一组与先天性代谢缺陷病相关的基因新突变及检测试剂盒的制作方法
本发明涉及生物
技术领域:
,尤其涉及一组与先天性代谢缺陷病相关的基因新突变及检测试剂盒。
背景技术:
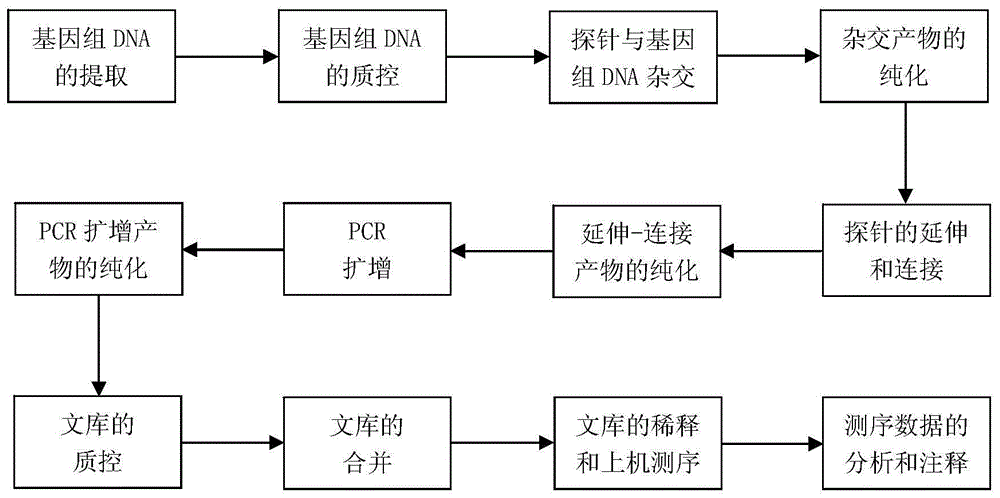
:先天性代谢缺陷病(Inbornerrorsofmetabolism,IEM)是指由于基因突变而引起酶缺失、细胞膜功能异常或受体缺陷,从而导致机体生化代谢紊乱,造成中间或旁路代谢产物蓄积,或终末代谢产物缺乏,引起一系列临床症状的一组疾病。先天性代谢缺陷病多为常染色体隐性遗传病,少数为常染色体显性遗传或X、Y连锁伴性遗传及线粒体遗传等。自1908年Garrod提出先天代谢缺陷病的概念以来,迄今发现的疾病已有500余种,并随着新的诊断技术的增加而逐渐增多。先天性代谢缺陷病虽单一疾病发病率低,但累积发病率高,5000个新生儿中约有1个患有先天性代谢缺陷病。先天性代谢缺陷病按其所涉及的代谢底物异常可直接分为氨基酸病(如苯丙酮尿症)、有机酸血症(如异戊酸血症)、脂肪酸氧化缺陷(如中链酰基辅酶A脱氢酶缺乏症)、过氧化酶体病(如Zellweger综合症)、糖代谢病(如半乳糖血症)、核酸代谢异常症(如腺嘌呤脱氧酶缺乏症)、溶酶体病(如粘多糖病)和金属代谢障碍(如Wilson病)等。根据异常代谢物的分子大小,可将先天性代谢病分为小分子病(如氨基酸病、有机酸代谢异常)和细胞器病(如脂类代谢病)。由于遗传的异质性,临床表现复杂,重者在新生儿期、疾病的急性发作期死亡,存活者多留有神经系统及其他脏器损伤的症状及体征。先天性代谢缺陷病患者在任何年龄均可发病。发病年龄不同,临床表现各异。一般发病年龄越早、预示病情重、死亡率高。由于新生儿对疾病的反应能力弱,临床主要表现为拒奶、呕吐、腹泻、脱水、嗜睡、肌张力异常、惊厥、呼吸困难等非特异性的表现。部分先天性代谢缺陷病可以进行治疗。新生儿及早发现先天性代谢缺陷病后,可对症治疗,挽救生命,减少神经系统后遗症。如婴儿患有甲基丙二酸尿症,可进行肌肉注射1mg的维生素B12进行治疗,并选择低蛋白饮食。先天性代谢缺陷病的疾病种类多,大部分有多个致病基因。传统的检测方法有酶免疫分析法、液相串联质谱仪(LC-MS/MS)技术、PCR技术、Sanger测序技术等。酶免疫分析法检测成本低、但一次只能检测一种疾病,且容易受样本采集的影响,具有一定假阳性和假阴性率。液相串联质谱仪技术检测成本低,能一次检测30多种先天性代谢缺陷病,但无法确定疾病相关的致病突变。PGR技术、Sanger测序技术能确定疾病相关的致病突变,但一次只能检测一个基因的一个或几个突变,通量低。因此采用上述方法很难对多种先天性代谢缺陷病同时进行检测并确定致病突变,检出率低,临床上很难对疾病进行确诊。自人类基因组计划后发展起来的新一代高通量测序技术,因其通量高,迅速占领了市场。近几年,新一代高通量测序技术不断完善,检测变异的准确性也逐步提高,与此同时,衍生起来的靶向重测序技术大大降低了检测成本,在遗传性疾病检测应用中极具前景。技术实现要素:本发明发现了编码甲基丙二酸单酰辅酶A变位酶的基因MUT基因的复合型杂合突变:错义突变c.1874A>C(p.Asp625Ala)和错义突变c.470T>A(p.Val157Asp)。所要解决的技术问题是如何诊断先天性代谢缺陷病或筛查先天性代谢缺陷病患者。为解决上述技术问题,本发明首先提供了下述任一产品:1)MUT突变蛋白质,为将序列2(MUT蛋白氨基酸序列)的625位的Asp突变为Ala,其将序列2的第157为的Val突变为Asp,其它氨基酸残基不变得到的蛋白质;2)编码所述MUT突变蛋白质的DNA分子或cDNA分子;3)由所述1)与X1组成的成套蛋白质;所述X1为35个与先天性代谢缺陷病相关的蛋白质发生突变得到的蛋白质中的至少一个;所述35个与先天性代谢缺陷病的蛋白质为ACADM、ARG1、ASL、ASS1、BCKDHA、BCKDHB、CPS1、DBT、DLD、ETFA、ETFB、ETFDH、FAH、GCDH、GCH1、HPD、IVD、LMBRD1、MCEE、MMAA、MMAB、MMACHC、MMADHC、MUT、NAGS、OTC、PAH、PCBD1、PCCA、PCCB、PTS、QDPR、SLC25A13、SLC25A14和TAT蛋白质;4)由所述2)与X2组成的成套DNA分子或cDNA分子;所述X2为编码所述X1的DNA分子或cDNA分子。编码所述MUT突变蛋白质的DNA分子或cDNA分子将编码MUT蛋白质的DNA分子的核苷酸序列第1874位的A突变为C,且将序列1第470位的T突变为A,其它核苷酸不变得到的DNA分子;编码MUT蛋白质的DNA分子的核苷酸序列为序列1。为解决上述技术问题,本发明还提供了诊断或辅助诊断先天性代谢缺陷病患者的成套试剂,为成套试剂1。本发明所提供的成套试剂1,由名称为G1的检测MUT基因c.1874A>C突变的物质、名称为G2的检测MUT基因c.470T>A突变的物质;所述MUT基因c.1874A>C突变为MUT基因编码区第1874位核苷酸由A突变成C得到的DNA分子;所述MUT基因c.470T>A突变为MUT基因编码区的第470位核苷酸由T突变成A所得到的DNA分子。