司坦唑醇糖精盐及其制备方法和应用与流程
本发明属于化学制药技术领域,具体地讲,涉及一种司坦唑醇糖精盐及其制备方法和应用。
背景技术:
药物活性成分通常以多种固体存在形式,如多晶型、水合物、溶剂化物、无定型、盐、共晶等。对同一种活性药物成分而言,不同的固体存在形式拥有不同的理化性质。因此,在制药行业中,获取适宜的药物固体存在形式具有很大的意义。药物以盐的形式存在,可在提高药物活性成分稳定性、增加溶解性、调节溶出度和改善引湿性等方面有着显著的作用;所以,在改善药物活性成分理化性质方面,药物盐通常是第一选择。
司坦唑醇,化学名为17β-羟基-17α-甲基雄甾烷醇[3,2-c]吡唑,是最常用的一类合成类固醇药物。但是,司坦唑醇在水中几乎不溶,且在高湿度条件下容易吸潮转化成一水合物;低溶解度会导致生物利用度欠佳,潮气不稳定性也会给药物的保存和质量控制带来严重问题。因此,有必要改善司坦唑醇这些理化学性质存在的问题,以使司坦唑醇能够获得更好的应用。
技术实现要素:
为解决上述现有技术存在的问题,本发明提供了一种司坦唑醇糖精盐及其制备方法和应用,该司坦唑醇糖精盐可有效地提高司坦唑醇的溶出度,并有效改善了司坦唑醇在潮气环境下稳定性差的问题。
为了达到上述发明目的,本发明采用了如下的技术方案:
一种司坦唑醇糖精盐,包括等物质的量的司坦唑醇阳离子和糖精阴离子;其中,所述司坦唑醇阳离子具有如下的结构式(1),所述糖精阴离子具有如下的结构式(2):
进一步地,所述司坦唑醇糖精盐属于正交晶系,p212121空间群;所述司坦唑醇糖精盐的晶胞参数为:轴长
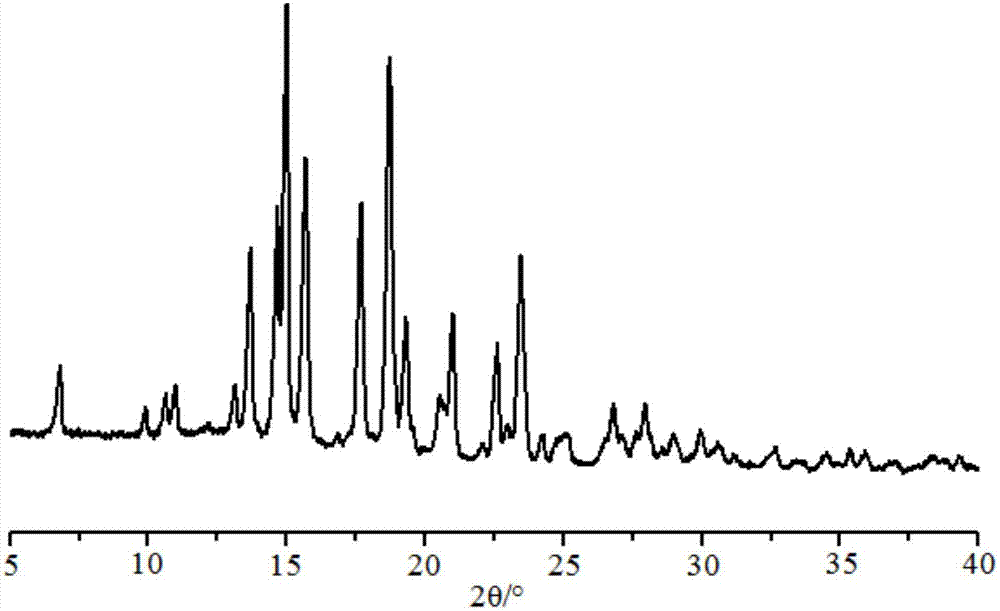
进一步地,所述司坦唑醇糖精盐具有用cu-kα射线得到的粉末x射线衍射谱图,所述粉末x射线衍射图谱中至少在衍射角2θ为6.9°、10.0°、10.8°、11.1°、13.3°、13.8°、14.8°、15.1°、15.9°、17.8°、18.9°、19.5°、19.7°、20.2°、20.7°、20.9°、22.8°、22.3°、23.7°、23.8°、24.4°、25.0°、25.4°、27.0°、28.1°、31.0°、33.6°、35.6°处具有特征峰。
进一步地,在所述司坦唑醇糖精盐的13c固体核磁共振图谱中,至少在化学位移为82.3ppm、120.1ppm、121.0ppm、124.1ppm、131.9ppm、133.6ppm、134.6ppm、141.5ppm、144.1ppm、170.3ppm处具有特征峰。
本发明的另一目的在于提供了一种如上任一所述的司坦唑醇糖精盐的制备方法,包括:将司坦唑醇和糖精混合,获得混合物;将所述混合物溶解于第一有机溶剂中,获得第一混合溶液;挥发所述第一混合溶液中的第一有机溶剂,获得所述司坦唑醇糖精盐。
进一步地,所述司坦唑醇与所述糖精的物质的量之比为1:1~1:2.5。
进一步地,所述第一有机溶剂选自丙酮、乙腈、乙酸乙酯中的至少一种。
进一步地,在室温~80℃的温度下,将所述混合物溶解于所述第一有机溶剂中;在室温~80℃的温度下,挥发所述混合溶液中的第一有机溶剂。
本发明的另一目的还在于提供了另一种如上任一所述的司坦唑醇糖精盐的制备方法,包括:将糖精溶解在第二有机溶剂中,获得糖精溶液;向所述糖精溶液中加入司坦唑醇,形成悬浊液;搅拌所述悬浊液,获得第二混合溶液,对所述第二混合溶液进行固液分离,获得滤渣和滤液,所述滤渣经干燥,获得所述司坦唑醇糖精盐。
