一种具有高电导率的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜及其制备方法与流程
本发明属于燃料电池材料技术领域,具体涉及一种具有高电导率的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜及其制备方法。
背景技术:
质子交换膜燃料电池(PEMFC)是一种高效、清洁、环境友好的发电装置,是电动汽车的理想动力源,亦可作为分散电站、潜艇及航天器等军用电源或便携式电源等,具有十分广阔的应用前景。然而目前广泛使用的是以Nafion®为代表的全氟型磺酸膜燃料电池,但这类质子交换膜的质子导电能力受膜内水含量和温度的影响极大,阻醇性能差,PEMFC的工作温度不能超过80℃。由于PEMFC受工作温度的限制,使得它在实际应用时面临CO耐受性差、系统的水热管理困难等问题。因此将PEMFC运行温度提高到100℃以上,就能有效地克服传统Nafion基PEMFC的上述问题,这一类型的燃料电池(FC)通常称之为高温质子交换膜燃料电池(HT-PEMFC),是PEMFC技术的一个重要的发展方向。
HT-PEMFC系统有如下优点:1)电化学反应速率提高,有效降低了阴极电化学极化过电位,允许降低催化剂担量,允许使用非铂催化剂;2)对反应气体的增湿要求降低;3)电池内水以气相存在简化了水热管理;此外,HT-PEMFC在一定程度上简化了FC冷却系统。鉴于HT-PEMFC诱人的发展前景,国内外广泛开展了HT-PEMFC关键材料的研制,包括高温质子交换膜、催化剂和载体等,并取得了较好的初步结果,其中高温质子交换膜是研究的热点之一。
目前对于HT-PEMFC质子交换膜的研究主要集中在聚苯并咪唑(PBI)上,它于1959年在美国专利上首次被报道,1988年美国Hoechst Celanese公司将PBI膜产品推向市场。如今,PBI作为工程热塑性塑料里最为出众的聚合物基材料,在用作HT-PEMFC的高温质子交换膜方面展现出巨大的有效性和可行性。但是PBI型膜材料在高温运行时(T≥150℃),会不可避免地发生降解。
研究发现,燃料电池运行过程中,氧气经过膜渗透到阳极侧,在阳极Pt及微量过渡金属离子的催化作用下,形成•OH和HOO•等自由基,•OH自由基进攻PBI主链上的含氮基团,HOO•自由基攻击苯环上的碳氢键,使PBI主链断裂;同时高温氧化环境还容易使PBI主链上的两个端氨基氧化,端羧基发生脱羧反应产生亚苯基自由基。产生的这些自由基会加剧PBI膜的降解,导致电池性能大幅下降。房建华等采用环氧化物(CN 200710171866.9)、二卤(多卤)烷烃(CN 200710171865.4)和马来酸酐(CN 200710171867.3)对PBI主链上的一个端氨基进行交联保护,从而减缓膜的降解;李忠芳等人采用尿素(CN 101768270 A),作为端氨基的保护性试剂对PBI进行了改性。变价金属类自由基淬灭剂(如CeO2、MnO2等)、或者改变聚合物 结构可以有效淬灭HT-PEMFC运行过程中产生的自由基,从而减缓PBI膜的降解。
综上所述,目前得到该类质子交换膜聚合物基底容易受自由基攻击而降解等难题制约其商业化的应用,通过将膜内添加自由基淬灭剂、在膜内构建交联结构,可以有效的避免自由基攻击而降解,提升膜的使用寿命,并提高膜的尺寸稳定性。
技术实现要素:
针对现有技术的不足,本发明的目的在于提供一种具有高电导率的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜及其制备方法,通过以聚苯并咪唑为聚合物骨架,以乙烯基-1,2,4-三氮唑为交联剂,通过自交联形成交联型高温质子交换膜,从而提高聚苯并咪唑高温质子交换膜的电导率以及尺寸稳定性。
为了实现上述目的,本发明采用的技术方案为:
一种具有高电导率的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜的制备方法,包括以下步骤:
(1)将0.2~1 g聚苯并咪唑溶解于5~10 mL高沸点溶剂中,搅拌溶解后,加入相同质量的三氮唑基聚乙烯,搅拌直至溶解;
(2)再将溶液放入油浴中,50~100 ℃下搅拌,反应12~48 h;