上述成套试剂1中,所述MUT基因c.1874A>C突变后的核苷酸序列为序列21;所述MUT基因c.470T>A突变后的核苷酸序列为序列23。上述成套试剂1中,所述MUT基因c.1874A>C突变可为纯合突变;所述MUT基因c.470T>A突变可为纯合突变。上述成套试剂1中,所述G1可为检测MUT基因c.1874A>C突变的探针对和/或引物对,所述探针对中的两条探针可分别与MUT基因c.1874A>C突变位点的上游和下游特异结合,所述引物对中的两条引物可分别与MUT基因c.1874A>C突变位点的上游和下游特异结合;所述G2可为检测MUT基因c.470T>A突变的探针对和/或引物对,所述探针对中的两条探针可分别与MUT基因c.470T>A突变位点的上游和下游特异结合,所述引物对中的两条引物可分别与MUT基因c.470T>A突变位点的上游和下游特异结合;上述成套试剂1中,所述检测MUT基因c.1874A>C突变的探针对的两个探针的序列分别为序列表中序列3和序列4;所述检测MUT基因c.470T>A突变的探针对的两个探针的序列分别为序列表中序列5和序列6;所述检测MUT基因c.1874A>C突变的引物对中的两条引物的序列分别为序列表中序列1和序列2;所述检测MUT基因c.470T>A突变的引物对中的两条引物的序列分别为序列表中序列13和序列14。为解决上述技术问题,本发明还提供了下述A1)-A3)中的任一种诊断或辅助诊断先天性代谢缺陷病患者的成套试剂,该成套试剂为成套试剂2:A1)所述G1;A2)所述G2;A3)由所述G1和所述G2这两种中的至少两种组成的成套试剂。为解决上述技术问题,本发明还提供了诊断或辅助诊断先天性代谢缺陷病患者的成套试剂,该成套试剂为成套试剂3。本发明所提供的成套试剂3,由所述成套试剂1或所述成套试剂2与检测35个与先天性代谢缺陷病相关基因突变的物质组成;所述35个与先天性代谢缺陷病相关基因为ACADM、ARG1、ASL、ASS1、BCKDHA、BCKDHB、CPS1、DBT、DLD、ETFA、ETFB、ETFDH、FAH、GCDH、GCH1、HPD、IVD、LMBRD1、MCEE、MMAA、MMAB、MMACHC、MMADHC、MUT、NAGS、OTC、PAH、PCBD1、PCCA、PCCB、PTS、QDPR、SLC25A13、SLC25A14和TAT基因。上述成套试剂3中,所述检测DLD基因突变的物质为由核苷酸序列分别为序列7和序列8的两个探针组成的探针对;所述检测HPD基因突变的物质为由核苷酸序列分别可为序列9和序列10的两个探针组成的探针对;所述检测LMBRD1基因突变的物质为由核苷酸序列分别可为序列11和序列12的两个探针组成的探针对。为解决上述技术问题,本发明还提供了诊断或辅助诊断先天性代谢缺陷病患者的试剂或试剂盒。本发明所提供的诊断或辅助诊断先天性代谢缺陷病患者的试剂或试剂盒,包括所述成套试剂1、所述成套试剂2或所述成套试剂3。具体来说,所述诊断或辅助诊断先天性代谢缺陷病患者的试剂或试剂盒可由所述成套试剂1、所述成套试剂2或所述成套试剂3与进行基因测序所需的试剂组成。所述进行基因测序所需的试剂可为illumina公司生产的TruSeqCustomAmpliconKit试剂。为解决上述技术问题,本发明还提供了检测与先天性代谢缺陷病相关基因突变的方法。本发明所提供的检测与先天性代谢缺陷病相关基因突变的方法,包括利用所述成套试剂1、所述成套试剂2或所述成套试剂3检测待测样本的MUT基因和/或所述35个与先天性代谢缺陷病相关基因这35个基因中的至少一种基因的序列,与相应的基因相比,确定上述基因是否突变。上述方法中,所述检测与先天性代谢缺陷病相关基因突变可为检测待测样本与先天性代谢缺陷病相关基因中是否存在无义突变(nonsensevariant)、移码突变(frameshift)、选择性剪切变异(Splicingvariant)、错义突变(missensevariant)、氨基酸截断或插入变异(nonframeshift)和/或密码子丢失(stoplost)。所述检测与先天性代谢缺陷病相关基因突变具体可为检测待测样本MUT基因、DLD基因、HPD基因、和/或LMBRD1基因是否突变。所述MUT基因突变可为MUT基因c.1874A>C突变和c.470T>A突变。与先天性代谢缺陷病相关基因的突变可导致出现相应基因的单核苷酸多态性,如MUT基因c.1874A>C突变可导致MUT基因编码区的第1874位出现二等位多态性,该位点为A或C;MUT基因c.470T>A突变可导致MUT基因编码区第470位出现二等位多态性,该位点为T或A。上述方法中,检测MUT基因c.1874A>C突变的方法可包括:a1)检测待测样本中MUT基因的cDNA序列,a2)将步骤a1)的序列与MUT基因的cDNA序列相比,步骤a1)的序列对应于MUT基因cDNA序列的第1874位为C或A/C杂合,MUT基因存在c.1874A>C突变。检测MUT基因c.470T>A突变的方法可包括:b1)检测待测样本中MUT基因的cDNA序列,b2)将步骤b1)的序列与MUT基因的cDNA序列相比,步骤b1)的序列对应于MUT基因cDNA序列的第470位为A或T/A杂合,MUT基因存在c.470T>A突变。