行转化,从而得到含有sgRNA的lentiCRISPRv2载体。用菌液测序鉴定sgRNA插入的正确性。
(3)制备表达sgRNA的慢病毒:
病毒包装质粒选用psPAX2和pMD2.G。将含有sgRNA的lentiCRISPRv2、psPAX2和pMD2.G转染293FT细胞,转染后培养48h,吸取细胞培养基,收获病毒,继续培养24h后第二次收获病毒。将两次收取的病毒合并,用0.45μm的滤器过滤,去除细胞碎片。在293FT细胞中鉴定病毒感染效率。在6孔板中培养293FT,18h后,每个孔分别用0μL、50μL、100μL、200μL、400μL、600μL的病毒,同时加入终浓度为10μg/ml的polybrene加强病毒感染效率。感染48后,用1.5μg/ml的puromycin筛选72h,此时对照组(0μL病毒组)的细胞全部死亡,选用能够使细胞100%存活的最小病毒量作进行后续实验。
(4)鉴定sgRNA效率:
采用两种方法鉴定sgRNA效率。
一种为T7E1核酸酶酶切实验。具体方案为:PCR扩增含有预期切割位点的DNA片段,使DNA片段长度为700bp左右,PCR产物进行切胶纯化。检测纯化后的DNA浓度,实验组和对照组取相同DNA量,用T7E1对DNA片段进行酶切。其中实验组为感染含有待检sgRNA的病毒的293FT,对照组为野生型293FT。酶切后用琼脂糖凝胶电泳检测,若实验组DNA产生小于700bp的两条条带,则说明sgRNA有效。
另一种为PCR结合一代测序。具体方案为:用PCR扩增含有预期切割位点的DNA片段,送公司测序,以分析结果PAM序列(NGG)前3个碱基附近起出现明显杂峰为sgRNA有效的标准目前对肝癌原代细胞进行癌症相关基因的筛选还没有常规方法。
建立NCOA1带有Flag标签的HepG2细胞模型
以NCOA1的Flag标记为例。用CRISPR/Cas9技术在NCOA1的转录终止位点(stop codon site)处插入Flag标签(Flag-tag),插入位置和donor的设计如图2所示。用lonza电转仪将含有靶向NCOA1的sgRNA的lentiCRISPRv2,转染到含有dox诱导ERα537位氨基酸突变的HepG2细胞,培养24h后,用puromycin筛选24h,用PCR结合一代测序检测NCOA1基因3’端是否有Flag-tag的插入。若有Flag-tag的插入,则用细胞稀释的方法分离和培养单个细胞,建立单克隆细胞株。并再次用PCR结合一代测序鉴定Flag-tag插入的正确性。使用Flag-tag抗体,采用Western blot检测Flag蛋白的表达,确认内源标记成功。
(5)Chip-seq文库构建与测序:
文库按照illumina公司的建库试剂盒说明书构建,将建好文库进行Hiseq2000的50SE进行高通量测序,结果进行生物信息分析。
目前对肝癌细胞抗体质量差的转录因子还没有成熟的常规方法。
本实施例结合现有的CRISPR/Cas9同源重组方法,结合Chip-seq得到了一个新的方法。与常规sgRNA设计方法相比,步骤(1)的使用染色质开放程度作为设计参数的方法能够更加有效的设计出sgRNA,其sgRNAu有效率为80%以上,而常规方法设计的sgRNA有效率小于30%。本发明步骤(4)中运用两种方法鉴定sgRNA效率,保证了实验的可信性,其中另一种为PCR结合一代测序。具体方案为:用PCR扩增含有预期切割位点的DNA片段,送公司测序,以分析结果PAM序列(NGG)前3个碱基附近起出现明显杂峰为sgRNA有效的标准。此方法操作简单用时短(PCR仅需2个小时),测序结果第二天可以得到。
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!