(3)将溶液中继续加入5~10 mL高沸点溶剂,搅拌30 min后,倒入铸膜板中,60~80 ℃下将溶剂蒸干,将得到的高温质子交换膜小心揭下,浸泡在乙醇中,洗涤后干燥;
(4)将干燥后的交联型高温质子交换膜浸泡在磷酸溶液中,60~120 ℃下浸渍24~48 h,后取出,得到高温质子交换膜。
优选地,步骤(1)、(3)中所述高沸点溶剂为N,N-二甲基乙酰胺或N-甲基吡咯烷酮。
优选地,步骤(1)中所述的三氮唑基聚乙烯的制备方法,包括如下步骤:
(1)将0.5~2 g1-乙烯基-1,2,4-三氮唑溶于10~20 mL二甲基甲酰胺中,再加入链引发剂偶氮二异丁腈,其中链引发剂的质量含量为高沸点溶剂的20~50%;
(2)将溶液搅拌溶解后,放入油浴中,反应24~48 h,反应温度为50~80 ℃;
(3)反应结束后,将溶液倒入乙醇中,得到白色絮状物,用乙醇洗涤后干燥。
优选地,步骤(4)中所述磷酸的质量分数为50%~85%。
采用上述制备方法得到的具有高电导率的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜。
本发明提供的具有高电导率的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜及其制备方法,与现有技术相比具有以下有益效果:
1. 本发明采用聚苯并咪唑为聚合物骨架,以乙烯基-1,2,4-三氮唑为交联剂,通过自交联制得的复合膜结构均匀,适用于无水体系,工作温度区间为120~200 ℃;
2. 本发明采用聚苯并咪唑/聚乙烯三氮唑交联结构,可以有效降低膜浸渍磷酸后的溶胀,并大幅提升高温质子交换膜的电导率,交联结构的构筑同时可以有效提升复合膜的抗氧化能力;
3. 本发明所采用的交联剂聚乙烯三氮唑,可以有效吸附磷酸分子,提高膜在电池运行过程中电导率的长期稳定性。
附图说明
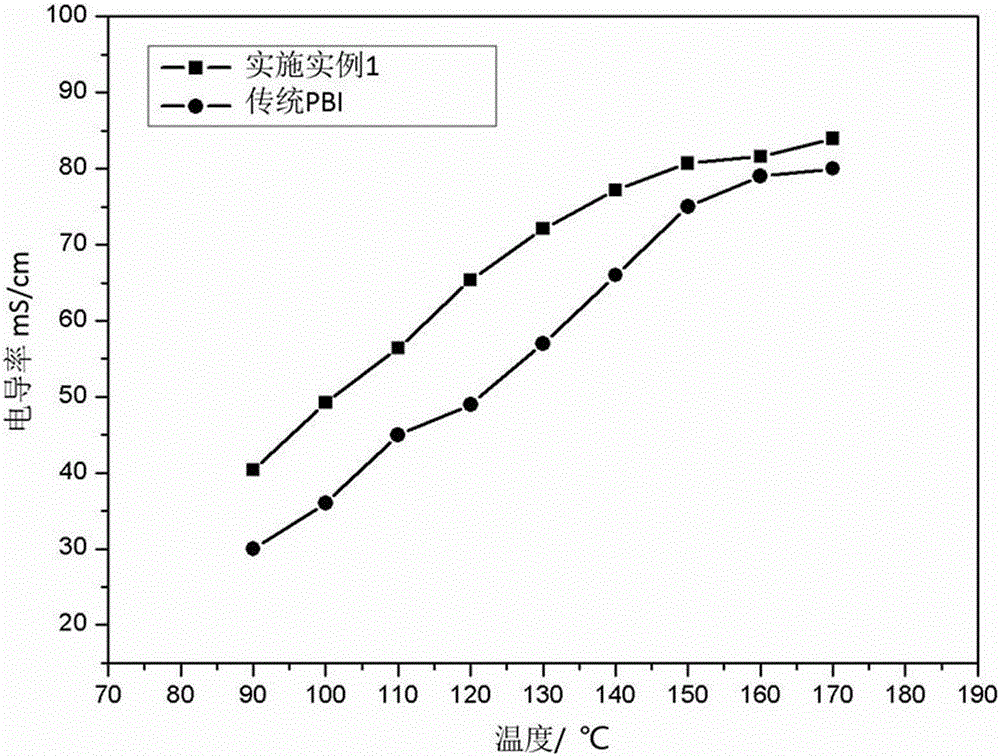
图1 是实施例1制得的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜与传统PBI复合膜的电导率随温度的变化图;
图2 是实施例2制得的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜与传统PBI复合膜的电池极化曲线图;
图3是实施例3制得的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜与传统PBI复合膜的电导率稳定性图。
具体实施方式
下面实施例中所用聚苯并咪唑(PBI,CAS:25928-81-8)购自FuMA-Tech公司;二甲基甲酰胺(DMF,CAS:68-12-2)、二甲基乙酰胺(DMAC,CAS:127-19-5)、N-甲基吡咯烷酮(NMP ,CAS 872-50-4)购自天津市大茂化学试剂公司;1-乙烯基-1,2,4-三唑(CAS: 2764-83-2)购自上海源叶生物科技有限公司;偶氮二异丁腈(AIBN,CAS: 78-67-1)购自天津市大茂化学试剂公司;磷酸购自上海阿拉丁生化科技股份有限公司。
为更进一步阐述本发明所采取的技术手段及其效果,以下结合本发明的优选实施例,并配合附图进行详细描述。
实施例1
一种具有高电导率的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜的制备方法,包括以下步骤:
(1)将0.2 g1-乙烯基-1,2,4-三氮唑溶于10 mL二甲基甲酰胺中,再加入链引发剂偶氮二异丁腈,其中链引发剂的质量含量为高沸点溶剂的20%;
(2)将溶液搅拌溶解后,放入油浴中,反应24 h,反应温度为60 ℃;
(3)反应结束后,将溶液倒入乙醇中,得到白色絮状物,用乙醇洗涤后干燥,得三氮唑基聚乙烯;
(4)将0.2 g聚苯并咪唑溶解于10 mL N-甲基吡咯烷酮中,搅拌溶解后,加入相同质量的三氮唑基聚乙烯,搅拌直至溶解;
(5)再将溶液放入油浴中,80 ℃下搅拌,反应12 h;
(6)将溶液中继续加入10 mL N-甲基吡咯烷酮,搅拌30 min后,倒入铸膜板中,80 ℃下将溶剂蒸干,将得到的高温质子交换膜小心揭下,浸泡在乙醇中洗涤后干燥;