上述方法中,所述待测样本可来自于待测个体(如新生儿)的血液或组织。为解决上述技术问题,本发明还提供了下述任一应用:I、所述成套试剂或所述试剂或试剂盒的下述E1)-E10)中任一应用:E1)在制备诊断或辅助诊断先天性代谢缺陷病患者产品中的应用;E2)在诊断或辅助诊断先天性代谢缺陷病患者中的应用;E3)在制备筛查或辅助筛查先天性代谢缺陷病患者产品中的应用;E4)在筛查或辅助筛查先天性代谢缺陷病患者中的应用;E5)在制备预测先天性代谢缺陷病患病风险产品中的应用;E6)在预测先天性代谢缺陷病患病风险中的应用;E7)在制备检测与先天性代谢缺陷病相关基因是否突变产品中的应用;E8)在检测与先天性代谢缺陷病相关基因是否突变中的应用;E9)在制备检测与先天性代谢缺陷病相关蛋白质是否突变产品中的应用;E10)在检测与先天性代谢缺陷病相关蛋白质是否突变中的应用;II、权利要求1所述产品的所述E1)-E6)中任一应用。上述应用中,所述患者可为新生儿、婴幼儿或胎儿。上述应用中,所述与先天性代谢缺陷病相关基因突变可为MUT基因、DLD基因、HPD基因、LMBRD1基因的突变,如MUT基因c.1874A>C突变、MUT基因c.470T>A突变。所述与先天性代谢缺陷病相关蛋白质突变可为MUT、DLD、HPD、和/或LMBRD1蛋白质的突变,如MUT蛋白质p.Asp625Ala突变和/或MUT蛋白质p.Val157Asp突变。c.1874A>C突变和c.470T>A突变均为杂合突变。本发明的发明人通过调研疾病数据库和文献,并根据与现有临床检验技术互补的原则,确定了与先天性代谢缺陷病下相关的35个致病基因,对35个基因的编码外显子和选择性剪切位点设计多重探针序列,然后采用新一代高通量靶向重测序技术,对导致先天性代谢缺陷病的35个致病基因的编码外显子和选择性剪切位点进行测序,通过临床样本验证和生物信息分析,基于人群和疾病数据库系统,筛选出已知的致病基因突变,对数据库没有收录的基因突变,进行生物信息学功能预测筛查出新的致病基因突变,并在家系及正常人群中验证排除,发现了新发的复合型杂合突变-MUT基因的错义突变c.1874A>C和错义突变c.470T>A与甲基丙二酸尿症相关。本发明的两个新发突变对开展先天性代谢缺陷病的分子诊断具有重要意义。附图说明图1为高通量测序的文库制备和数据分析流程。图2为对MUT基因突变的家系分析结果。其中,A为患者MUT基因c.1874A>C杂合突变和c.470T>A杂合突变的Sanger验证结果;B为患者父亲MUT基因c.1874A>C杂合突变的Sanger验证结果,C为患者母亲MUT基因c.470T>A杂合突变的Sanger验证结果。图3为变异质量控制的步骤和条件。具体实施方式下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。所需原料均在下文中有说明,一部分购买于illumina的illuminaTruSeqCustomAmpliconKit,一部分购于其他厂家。下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。实施例1、与先天性代谢缺陷病有关的基因突变的发现本实施例用新一代高通量靶向重测序技术,对有先天性代谢缺陷病表型的62例临床样本和6例无先天性代谢缺陷病表型的正常人样本进行了已知35个致病基因的外显子和选择性剪切位点的高通量测序,结合生物信息分析发现了MUT基因的新发复合型杂合突变:错义突变c.1874A>C和错义突变c.470T>A与甲基丙二酸尿症有关,并通过正常人样本筛选和Sanger测序验证了这两个变异。MUT基因野生型的核苷酸序列为序列1,其编码的蛋白MUT的氨基酸序列为序列2;发生错义突变c.1874A>C和错义突变c.470T>A后的MUT突变基因的核苷酸序列为将序列1第1874位的A突变为C,且将序列1第470位的T突变为A,其它核苷酸不变。MUT突变基因编码的MUT突变蛋白的氨基酸序列为将序列2的625位的Asp突变为Ala,且将序列2的第157为的Val突变为Asp,其它氨基酸残基不变。一、与先天性代谢缺陷病有关的基因突变的发现流程图如图1所示。1.样本采集采集了62例临床具有先天性代谢缺陷病表型的婴幼儿的外周血和干血片样本,并采集了6例无先天性代谢缺陷病表型的正常人外周血样本,EDTA抗凝,-80摄氏度保存。62例临床具有先天性代谢缺陷病表型的样本大部分经过串联质谱筛查,并检测出相应的代谢异常。2.文库制备和测序采用illumia的DesignStudio平台针对35个基因的编码外显子和选择性剪切位点设计探针,得到成套探针,这35个基因为:基因ACADM、ARG1、ASL、ASS1、BCKDHA、BCKDHB、CPS1、DBT、DLD、ETFA、ETFB、ETFDH、FAH、GCDH、GCH1、HPD、IVD、LMBRD1、MCEE、MMAA、MMAB、MMACHC、MMADHC、MUT、NAGS、OTC、PAH、PCBD1、PCCA、PCCB、PTS、QDPR、SLC25A13、SLC25A14和TAT基因。