进一步地,所述司坦唑醇与所述糖精溶液中糖精的物质的量之比为1:1~1:2.5。
进一步地,所述第二有机溶剂选自丙酮、乙腈、乙酸乙酯中的至少一种。
进一步地,在室温~40℃的温度下,将所述糖精溶解在所述第二有机溶剂中;在室温~40℃的温度下,搅拌所述悬浊液48h以上。
本发明的另一目的还在于提供了如上任一所述的司坦唑醇糖精盐在固醇药物中的应用。
本发明通过将司坦唑醇转化为一种全新的司坦唑醇糖精盐,该司坦唑醇糖精盐较司坦唑醇具有更大的表观溶解度,有利于提高该类药物的生物利用度,以及优异的耐湿性和良好的热稳定性。同时,本发明的司坦唑醇糖精盐的辅料为糖精,其作为一种甜味剂,可以改善司坦唑醇类药物的口感,能够应用于制药工业中。另外,本发明公开的两种上述司坦唑醇糖精盐的制备方法工艺简单、易于操作、原料易得、成本低廉,且无有毒溶剂残留。
附图说明
通过结合附图进行的以下描述,本发明的实施例的上述和其它方面、特点和优点将变得更加清楚,附图中:
图1是根据本发明的实施例1的司坦唑醇盐的晶胞结构图。
图2是根据本发明的实施例1的司坦唑醇盐的粉末x射线衍射图谱。
图3是根据本发明的实施例1的司坦唑醇盐的13c固体核磁共振图谱。
图4是根据本发明的实施例1的司坦唑醇盐的差示扫描量热曲线。
图5是司坦唑醇与根据本发明的实施例1的司坦唑醇盐的溶解度对比图。
图6是司坦唑醇与根据本发明的实施例1的司坦唑醇盐在25℃、95%相对湿度的条件下分别加速20天后的粉末x射线衍射图谱对比图。
具体实施方式
以下,将参照附图来详细描述本发明的实施例。然而,可以以许多不同的形式来实施本发明,并且本发明不应该被解释为限制于这里阐述的具体实施例。相反,提供这些实施例是为了解释本发明的原理及其实际应用,从而使本领域的其他技术人员能够理解本发明的各种实施例和适合于特定预期应用的各种修改。
将理解的是,尽管在这里可使用术语“第一”、“第二”等来描述各种物质,但是这些物质不应受这些术语的限制。这些术语仅用于将一个物质与另一个物质区分开来。
实施例1
本实施例公开了一种全新的物质,该物质为一种司坦唑醇糖精盐,该司坦唑醇糖精盐由等物质的量的司坦唑醇阳离子和糖精阴离子组成;其中,司坦唑醇阳离子及糖精阴离子分别具有如下的结构式(1)和(2):
本实施例的司坦唑醇糖精盐以司坦唑醇和糖精为原料,采用溶剂挥发法或混悬法进行制备。
分别对本实施例的司坦唑醇糖精盐的结构及性质等进行了分析检测,包括如下几个方面:
(1)晶体结构测定
具体来讲,采用brukerapexiiccd衍射仪,在273(2)k(即273k±0.2k)下,用经石墨单色化的mokα射线(λ=0.71073nm)以ω扫描方式收集衍射点,收集的数据通过saint程序还原并用sadabs方法进行半经验吸收校正。最终司坦唑醇糖精盐的结构使用直接法解得,通过全矩阵最小二乘方法对f2进行修正得到全部非氢原子的坐标及各向异性参数。碳上的氢原子在结构精修过程中被理论固定在母原子上,氮原子或氧原子上连接的氢原子是通过差值傅立叶图来确定的;获得如图1所示的晶胞结构图。通过对司坦唑醇糖精盐的晶体结构的测定,获知该司坦唑醇糖精盐属于正交晶系,p212121空间群;且司坦唑醇糖精盐的晶胞参数为:轴长