(7)将干燥后的交联型高温质子交换膜浸泡在质量分数为85 wt%磷酸溶液中,60 ℃下浸渍24 h后取出,得到交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜。
为了与上述制得的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜进行对比,采用溶液浇铸法制备厚度为50 μm的PBI膜,再浸渍磷酸,制得PBI/磷酸膜,具体制备步骤为:将PBI的N-甲基吡咯烷酮溶液倒在玻璃模具中,于80 ℃下真空干燥25 h,得到PBI膜;将PBI膜浸没于60 ℃的磷酸溶液中60 h,取出并在80 ℃干燥24 h,记作传统PBI复合膜;其中,PBI的N-甲基吡咯烷酮溶液中PBI的质量分数为2%,磷酸溶液的浓度为70 wt%。
实施例1制得的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜及传统PBI复合膜,测试电导率随温度的变化图。将实施例1制得的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜及传统PBI复合膜均裁成尺寸为40 mm×10 mm的矩形片,然后将实施例1制得的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜及传统PBI复合膜分别置于电导率夹具中,将夹具放于真空干燥箱中,在90 ℃下保持3 h,然后温度从 90 ℃升至170 ℃,每隔10 ℃测试下膜的电导率,结果如图1所示。
从图1可以看出,实施例1制得的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜相比传统PBI复合膜,电导率大幅提升,这是由于交联结构中三氮唑的存在,提供了更多的磷酸吸附位点,从而提升了膜的电导率。
实施例2
一种具有高电导率的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜的制备方法,包括以下步骤:
(1)将1 g1-乙烯基-1,2,4-三氮唑溶于10 mL二甲基甲酰胺中,再加入链引发剂偶氮二异丁腈,其中链引发剂的质量含量为高沸点溶剂的50%;
(2)将溶液搅拌溶解后,放入油浴中,反应48 h,反应温度为80 ℃;
(3)反应结束后,将溶液倒入乙醇中,得到白色絮状物,用乙醇洗涤后干燥,得三氮唑基聚乙烯;
(4)将1 g聚苯并咪唑溶解于8 mL N-甲基吡咯烷酮中,搅拌溶解后,加入相同质量的三氮唑基聚乙烯,搅拌直至溶解;
(5)再将溶液放入油浴中,50 ℃下搅拌,反应48 h;
(6)将溶液中继续加入8 mL N-甲基吡咯烷酮,搅拌30 min后,倒入铸膜板中,60 ℃下将溶剂蒸干,将得到的高温质子交换膜小心揭下,浸泡在乙醇中洗涤后干燥;
(7)将干燥后的交联型高温质子交换膜浸泡在质量分数为60 wt%磷酸溶液中,120 ℃下浸渍48 h后取出,得到交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜。
为了与上述制得的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜进行对比,采用溶液浇铸法制备厚度为50 μm的PBI膜,再浸渍磷酸,制得PBI/磷酸膜,具体制备步骤为:将PBI的N-甲基吡咯烷酮溶液倒在玻璃模具中,于80 ℃下真空干燥25 h,得到PBI膜;将PBI膜浸没于60 ℃的磷酸中60 h,取出并在80 ℃干燥24 h,即得,记作传统PBI复合膜;其中,PBI的NMP溶液中PBI的质量分数为2%,磷酸的浓度为70 wt%。
将实施例2制得的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜及传统PBI复合膜,在单电池评价装置上测试其不同温度下的电池性能。其中,电池负极采用碳(C)载镍(Ni)催化剂与PBI溶液混合均匀后涂抹于碳纸上制得(采用专利CN02127802.4中公开的制备方法),Ni占C/Ni催化剂的质量百分比为40%,C/Ni与PBI的质量比为1:1;电池正极采用碳(C)载铂(Pt)催化剂与PBI溶液混合均匀后涂抹于碳纸上制得(采用专利CN02127802.4中公开的制备方法),Pt占C/ Pt催化剂的质量百分比为70%,正极中Pt载量为0.5 mg/cm2;电池操作环境分别设定为:电池温度为150 ℃;氢气及氧气流速分别为50 mL/min、100 mL/min,气体无增湿,压强为0.05 MPa,测试结果如图2所示。
