一种用于植物基因组定点碱基替换的载体的制作方法
本发明涉及生物技术领域,具体地,涉及一种用于植物基因组定点碱基替换的载体以及植物基因组定点碱基替换的方法。
背景技术:
锌指核酸技术(Zinc finger nuclease,ZFN)、转录激活因子样效应物核酸酶技术(transcription activator-like(TAL)effector nucleases,Talen)和CRISPR/Cas9技术是这几年基因组编辑领域的几项突破性技术1-3。这三种技术都可以在生物体基因组指定位点特异性的切割DNA产生双链断裂,从而利用生物的非同源末端连接或同源重组,进行定点编辑。ZFN和Talen技术用特异性的蛋白引导,蛋白元件的构建相对较复杂,编辑效率有待提高。CRISPR/Cas9技术以RNA引导,体外构建简单,编辑效率较高。目前,这三种技术已成功的应用于多种生物,包括大肠杆菌、酵母、水稻、拟南芥、大豆、玉米、小鼠和人类细胞等4。
基因组编辑技术在生物体基因组的指定位点引入双链断裂后,其编辑的结果都是随机的碱基插入或缺失(Indel)4。到目前为止,在植物基因组中进行精确的碱基替换一直是一项难题,其实现途径主要是同源重组,即通过外源供体DNA来实现碱基替换,但是其效率非常低,至今还尚未有效应用于植物研究和育种。现有的几个发表在国际顶级期刊的碱基替换的报道都还只是瞬时表达实验5,6,或者其编辑位点本身就是筛选标签7,例如除草剂双草醚的靶基因乙酸乳酸合成酶ALS等8。另一方面,大规模的基因测序表明,重要的农艺性状位点大多都是单碱基改变9,10。因此,在植物研究和育种领域,迫切需要一种能够进行高效的碱基替换的方法。
因此,本领域迫切需要在植物领域建立一套适用于植物细胞的高效的定点碱基替换工具,并且可获得稳定植株,从而有效的应用于植物研究和育种。
技术实现要素:
本发明的目的在于提供一种适用于植物细胞的高效的定点碱基替换工具,并且可获得稳定植株,从而有效的应用于植物研究和育种。
本发明第一方面提供了一种用于对植物进行基因编辑的核酸构建物,所述的核酸构建物包括:
第一表达盒;
第二表达盒;和
任选的第三表达盒;
其中,所述第一表达盒为用于表达gRNA的gRNA表达盒;
所述第二表达盒为用于表达融合蛋白的融合蛋白表达盒,所述的融合蛋白具有式Ia或Ib结构:
A-L1-B-L2-C (Ia)
B-L1-A-L2-C (Ia)
各式中,
A为胞嘧啶脱氨基酶;
L1为无或第一连接肽(如Xten)
B为无切割活性的Cas9或仅具有单链切割活性的Cas9;
L2为无或第二连接肽(如Xten);
C为无或Uracil DNA glycosylase inhibitor(UGI);
所述第三表达盒为用于表达筛选标记的筛选标记表达盒;
并且,所述的第一表达盒、第二表达盒、和第三表达盒各自独立地位于相同或不同的载体上。
在另一优选例中,所述的第一表达盒具有式A结构:
X1-X2-X3 (A)
式中,
X1为植物启动子,优选植物聚合酶III驱动的启动子(如U6、U3启动子);
X2为gRNA的编码序列;
X3为polyT。
在另一优选例中,所述的第二表达盒具有式B结构:
Y1-Y2-Y3 (B)
式中,
Y1为植物启动子,优选UBI、CaMV35S、Actin启动子;
Y2为融合蛋白的编码序列,所述融合蛋白具有如上所述的式Ia或Ib结构;
Y3为终止子。
在另一优选例中,所述的第三表达盒具有式C结构:
Z1-Z2-Z3 (C)
式中,
Z1为植物启动子,优选35S、UBI、Actin启动子;
Z2为编码筛选标记的编码序列;
Z3为终止子。
在另一优选例中,所述的筛选标记选自下组:潮霉素的抗性基因(HYG)、G418和卡那霉素的抗性基因(NPTII)、Basta的抗性基因(BAR)、嘌呤霉素抗性基因(PAC)、新霉素抗性基因(NEO)、或其组合。