成套探针如下:检测MUT基因c.1874A>C突变的探针序列为序列3和序列4:序列3(检测MUT基因c.1874A>C突变的上游探针序列)5’-TGCTCAATGTCTGTCATCATTTTACTAC-3’序列4(检测MUT基因c.1874A>C突变的下游探针序列)5’-GGGTTAATGTGCAGGCTTGTTACATAG-3’检测MUT基因c.470T>A突变的探针序列为序列5和序列6:序列5(检测MUT基因c.470T>A突变的上游探针序列)5’-ATCGTGGCTATGATTCAGACAACCCTC-3’序列6(检测MUT基因c.470T>A突变的下游探针序列)5’-GGAATTTTTACAACATGCTATAACTGAATT-3’检测DLD基因突变的探针序列为序列7和8:序列7(检测DLD基因突变的上游探针序列)5’-CTCAGGAAAAAGTTGGCAAAGGAACAG一3’序列8(检测DLD基因突变的下游探针序列)5’-AAACAGACTTCAAATAATCAAAAAGCAC-3’检测HPD基因突变的探针序列为序列9和10:序列9(检测HPD基因突变的上游探针序列)5’-TGAGTTGACAAAATAAAATGTCCGGCC-3’序列10(检测HPD基因突变的上游探针序列)5’-AAGATGACGGGTCCTCACGGTGGACT-3’检测LMBRD1基因突变的探针序列为序列11和12:序列11(检测LMBRD1基因突变的上游探针序列)5’-TGTTCCACAGTCTGAAAATAGCATTCC-3’序列12(检测LMBRD1基因突变的上游探针序列)5’-GGAAAAGAATTTCCTGGCATTTTGCTG-3’采用illuminaTruSeqCustomAmpliconKit试剂对62例临床样本和6例正常对照样本进行文库制备和合并。合并后的文库采用MiSeq测序仪进行测序,读长2×300bp。下述方法中的步骤(c)、(d)、(e)、(f)、(g)和(h)所用试剂均是illumina公司生产的TruSeqCustomAmpliconKit试剂。(a)基因组DNA的提取提取外周血样本的基因组DNA,提取方法可参照《分子克隆实验指南》中提供的基因组DNA提取方法,也可购买商品化试剂盒(如QIAGEN公司)按照说明书操作。(b)基因组DNA的质控对提取的基因组DNA进行质控,Nanodrop2000分光光度计测纯度,要求OD260/0D280在1.8~2.0之间;Qubit荧光光度计测定浓度,要求浓度不低于3.5ng/μL,总量不低于50ng。(c)探针与基因组DNA杂交将上述成套探针和杂交缓冲液依次加到稀释后的基因组DNA中,95℃孵育1min,然后缓慢降温到40℃,得到杂交混合液。(d)杂交产物的纯化组装过滤板,用缓冲液预洗过滤板孔。将杂交混合液加入到过滤膜中,离心,过滤掉没有与基因组DNA结合的探针,然后用洗涤buffer进行洗涤,离心,得到探针-基因组DNA杂交复合物。(e)探针的延伸-连接向纯化后的探针-基因组DNA杂交复合物中,加入延伸-连接酶和buffer混合液,37℃孵育45min。(f)延伸-连接产物的纯化孵育结束后,将得到的延伸-连接产物采用AMPureXP磁珠(美国Beckman公司)进行纯化,去除未反应完的酶、buffer和dNTPMixture。(g)PCR扩增取96孔PCR板,将i5接头管按PCR板A-H的方向排列,将i7接头管按PCR板1-12的方向排列,向PCR板的每行(1-12)加入相同的i5接头,向PCR板每列(A-H)加入相同的i7接头;向PCR板孔中加入PCRmastermix;再向每个孔中加入纯化后的延伸-连接产物。短暂离心,进行PCR扩增,扩增条件:95℃预变性3min;95℃变性30s,66℃退火30s,72℃延伸60s共扩增25~30个循环;最后72℃延伸5min。(h)PCR扩增产物的纯化扩增后得到的PCR产物,使用AMPureXP磁珠(美国Beckman公司)进行纯化,并按照产品推荐的方案进行实验操作。(i)文库的质控纯化后的PCR产物进行浓度检测和扩增片段大小检测。使用Qubit荧光光度计用BR试剂粗略测定浓度,使用QPCR仪准确测定浓度。用Agilent2100生物分析仪检测文库大小分布,上样量luL,文库大小分布要求:单峰、片段大小在600bp左右。(j)文库的合并对质控合格的文库进行合并,根据每个文库的浓度计算每个文库合并时加入的体积,等摩尔合并各个文库;根据测序仪的数据通量、目标区域大小和需要的覆盖深度,计算合并的文库个数。(k)文库的稀释和上机测序用杂交稀释buffer(illumina公司产品,货号:为MS-102-3003)稀释合并后的文库,然后按照测序仪的操作说明进行簇生成和测序。3.数据分析采用MiSeq测序仪配套的数据分析软件MiSeqReporter进行原始数据的处理、分析,检测SNP和Indel变异。对检测的SNP和Indel变异使用SnpEff软件进行注释。