值得说明的是,在图1中,方框中表示司坦唑醇糖精盐的一个晶胞;同时,图1中坐标系上的a、b即代表上述轴长a、b的方向,而上述轴长c的方向由于垂直于纸面,因此未示出。
(2)粉末x射线衍射测定
具体来讲,采用brukerd8advance衍射仪测定,测定条件如下:辐射源为cukα,管电压为40kv,管电流为40ma,步长为0.01°,扫描范围为3°~40°;获得如图2所示的粉末x射线衍射图谱。通过对司坦唑醇糖精盐的粉末x射线衍射图谱的测定,获知该司坦唑醇糖精盐至少在衍射角2θ为6.9°、10.0°、10.8°、11.1°、13.3°、13.8°、14.8°、15.1°、15.9°、17.8°、18.9°、19.5°、19.7°、20.2°、20.7°、20.9°、22.8°、22.3°、23.7°、23.8°、24.4°、25.0°、25.4°、27.0°、28.1°、31.0°、33.6°、35.6°处具有特征峰;测量误差为±0.1°。
(3)13c固体核磁共振测定
具体来讲,采用brukeravanceiii-500核磁共振波谱仪测定,测定条件如下:4mm双共振魔角旋转探头,磁场强度为11.7t,魔角自旋速度为8khz,以四甲基硅烷定标(0ppm);获得如图3所示的13c固体核磁共振图谱。通过对司坦唑醇糖精盐的13c固体核磁共振的测定,获知该司坦唑醇糖精盐至少在化学位移82.3ppm、120.1ppm、121.0ppm、124.1ppm、131.9ppm、133.6ppm、134.6ppm、141.5ppm、144.1ppm、170.3ppm处具有特征峰;测量误差为±0.1ppm。
与此同时,结合上述图2和图3的结果,可以看出,本实施例的司坦唑醇糖精盐的晶型纯度高,无明显的原料杂质。
(4)差示扫描量热测试
具体来讲,采用taq2000差示扫描量热仪测定,测定条件如下:3mg左右的司坦唑醇糖精盐作为样品,用铝盘封装,加热温度范围未25℃~200℃,升温速率为10.0℃/min,吹扫气为50ml/min的n2,温度校准使用nist铟金属进行;获得如图4所示的差示扫描量热测试曲线。通过对司坦唑醇糖精盐的差示扫描量热进行测试,可知该司坦唑醇糖精盐的熔点为216.1℃。
(5)溶解度测试
为了更好地说明本实施例的司坦唑醇糖精盐的表观溶解度,在相同的条件下,对现有技术中的司坦唑醇的溶解度也进行了测定,二者的溶解度对比如图5所示。
在图5中,1线表示司坦唑醇的溶解度随时间的变化,2线表示司坦唑醇糖精盐的溶解度随时间的变化。从图5中可以看出,根据本实施例的司坦唑醇糖精盐较司坦唑醇的表观溶解度有了大幅度的提升。
(6)耐湿性测试
具体地,在25℃、95%相对湿度的条件下,进行加速测试,加速时间为20d,并在20d后测定加速前后该司坦唑醇糖精盐的粉末x射线衍射图谱。同时,为了更好地说明本实施例的司坦唑醇糖精盐的耐湿性,在相同的条件下,对现有技术中的司坦唑醇的耐湿性也进行了测定,二者的耐湿性变化对比如图6所示。
在图6中,1线表示司坦唑醇在加速前的粉末x射线衍射图谱,2线表示司坦唑醇在加速后的粉末x射线衍射图谱,3线表示司坦唑醇糖精盐在加速前的粉末x射线衍射图谱,4线表示司坦唑醇糖精盐在加速后的粉末x射线衍射图谱。从图6中可以看出,本实施例的司坦唑醇糖精盐在上述较为严苛的条件下加速测试结果为其结构并未改变,而现有技术中的司坦唑醇的结构却发生了变化;说明本实施例的司坦唑醇糖精盐具有优异的耐湿性。