在另一优选例中,所述的第一表达盒和第二表达盒位于同一的载体上。
在另一优选例中,所述的第一表达盒、第二表达盒和第三表达盒位于同一的载体上。
在另一优选例中,所述的第二表达盒和第三表达盒位于同一的载体上。
在另一优选例中,所述的载体为可转染或转化植物细胞的表达载体。
在另一优选例中,所述的载体为农杆菌Ti载体。
在另一优选例中,当第一表达盒、第二表达盒和第三表达盒中的两个或三个位于同一载体时,所述的二个或三个表达盒是成簇排列的,从而构成表达盒簇(cluster)。
在另一优选例中,在所述表达盒簇的5’端外侧含有RB序列;并且在所述表达盒簇的3’端外侧含有LB序列。
在另一优选例中,所述的表达盒簇中,各表达盒的排列次序(从5’-3’)选自下组:
第一表达盒和第二表达盒;
第一表达盒、第二表达盒和第三表达盒;
第二表达盒、第一表达盒和第三表达盒;
第三表达盒、第二表达盒和第一表达盒;
第三表达盒、第一表达盒和第二表达盒;
第三表达盒、第二表达盒;
第二表达盒、第三表达盒。
在另一优选例中,所述的第一表达盒和第二表达盒位于不同的载体上。
在另一优选例中,当单个表达盒位于一载体时,则在所述表达盒的5’端外侧含有RB序列;并且在所述表达盒的3’端外侧含有LB序列。
本发明第二方面提供了一种表达载体,所述表达载体含有如本发明第一方面所述的核酸构建物,其中,所述的第一表达盒、第二表达盒、和第三表达盒各自独立地位于相同或不同的载体上。
在另一优选例中,所述的表达载体骨架为pCamiba1300。
在另一优选例中,所述的载体为可转染或转化植物细胞的表达载体。
在另一优选例中,所述的载体为农杆菌Ti载体。
在另一优选例中,所述载体是环状的或者是线性的。
本发明第三方面提供了一种宿主细胞,所述细胞含有本发明第二方面所述的表达载体。
在另一优选例中,所述的宿主细胞为农杆菌。
在另一优选例中,所述的宿主细胞为植物细胞。
本发明第四方面提供了一种基因工程细胞,所述的细胞含有本发明第一方面所述的构建物,或其基因组整合有一个或多个本发明第一方面所述的构建物。
在另一优选例中,所述的细胞为植物细胞。
在另一优选例中,所述的植物选自下组:禾本科植物、豆科植物、十字花科植物、或其组合。
在另一优选例中,所述的植物选自下组:水稻、大豆、番茄、玉米、烟草、小麦、高粱、拟南芥、大麦、燕麦、粟、花生、或其组合。
在另一优选例中,所述的基因工程细胞是用选自下组的方法将本发明第一方面所述的构建物导入细胞的:农杆菌转化法、基因枪法、显微注射法、电击法、超声波法、聚乙二醇(PEG)介导法、或其组合。
本发明第五方面提供了一种对植物进行基因编辑的方法,包括步骤:
(i)提供待编辑植物;
(ii)将本发明第二方面所述的表达载体导入所述待编辑植物。
在另一优选例中,所述导入为通过农杆菌导入。
在另一优选例中,所述导入为通过基因枪导入。
在另一优选例中,所述的基因编辑为定点碱基替换。
在另一优选例中,所述的定点突变包括将C突变为T或者C突变为G
在另一优选例中,所述的定点突变包括将PAM远端第4-8位中的C进行定点突变。
本发明第六方面提供了一种制备转基因植物细胞的方法,包括步骤:
(i)将本发明第一方面所述的构建物、或本发明第二方面所述的载体转染植物细胞,使得所述构建物与所述植物细胞中的染色体发生定点突变,从而制得所述转基因植物细胞。
在另一优选例中,所述的转染采用农杆菌转化法或基因枪轰击法。
本发明第七方面提供了一种制备转基因植物细胞的方法,包括步骤:
(i)将本发明第一方面所述的构建物、或本发明第二方面所述的载体转染植物细胞,使得所述植物细胞含有所述构建物,从而制得所述转基因植物细胞。
本发明第八方面提供了一种制备转基因植物的方法,包括步骤:
将本发明第六方面或本发明第七方面所述方法制备的所述转基因植物细胞再生为植物体,从而获得所述转基因植物。