尤其关注无义突变(nonsensevariant)、移码突变(frameshift)、选择性剪切变异(splicingvariant)、错义突变(missensevariant)、氨基酸截断或插入变异(nonframeshift)和密码子丢失(stoplost)六种变异类型。利用ClinVar等疾病数据库和其他突变数据库筛选出已知的致病突变,利用人群数据库,如dbSNP数据、千人基因组数据库、ESP(外显子组测序数据库)过滤掉已知变异。同时利用正常对照样本的测序结果对剩下的变异进行过滤。选取患者中存在的变异,而正常对照样本中不存在的变异。利用SIFT、Polyphen、MutationTaster等多款预测软件对SNP进行功能预测。预测结果为致病的SNP为潜在的致病突变。在这些突变中发现MUT基因发生了c.1874A>C的杂合突变和c.470T>A的杂合突变为潜在的致病突变。这两个变异在同一个患者样本中检测到,并经过Sanger测序验证。该患者有先天性代谢缺陷病的临床表现,经串联质谱筛查为甲基丙二酸尿症。对患者的父亲和母亲采取外周血,提取DNA,进行MUT基因的Sanger测序。结果发现患者父亲的MUT基因发生了c.1874A>C的杂合突变,未发生c.470T>A的杂合突变;患者母亲的MUT基因发生了c.470T>A的杂合突变,未发生c.1874A>C的杂合突变。由此可以确定MUT基因的c.1874A>C杂合突变和c.470T>A杂合突变为复合型杂合突变。患者为这两个杂合突变同时发生时致病,父母只是其中一个杂合突变的携带者,为正常人。MUT基因c.1874A>C杂合突变和c.470T>A杂合突变在6例正常对照样本中证实不存在。利用UCSC数据库的10000个脊椎动物的比较基因组分析发现MUT基因的c.1874A>C突变和c.470T>A位于高度保守位点。4.实验结果通过35个致病基因的靶向重测序及生物信息分析发现一名患者发生了两个新发突变,分别是MUT基因上c.1874A>C的杂合突变和c.470T>A的杂合突变,经过家系验证为复合型杂合突变。OMIM数据库注释MUT基因对应的疾病如表1。表1、OMIM数据库注释MUT基因对应的疾病通过家系内共分离实验及患者的临床表现与基因突变相关疾病的表型信息证明,MUT基因上c.1874A>C和c.470T>A的复合型杂合突变与甲基丙二酸尿症有关。5.变异的质量控制步骤和条件每个样本检测出的变异数目有几百个,按照MiSeqReporter软件默认的质量控制条件,通过质量控制的变异经Sanger测序法验证的一致率为61.42%,没有通过质量控制的变异经Sanger测序法验证的一致率为17.78%。由此可见按照默认的质量控制条件会有较高的假阳性和假阴性。为降低假阳性和假阴性,建立了一套基于MiSeq扩增子测序的变异质量控制步骤和条件:(a)过滤掉标记为“R8”的变异;(b)过滤掉标记为“LowVariantFrequency”的变异;(c)过滤掉标记为“LowGQX”的变异;(d)过滤掉QUAL值小于50的变异;(e)基因型杂合时,过滤掉测序深度小于20乘的变异;基因型为纯合时,过滤掉测序深度小于4乘的SNP和测序深度小于10乘的Indel。按照上述质量控制步骤和条件,通过质量控制的变异经Sanger测序法验证一致率为89.28%,没有通过质量控制的变异经Sanger测序法验证一致率为1.69%。由此可见,按照调整后的质量控制步骤和条件,大大的降低了假阳性和假阴性。变异质量控制的步骤和条件如图3所示。二、与先天性代谢缺陷病有关的基因突变的Sanger法测序验证对患者样本中检测到的突变进行Sanger测序验证,并进行家系验证。具体方法步骤如下:(1)基因组DNA提取对检测到上述突变的临床样本,采用QIAGENMiniKit从外周血中提取基因组DNA。对提取的基因组DNA进行质量控制,0D260/280在1.8~2.0之间,0D260/230在1.8~2.0之间。(2)引物设计及PCR反应参考人类基因组序列数据库GRCh37/hg19,采用Primer3.0分别针对MUT基因的c.1874A>C突变位点和c.470T>A突变位点设计特异性引物,引物序列见下表2:表2、MUT基因的c.1874A>C突变位点和c.470T>A突变位点设计特异性引物基因突变位点正向引物(5’>3’)反向引物(5’>3’)产物大小MUTc.1874A>CTGGGCAGGAATGGAGAGAAA(序列13)GGGGTGATCTATGCAGCTGTT(序列14)542bpMUTc.470T>ATCATGGATGGTTCTGGAGGAA(序列15)TGGCACAAGGGAAACAACTG(序列16)535bp(3)PCR反应体系按下表3向PCR管中加入各组分,反应总体积50μL。表3试剂体积/每个反应10×PCRBuffer(mg2+plus)5μLdNTPMixture(each10mM)1μLTaKaRaTaq(5U/μL)0.25μL正向引物2μL反向引物2μL基因组DNA100ngddH20Upto50μL(4)PCR热循环条件将PCR管短暂离心,然后于PCR仪上按照下表中的热循环条件进行扩增反应。表4(5)Sanger测序将PCR产物测序结果见图2。图2显示了患者样本Sanger测序和家系验证的结果。