根据本实施例的司坦唑醇糖精盐可作为司坦唑醇的不同形态的转化药物应用在固醇药物领域。如在临床上,其可用于遗传性血管神经性水肿的预防和治疗,以及严重创伤、慢性感染、营养不良等消耗性疾病的治疗。
与此同时,通过上述(5)溶解度测试和(6)耐湿性测试的结果,可以看出,本实施例的司坦唑醇糖精盐不仅能够克服司坦唑醇类药物的低溶解度的弊端,有利于提高司坦唑醇类药物的生物利用度,而且克服了司坦唑醇类药物的潮气不稳定性的弊端,从而更好地应用于医疗治疗中。
本发明还公开了上述实施例1中的司坦唑醇糖精盐的两种制备方法,以下分别以不同的实施例进行介绍。
实施例2
本实施例公开了如实施例1中的司坦唑醇糖精盐的其中一种制备方法,该制备方法为溶剂挥发法,具体包括如下步骤:
s1:将0.1mmol司坦唑醇和0.1mmol糖精混合,获得混合物。
一般地,司坦唑醇与糖精的物质的量之比控制为1:1~1:2.5的范围内均可。
s2:在室温~80℃的温度下,将混合物溶解于第一有机溶剂中,获得第一混合溶液。
优选地,第一有机溶剂为丙酮、乙腈、乙酸乙酯中的任意一种或至少两种的混合物;本实施例中,以5ml丙酮作为第一有机溶剂。
s3:在室温~80℃的温度下,挥发第一混合溶液中的第一有机溶剂,获得司坦唑醇糖精盐。
优选地,在挥发第一混合溶液中的第一有机溶剂之前,可将第一混合溶液先进行过滤操作,优选使用孔径为0.22μm的筛子进行,以去除第一混合溶液中的杂质、纯化第一混合溶液,从而获得晶型更好、纯度更高的司坦唑醇糖精盐。
值得注意的是,在步骤s2中溶解混合物时,所获得的第一混合溶液的浓度可以是任意的,但为了减少后续第一混合溶液中第一有机溶剂的挥发时间,优选第一混合溶液为饱和溶液。
鉴于本实施例的制备方法,所获得的司坦唑醇糖精盐为晶体状盐,产率为90%。
实施例3
本实施例提供了如实施例1中的司坦唑醇糖精盐的另外一种制备方法,该制备方法为混悬法,具体包括如下步骤:
q1:在室温~40℃的温度下,将0.5mmol糖精溶解在第二有机溶剂中,获得糖精溶液。
优选地,第二有机溶剂为丙酮、乙腈、乙酸乙酯中的任意一种或至少两种的混合物;在本实施例中,以5ml乙酸乙酯作为第二有机溶剂。
值得注意的是,此处配制糖精溶液时,其浓度可以是任意的,但为了减少后续固液分离时减少第二有机溶剂的浪费,优选该糖精溶液为饱和溶液。
q2:向糖精溶液中加入0.5mmol司坦唑醇,形成悬浊液。
一般地,司坦唑醇与糖精溶液中糖精的物质的量之比控制为1:1~1:2.5均可。
q3:在室温~40℃的温度下,搅拌悬浊液48h以上,获得第二混合溶液。
在本实施例中,悬浊液搅拌时间为3d。
q4:对第二混合溶液进行固液分离,获得滤渣和滤液,滤渣经干燥,获得司坦唑醇糖精盐。
在本步骤中,优选采用抽滤的方式进行固液分离,所获得的滤饼经真空干燥,即为所述司坦唑醇糖精盐。
鉴于本实施例的制备方法,所获得的司坦唑醇糖精盐为粉末状盐,产率为84%。
虽然已经参照特定实施例示出并描述了本发明,但是本领域的技术人员将理解:在不脱离由权利要求及其等同物限定的本发明的精神和范围的情况下,可在此进行形式和细节上的各种变化。
- 还没有人留言评论。精彩留言会获得点赞!