本发明第九方面提供了一种转基因植物,所述的植物是用本发明第八方面所述的方法制备的,或者所述植物的植物细胞中含有本发明第一方面所述的构建物或其基因组整合有一个或多个本发明第一方面所述的构建物。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
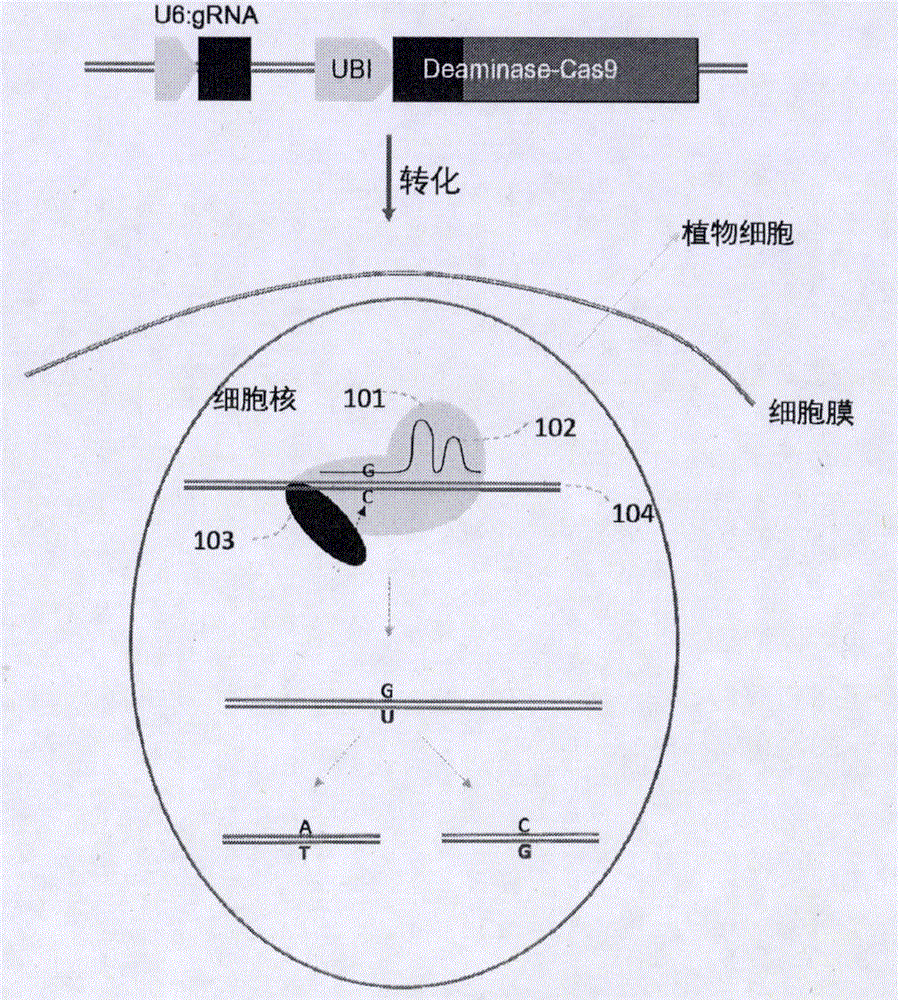
图1基于胞嘧啶脱氨酶的定点碱基替换示意图。将表达Cas的DNA序列与表达胞嘧啶脱氨基酶的DNA序列连接,转化植物受体,使转化的植物细胞中的DNA表达该融合蛋白和guide RNA。融合胞嘧啶脱氨基酶(103)的Cas蛋白(101)在其guide RNA(102)的引导下,将植物基因组(104)中靶点位置的胞嘧啶C脱氨基生成尿嘧啶U,然后在植物细胞的修复系统作用下,将U修复成T或G。
图2采用APOBEC1和CRISPR/Cas9的定点碱基替换载体。(A)载体pCSGAPO1的示意图。(B)脱氨基区域示意图。
图3水稻愈伤组织定点碱基替换结果。(A)水稻NRT1.1B基因碱基替换示意图。(B)水稻SLR1基因碱基替换示意图。(C)高通量测序检测结果。
图4水稻转基因植株中NRT1.1B基因的碱基替换结果。(A)Indel植株(#35)和碱基替换植株(#15)的Sanger测序结果。(B)碱基替换植株(#15)的PCR产物单菌落测序结果。
图5水稻转基因植株中SLR1基因的碱基替换结果。(A)StyI酶切检测示意图及结果。(B)Indel植株(#07)和碱基替换植株(#07、#10、#18、#23、#28和#37)的Sanger测序结果。(C)碱基替换植株(#07)的单菌落测序结果。(D)SLR1基因野生型(WT)、敲除(SLR#09)和碱基替换(SLR#07)的T0代植株表型。
具体实施方式
本发明人经过广泛而深入地研究,通过大量筛选,筛选出针对特异性位点的优化的启动子,通过采用特定结构的核酸构建物,本发明首次在植物中成功实现了RNA引导的碱基定点突变(如C突变为T或G),并且突变效率可高达11.5%,并且在一个突变植株内,有高达60%的细胞发生了替换。
具体地,本发明首次发现胞嘧啶脱氨基酶能够在CRISPR/Cas9的引导下,在植物细胞的基因组上定点修改靶点上的胞嘧啶,将其突变为T或G,并在此基础上完成了本发明。
如本文所用,术语“碱基替换”、碱基突变”可互换使用,均指将某一碱基突变为其他碱基,比如C突变为T或者C突变为G。
如本文所用,“筛选标记基因”指转基因过程中用来筛选转基因细胞或转基因动物的基因,可用于本申请的筛选标记基因没有特别限制,包括转基因领域常用的各种筛选标记基因,代表性例子包括(但并不限于):潮霉素抗性基因(Hyg)、卡那霉素抗性基因(NPTII)、新霉素基因、嘌呤霉素抗性基因、潮霉素的抗性基因(HYG)、G418和卡那霉素的抗性基因(NPTII)、Basta的抗性基因(BAR)、嘌呤霉素抗性基因(PAC)、和/或新霉素抗性基因(NEO)。
如本文所用,术语“表达盒”是指含有待表达基因以及表达所需元件的序列组件的一段多聚核苷酸序列。例如,在本发明中,术语“筛选标记表达盒”指含有编码筛选标记的序列以及表达所需元件的序列组件的多聚核苷酸序列。表达所需的组件包括启动子和聚腺苷酸化信号序列。此外,筛选标记表达盒还可以含有或不含有其他序列,包括(但并不限于):增强子、分泌信号肽序列等。
在本发明中,适用于外源基因表达盒和筛选标记基因表达盒的启动子可以是任何一种常见的启动子,它可以是组成型启动子或诱导型启动子。较佳地,该启动子是组成型启动子,例如UBI启动子等其它适用于真核表达的植物启动子。
如本文所用,术语“植物启动子”指能够在植物细胞中启动核酸转录的核酸序列。该植物启动子可以是来源于植物、微生物(如细菌、病毒)或动物等,或者是人工合成或改造过的启动子。