图2中A为患者MUT基因c.1874A>C的杂合突变和c.470T>A杂合突变的测序峰图,两个位点都发生了杂合突变。图2中B为患者父亲MUT基因c.1874A>C的杂合突变和c.470T>A杂合突变的测序峰图,只有MUT基因编码区第1874位发生了A到C的杂合突变。图2中C为患者母亲MUT基因c.1874A>C的杂合突变和c.470T>A杂合突变的测序峰图,只有MUT基因编码区第470位发生了T到A的杂合突变。箭头指示的位置为突变位点。序列表<110>中国人民解放军陆军总医院博奥生物集团有限公司深圳市第三人民医院<120>一组与先天性代谢缺陷病相关的基因新突变及检测试剂盒<160>16<170>PatentInversion3.5<210>1<211>2253<212>DNA<213>人工序列<220><223><400>1atgttaagagctaagaatcagctttttttactttcacctcattacctgaggcaggtaaaa60gaatcatcaggctccaggctcatacagcaacgacttctacaccagcaacagccccttcac120ccagaatgggctgccctggctaaaaagcagctgaaaggcaaaaacccagaagacctaata180tggcacaccccggaagggatctctataaaacccttgtattccaagagagatactatggac240ttacctgaagaacttccaggagtgaagccattcacacgtggaccatatcctaccatgtat300acctttaggccctggaccatccgccagtatgctggttttagtactgtggaagaaagcaat360aagttctataaggacaacattaaggctggtcagcagggattatcagttgcctttgatctg420gcgacacatcgtggctatgattcagacaaccctcgagttcgtggtgatgttggaatggct480ggagttgctattgacactgtggaagataccaaaattctttttgatggaattcctttagaa540aaaatgtcagtttccatgactatgaatggagcagttattccagttcttgcaaattttata600gtaactggagaagaacaaggtgtacctaaagagaagcttactggtaccatccaaaatgat660atactaaaggaatttatggttcgaaatacatacatttttcctccagaaccatccatgaaa720attattgctgacatatttgaatatacagcaaagcacatgccaaaatttaattcaatttca780attagtggataccatatgcaggaagcaggggctgatgccattctggagctggcctatact840ttagcagatggattggagtactctagaactggactccaggctggcctgacaattgatgaa900tttgcaccaaggttgtctttcttctggggaattggaatgaatttctatatggaaatagca960aagatgagagctggtagaagactctgggctcacttaatagagaaaatgtttcagcctaaa1020aactcaaaatctcttcttctaagagcacactgtcagacatctggatggtcacttactgag1080caggatccctacaataatattgtccgtactgcaatagaagcaatggcagcagtatttgga1140gggactcagtctttgcacacaaattcttttgatgaagctttgggtttgccaactgtgaaa1200agtgctcgaattgccaggaacacacaaatcatcattcaagaagaatctgggattcccaaa1260gtggctgatccttggggaggttcttacatgatggaatgtctcacaaatgatgtttatgat1320gctgctttaaagctcattaatgaaattgaagaaatgggtggaatggccaaagctgtagct1380gagggaatacctaaacttcgaattgaagaatgtgctgcccgaagacaagctagaatagat1440tctggttctgaagtaattgttggagtaaataagtaccagttggaaaaagaagacgctgta1500gaagttctggcaattgataatacttcagtgcgaaacaggcagattgaaaaacttaagaag1560atcaaatccagcagggatcaagctttggctgaacgttgtcttgctgcactaaccgaatgt1620gctgctagcggagatggaaatatcctggctcttgcagtggatgcatctcgggcaagatgt1680acagtgggagaaatcacagatgccctgaaaaaggtatttggtgaacataaagcgaatgat1740cgaatggtgagtggagcatatcgccaggaatttggagaaagtaaagagataacatctgct1800atcaagagggttcataaattcatggaacgtgaaggtcgcagacctcgtcttcttgtagca1860aaaatgggacaagatggccatgacagaggagcaaaagttattgctacaggatttgctgat1920cttggttttgatgtggacataggccctcttttccagactcctcgtgaagtggcccagcag1980gctgtggatgcggatgtgcatgctgtgggcataagcaccctcgctgctggtcataaaacc2040ctagttcctgaactcatcaaagaacttaactcccttggacggccagatattcttgtcatg2100tgtggaggggtgataccacctcaggattatgaatttctgtttgaagttggtgtttccaat2160gtatttggtcctgggactcgaattccaaaggctgccgttcaggtgcttgatgatattgag2220aagtgtttggaaaagaagcagcaatctgtataa2253<210>2<211>750<212>PRT<213>人工序列<220><223><400>2MetLeuArgAlaLysAsnGlnLeuPheLeuLeuSerProHisTyrLeu151015ArgGlnValLysGluSerSerGlySerArgLeuIleGlnGlnArgLeu202530LeuHisGlnGlnGlnProLeuHisProGluTrpAlaAlaLeuAlaLys354045LysGlnLeuLysGlyLysAsnProGluAspLeuIleTrpHisThrPro505560GluGlyIleSerIleLysProLeuTyrSerLysArgAspThrMetAsp65707580LeuProGluGluLeuProGlyValLysProPheThrArgGlyProTyr859095ProThrMetTyrThrPheArgProTrpThrIleArgGlnTyrAlaGly100105110PheSerThrValGluGluSerAsnLysPheTyrLysAspAsnIleLys115120125AlaGlyGlnGlnGlyLeuSerValAlaPheAspLeuAlaThrHisArg130135140GlyTyrAspSerAspAsnProArgValArgGlyAspValGlyMetAla145150155160GlyValAlaIleAspThrValGluAspThrLysIleLeuPheAspGly165170175IleProLeuGluLysMetSerValSerMetThrMetAsnGlyAlaVal180185190IleProValLeuAlaAsnPheIleValThrGlyGluGluGlnGlyVal195200205ProLysGluLysLeuThrGlyThrIleGlnAsnAspIleLeuLysGlu210215220PheMetValArgAsnThrTyrIlePheProProGluProSerMetLys225230235240IleIleAlaAspIlePheGluTyrThrAlaLysHisMetProLysPhe245250255AsnSerIleSerIleSerGlyTyrHisMetGlnGluAlaGlyAlaAsp260265270AlaIleLeuGluLeuAlaTyrThrLeuAlaAspGlyLeuGluTyrSer275280285ArgThrGlyLeuGlnAlaGlyLeuThrIleAspGluPheAlaProArg290295300LeuSerPhePheTrpGlyIleGlyMetAsnPheTyrMetGluIleAla305310315320LysMetArgAlaGlyArgArgLeuTrpAlaHisLeuIleGluLysMet325330335PheGlnProLysAsnSerLysSerLeuLeuLeuArgAlaHisCysGln340345350ThrSerGlyTrpSerLeuThrGluGlnAspProTyrAsnAsnIleVal355360365ArgThrAlaIleGluAlaMetAlaAlaValPheGlyGlyThrGlnSer370375380LeuHisThrAsnSerPheAspGluAlaLeuGlyLeuProThrValLys385390395400SerAlaArgIleAlaArgAsnThrGlnIleIleIleGlnGluGluSer405410415GlyIleProLysValAlaAspProTrpGlyGlySerTyrMetMetGlu420425430CysLeuThrAsnAspValTyrAspAlaAlaLeuLysLeuIleAsnGlu435440445IleGluGluMetGlyGlyMetAlaLysAlaValAlaGluGlyIlePro450455460LysLeuArgIleGluGluCysAlaAlaArgArgGlnAlaArgIleAsp465470475480SerGlySerGluValIleValGlyValAsnLysTyrGlnLeuGluLys485490495GluAspAlaValGluValLeuAlaIleAspAsnThrSerValArgAsn500505510ArgGlnIleGluLysLeuLysLysIleLysSerSerArgAspGlnAla515520525LeuAlaGluArgCysLeuAlaAlaLeuThrGluCysAlaAlaSerGly530535540AspGlyAsnIleLeuAlaLeuAlaValAspAlaSerArgAlaArgCys545550555560ThrValGlyGluIleThrAspAlaLeuLysLysValPheGlyGluHis565570575LysAlaAsnAspArgMetValSerGlyAlaTyrArgGlnGluPheGly580585590GluSerLysGluIleThrSerAlaIleLysArgValHisLysPheMet595600605GluArgGluGlyArgArgProArgLeuLeuValAlaLysMetGlyGln610615620AspGlyHisAspArgGlyAlaLysValIleAlaThrGlyPheAlaAsp625630635640LeuGlyPheAspValAspIleGlyProLeuPheGlnThrProArgGlu645650655ValAlaGlnGlnAlaValAspAlaAspValHisAlaValGlyIleSer660665670ThrLeuAlaAlaGlyHisLysThrLeuValProGluLeuIleLysGlu675680685LeuAsnSerLeuGlyArgProAspIleLeuValMetCysGlyGlyVal690695700IleProProGlnAspTyrGluPheLeuPheGluValGlyValSerAsn705710715720ValPheGlyProGlyThrArgIleProLysAlaAlaValGlnValLeu725730735AspAspIleGluLysCysLeuGluLysLysGlnGlnSerVal740745750<210>3<211>28<212>DNA<213>人工序列<220><223><400>3tgctcaatgtctgtcatcattttactac28<210>4<211>27<212>DNA<213>人工序列<220><223><400>4gggttaatgtgcaggcttgttacatag27<210>5<211>27<212>DNA<213>人工序列<220><223><400>5atcgtggctatgattcagacaaccctc27<210>6<211>30<212>DNA<213>人工序列<220><223><400>6ggaatttttacaacatgctataactgaatt30<210>7<211>27<212>DNA<213>人工序列<220><223><400>7ctcaggaaaaagttggcaaaggaacag27<210>8<211>28<212>DNA<213>人工序列<220><223><400>8aaacagacttcaaataatcaaaaagcac28<210>9<211>27<212>DNA<213>人工序列<220><223><400>9tgagttgacaaaataaaatgtccggcc27<210>10<211>26<212>DNA<213>人工序列<220><223><400>10aagatgacgggtcctcacggtggact26<210>11<211>27<212>DNA<213>人工序列<220><223><400>11tgttccacagtctgaaaatagcattcc27<210>12<211>27<212>DNA<213>人工序列<220><223><400>12ggaaaagaatttcctggcattttgctg27<210>13<211>20<212>DNA<213>人工序列<220><223><400>13tgggcaggaatggagagaaa<210>14<211>21<212>DNA<213>人工序列<220><223><400>14ggggtgatctatgcagctgtt<210>15<211>21<212>DNA<213>人工序列<220><223><400>15tcatggatggttctggaggaa<210>16<211>21<212>DNA<213>人工序列<220><223><400>16tggcacaagggaaacaactg当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1