从图2可以看出,实施例2制得的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜相比传统PBI复合膜,由于电导率的提升,降低了电池的反应内阻,使得电池放电性能也大为提升,最高功率密度可达1000 mW/cm2。
实施例3
一种具有高电导率的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜的制备方法,包括以下步骤:
(1)将0.5 g1-乙烯基-1,2,4-三氮唑溶于10 mL二甲基甲酰胺中,再加入链引发剂偶氮二异丁腈,其中链引发剂的质量含量为高沸点溶剂的40%;
(2)将溶液搅拌溶解后,放入油浴中,反应30 h,反应温度为50 ℃;
(3)反应结束后,将溶液倒入乙醇中,得到白色絮状物,用乙醇洗涤后干燥,得三氮唑基聚乙烯;
(4)将0.8 g聚苯并咪唑溶解于9 mL N-甲基吡咯烷酮中,搅拌溶解后,加入相同质量的三氮唑基聚乙烯,搅拌直至溶解;
(5)再将溶液放入油浴中,80 ℃下搅拌,反应32 h;
(6)将溶液中继续加入9 mL N-甲基吡咯烷酮,搅拌30 min后,倒入铸膜板中,60 ℃下将溶剂蒸干,将得到的高温质子交换膜小心揭下,浸泡在乙醇中洗涤后干燥;
(7)将干燥后的交联型高温质子交换膜浸泡在质量分数为70 wt%磷酸溶液中,90 ℃下浸渍48 h后取出,得到交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜。
为了与上述制得的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜进行对比,采用溶液浇铸法制备厚度为50 μm的PBI膜,再浸渍磷酸,制得PBI/磷酸膜,具体制备步骤为:将PBI的N-甲基吡咯烷酮溶液倒在玻璃模具中,于80 ℃下真空干燥25 h,得到PBI膜;将PBI膜浸没于60 ℃的磷酸中60 h,取出并在80 ℃干燥24 h,记作传统PBI复合膜;其中,PBI的N-甲基吡咯烷酮溶液中PBI的质量分数为2%,磷酸的浓度为70 wt%。
测试传统PBI复合膜、实施例2及实施例3制得的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜经过水浸泡后的电导率变化,具体方法如下:将2种膜分别浸泡在去离子水中5 min后取出,测试膜于150 ℃的电导率,重复此步骤,记录次数与电导率变化情况。
从图3可以看出,本发明制备的交联型聚苯并咪唑/聚乙烯三氮唑高温质子交换膜,不仅电导率有所提升,而且电导率在水中的稳定性更加突出,说明本发明的结构有效地降低了膜的离子流失率。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,本领域普通技术人员对本发明的技术方案所做的其他修改或者等同替换,只要不脱离本发明技术方案的精神和范围,均应涵盖在本发明的权利要求范围当中。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种具有阻燃功能的复合纱线的制作方法
- 一种具有阻燃功能的面料的制作方法
- 制动蹄自动铆压设备的密封橡胶圈的制作方法
- 一种混合脂肪基双(苯并咪唑啉季铵盐)及其制备方法与应用
- Artalbic acid的O?(苯并咪唑基)乙基与O?(二羟乙胺基)乙基衍生物的组合物在抗病毒 ...的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备抗骨质疏松药物的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备防治胰腺纤维化药物的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备抗缺氧药物的制作方法
- 二氮芴基芳香型二酸单体及其聚苯并咪唑聚合物的合成的制作方法
- 一种苯并三唑基?亚烷基双酚化合物的制备方法