如本文所用,术语“植物终止子”指能够在植物细胞中可使转录停止的终止子。该植物转录终止子可以是来源于植物、微生物(如细菌、病毒)或动物等,或者是人工合成或改造过的终止子。代表性的例子包括(但并不限于):Nos终止子。
如本文所用,术语“Cas蛋白”指一种核酸酶。一种优选的Cas蛋白是Cas9蛋白。典型的Cas9蛋白包括(但并不限于):来源于酿脓链球菌(Streptococcuspyogenes)的Cas9。在本发明中,Cas9蛋白为突变的Cas9蛋白,具体地,是无切割活性或只具有单链切割活性的突变的Cas9蛋白。
如本文所用,术语“Cas蛋白的编码序列”指编码Cas蛋白的核苷酸序列。在插入的多聚核苷酸序列被转录和翻译从而产生功能性Cas蛋白的情况下,技术人员会认识到,因为密码子的简并性,有大量多聚核苷酸序列可以编码相同的多肽。另外,技术人员也会认识到不同物种对于密码子具有一定的偏好性,可能会根据在不同物种中表达的需要,会对Cas蛋白的密码子进行优化,这些变异体都被术语“Cas蛋白的编码序列”所具体涵盖。此外,术语特定地包括了全长的、与Cas基因序列基本相同的序列,以及编码出保留Cas蛋白功能的蛋白质的序列。
如本文所用,术语“植物”包括全植株、植物器官(如叶、茎、根等)、种子和植物细胞以及它们的子代。可用于本发明方法的植物的种类没有特别限制,一般包括任何可进行转化技术的高等植物类型,包括单子叶、双子叶植物和裸子植物。
本发明的构建物
在本发明中,提供了一种核酸构建物,用于对植物进行基因编辑,所述的核酸构建物包括:
第一表达盒;
第二表达盒;和
任选的第三表达盒;
其中,第一表达盒为用于表达gRNA的gRNA表达盒;
第二表达盒为用于表达融合蛋白的融合蛋白表达盒,所述的融合蛋白具有式Ia或Ib结构:
A-L1-B-L2-C (Ia)
B-L1-A-L2-C (Ia)
各式中,
A为胞嘧啶脱氨基酶;
L1为无或第一连接肽(如Xten)
B为无切割活性的Cas9或仅具有单链切割活性的Cas9;
L2为无或第二连接肽(如Xten);
C为无或Uracil DNA glycosylase inhibitor(UGI);
第三表达盒为用于表达筛选标记的筛选标记表达盒;
并且,所述的第一表达盒、第二表达盒、和第三表达盒各自独立地位于相同或不同的载体上。
本发明的构建物中所用的各种元件都是本领域中已知的,因此本领域技术人员可以用常规方法,如PCR方法、全人工化学合成法、酶切方法获得相应的元件,然后通过熟知的DNA连接技术连接在一起,就形成了本发明的构建物。
将本发明的构建物插入外源载体(尤其是适合转基因植物操作的载体),就构成了本发明的载体。
将本发明的载体转化植物细胞从而介导本发明的载体对植物细胞染色体进行整合,制得转基因植物细胞。
将本发明的转基因植物细胞再生为植物体,从而获得转基因植物。
将本发明构建好的上述核酸构建物,通过常规的植物重组技术(例如农杆菌转让技术),可以导入植物细胞,从而获得携带所述核酸构建物(或带有所述核酸构建物的载体)的植物细胞,或获得基因组中整合有所述核酸构建物的植物细胞。
载体构建
该载体的主要特征是将胞嘧啶脱氨基酶与CRISPR/Cas系统中的Cas蛋白连接形成融合蛋白,并由guide RNA引导至基因组中的靶点位置。由于胞嘧啶脱氨基酶直接将胞嘧啶C脱氨基生成U,并不需要Cas蛋白的DNA双链切割活性。因此,在本发明中Cas蛋白是无切割活性的突变的Cas蛋白。胞嘧啶C脱氨基生成U后,植物细胞启动碱基错配修复,将U-G修复为T-A或者C-G。为了增加U-G转化为T-A的比例,可以保留Cas蛋白的DNA单链切刻活性,即在非编辑单链(编辑靶点C的匹配链)造成单链切刻。因此,优选的,将胞嘧啶脱氨基酶与具有单链切刻活性的Cas蛋白融合,形成融合蛋白。一般的,为了增加融合蛋白的活性,蛋白间一般通过一些柔性短肽连接,即Linker。优选的,该Linker可以选用XTEN。
为了增加修复效率,一般选择适用于植物细胞的强启动子,比如CaMV 35S启动子或者UBI启动子等。选择适用于植物细胞的guide RNA的表达框,并将其与上述融合蛋白表达框构建在同一载体。显而易见的,这两个表达框也可以在不同的DNA分子上。
靶点设计
胞嘧啶脱氨基酶通过CRISPR/Cas9系统引导至靶点位置后,脱氨基酶的作用区域一般固定。例如,将胞嘧啶脱氨基酶APOBEC1蛋白通过16个氨基酸的XTEN Linker连接至Cas9的N端后,其脱氨基的作用区域为guilde RNA的第4-8个碱基区域,该区域即为“脱氨基窗口区”(图2)。根据这一原则,在设计靶点时,需将待编辑的胞嘧啶处于“脱氨基窗口区”。
遗传转化
将上述载体通过合适的方法导入到植物受体中。导入方法包括但不局限于:基因枪法、显微注射法、电击法、超声波法和聚乙二醇(PEG)介导法等。受体植物包括但不限于水稻、大豆、番茄、玉米、烟草、小麦、高粱等。上述DNA载体或片段导入植物细胞后,使转化的植物细胞中的DNA表达该融合蛋白和guide RNA。融合胞嘧啶脱氨基酶的Cas蛋白在其guide RNA的引导下,将靶点位置的胞嘧啶C脱氨基生成尿嘧啶U,然后在植物细胞的修复系统作用下,将U修复成T或G。进一步的,通过组织培养获得碱基替换后的植株。
应用
本发明可以用于植物基因工程领域,用于植物研究和育种,尤其是具有经济价值的农作物和林业作物的遗传改良。
本发明的主要优点包括:
(1)本发明首次提供了一种在植物中实现定点碱基突变的高效方法,可以广泛的用于植物研究和育种。
(2)本发明的定点碱基突变的方法可高效地在植物细胞的特定位置进行碱基突变(如C突变为T或G)。
(3)用本发明的方法在植物细胞中进行碱基突变的效果高达11.5%,并且在一个突变植株内,有高达60%的细胞发生了碱基替换。
(4)本发明的方法在对植物细胞进行精准碱基替换时,无需额外引入供体DNA,能有效避免外源DNA的随机整合。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件如Sambrook等人,分子克隆:实验室手册(New York:Cold Spring Harbor Laboratory Press,1989)中所述的条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。本发明中所涉及的实验材料和试剂如无特殊说明均可从市售渠道获得。
通用方法
体外合成小鼠胞嘧啶脱氨基酶APOBEC1,并将其连接至突变的Cas9(D10A)的N端,通过16aa的XTEN柔性短肽连接形成融合蛋白,其核苷酸序列如SEQ ID NO.:1所示:
其蛋白序列如SEQ ID NO.:8所示:
将该序列构建至水稻pCambia1300载体(购自澳大利亚Camiba研究所),并由植物强启动子UBI启动表达。同时,将guide RNA的表达框也构建至该载体,生成载体pCSGAPO1(图2A)。将该载体应用于下列实施例。
实施例1在水稻愈伤组织中实现定点碱基替换
首先尝试应用该发明在水稻愈伤中实现碱基替换。我们选择水稻两个重要的基因NRT1.1B和SLR1。NRT1.1B可以控制水稻营养元素氮的吸收,增加产量;SLR1控制赤霉素的合成,影响水稻植株高度。利用本发明,我们设计相应的靶点,通过高通量测序,成功地在水稻愈伤中检测到定点碱基替换,其效率可达11.5%,远远高于以前的在植物原生质体中的运用同源重组的瞬时实验。具体操作流程如下:
1.1载体构建与遗传转化
水稻NRT1.1B基因待编辑的氨基酸为Thr327,而SLR1基因待编辑的氨基酸为Ser97。针对这两个位点,我们分别设计了靶点。如图3A和3B所示,待替换的胞嘧啶C都处于靶点的“脱氨基窗口”内。因此,我们分别将这两个靶点的guide RNA表达框(SEQ ID NO.:2和SEQ ID NO.:3)构建至载体pCSGAPO1(获自中国科学院上海生命科学研究院),获得碱基替换载体pCSGAPO1-NRT和pCSGAPO1-SLR1。
将上述两个载体转化农杆菌EAH105,然后转化水稻中花11的愈伤组织。经过4周筛选培养后,挑选100-200颗抗性愈伤颗粒至新的培养基上继续筛选2周。最后,挑出全部的抗性愈伤颗粒,混合后提取基因组DNA,进行碱基替换检测。
1.2碱基替换效率检测
在获得总的基因组DNA后,分别设计引物(表1),通过PCR扩增NRT1.1B和SLR1,然后将PCR产物进行高通量测序分析。如图3C所示,在NRT1.1B的靶点位置上,其第7位的胞嘧啶被替换成了T或者G,其效率分别为1.4%和1.6%;在SLR1的靶点位置上,其guide RNA的第4位置产生了C→T碱基替换,其效率为2.9%,而第6位置的胞嘧啶产生了C→T和C→G的碱基替换,其效率分别为11.5%和3.9%。同时,我们也检测到了一定比率的Indel,其由单链切刻活性的Cas9(D10A)导致。由于NRT1.1B所设计的guide RNA的靶向效率低于SLR1,其碱基替换效率也比SLR1低。这些实验结果表明,该发明可有效的应用于植物基因组的定点碱基替换。
表1.扩增靶点所用的引物
实施例2获得定点碱基替换的水稻
将实施例1中的两个碱基替换载体pCSGAPO1-NRT和pCSGAPO1-SLR1转化农杆菌EHA105,通过常规的水稻遗传转化,分别获得了37株编辑NRT1.1B的转基因植株和45株编辑SLR1的转基因植株(表2)。
表2.水稻转基因植株的碱基替换效率统计
针对37株编辑NRT1.1B的转基因植株,我们采用Sanger测序进行DNA测序分析。测序结果表明,其中4株在靶点位置产生了Indel突变,有1株植株(NRT#15)在其guide RNA的第7位置的胞嘧啶产生了C→G碱基替换。将该转基因植株的PCR扩增产物克隆至T载体,挑选17个单克隆进行测序,结果表明该植株中的C→G碱基替换效率为23.5%(4/17)。
对于SLR1的编辑,由于C→T碱基替换可以产生StyI的酶切位点,因此可以通过酶切多态性检测该位点的碱基替换。如图5A所示,对于C→T碱基替换的PCR产物(404bp),StyI可以将其酶切成90bp+314bp两个条带。实验结果表明,45株编辑SLR1的转基因植株中,有6株在其guide RNA的第6位置产生了C→T的碱基替换。对这45株植株进行Sanger测序分析,进一步验证了该实验结果。我们将转基因植株SLR#07的PCR扩增产物克隆至T载体,挑选10个单克隆进行测序,结果表明该植株中的C→T碱基替换效率为60%(6/10)。该结果与NRT1.1B的编辑结果类似,即SLR1靶点上的胞嘧啶也未被彻底的替换,也就是说转基因植株NRT#15和SLR#07中的碱基替换位点是嵌合的或者杂合的。但是,这些转基因植株中,靶位点上高达23.5%-60%的碱基替换率使其足够遗传至下一代。
根据以前的研究,SLR1基因在该位点的碱基替换为显性突变,可以影响植株生长高度。通过对SLR1的T0代植株的观察发现,SLR1敲除植株的植株茎叶纤细,而发生碱基替换的转基因植株的植株高度明显矮于野生型的T0代植株,呈现又矮又壮的表型(图5D)。这些结果进一步证明了,应用本发明,在该位点成功进行了定点碱基替换,并且替换效率很高。
在该实施例中,我们只采取了常规的遗传转化工作,便获得了定点碱基替换的植株,说明本发明在植物研究和育种中广泛推广和应用的巨大潜力。
并且,经过大量实验发现,动物启动子不能对本发明的植物(如水稻)进行定点碱基替换,只有本发明的植物启动子(尤其是水稻特异性的强启动子)才可对本发明的植物(如水稻)进行定点碱基替换,并且碱基替换效率可达11.5%,并且在一个突变植株内,有高达60%的细胞发生了碱基替换。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
参考文献
1 Carroll,D.Genome engineering with zinc-finger nucleases.Genetics 188,773-782,doi:10.1534/genetics.111.131433(2011).
2 Cong,L.et al.Multiplex genome engineering using CRISPR/Cas systems.Science 339,819-823,doi:10.1126/science.1231143(2013).
3 Mahfouz,M.M.et al.De novo-engineered transcription activator-like effector(TALE)hybrid nuclease with novel DNA binding specificity creates double-strand breaks.Proceedings of the National Academy of Sciences of the United States of America 108,2623-2628,doi:10.1073/pnas.1019533108(2011).
4 Hsu,P.D.,Lander,E.S.&Zhang,F.Development and Applications of CRISPR-Cas9 for Genome Engineering.Cell 157,1262-1278,doi:10.1016/j.cell.2014.05.010(2014).
5 Mao,Y.et al.Application of the CRISPR-Cas System for Efficient Genome Engineering in Plants.Molecular plant,doi:10.1093/mp/sst121(2013).
6 Li,J.F.et al.Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9.Nature biotechnology 31,688-691,doi:10.1038/nbt.2654(2013).
7 Shukla,V.K.et al.Precise genome modification in the crop species Zea mays using zinc-finger nucleases.Nature 459,437-441,doi:10.1038/nature07992(2009).
8 Sun,Y.et al.Engineering Herbicide-Resistant Rice Plants through CRISPR/Cas9-Mediated Homologous Recombination of Acetolactate Synthase.Molecular plant 9,628-631,doi:10.1016/j.molp.2016.01.001(2016).
9 Huang,X.et al.Genomic architecture of heterosis for yield traits in rice.Nature 537,629-633,doi:10.1038/nature19760(2016).
10 Huang,X.et al.Genome-wide association studies of 14 agronomic traits in rice landraces.Nature genetics 42,961-967,doi:10.1038/ng.695(2010).
- 还没有人留言评论。精彩留言会获得点赞!