原毒素‑II变体及使用方法与流程
相关申请的交叉引用本申请根据35u.s.c.§119(e)要求2015年3月3日提交的美国临时申请62/127,339的优先权,其公开内容以引用方式全文并入本文。本发明涉及原毒素-ii变体、编码该变体的合成多核苷酸、以及制备和使用该变体和该合成多核苷酸的方法。
背景技术:
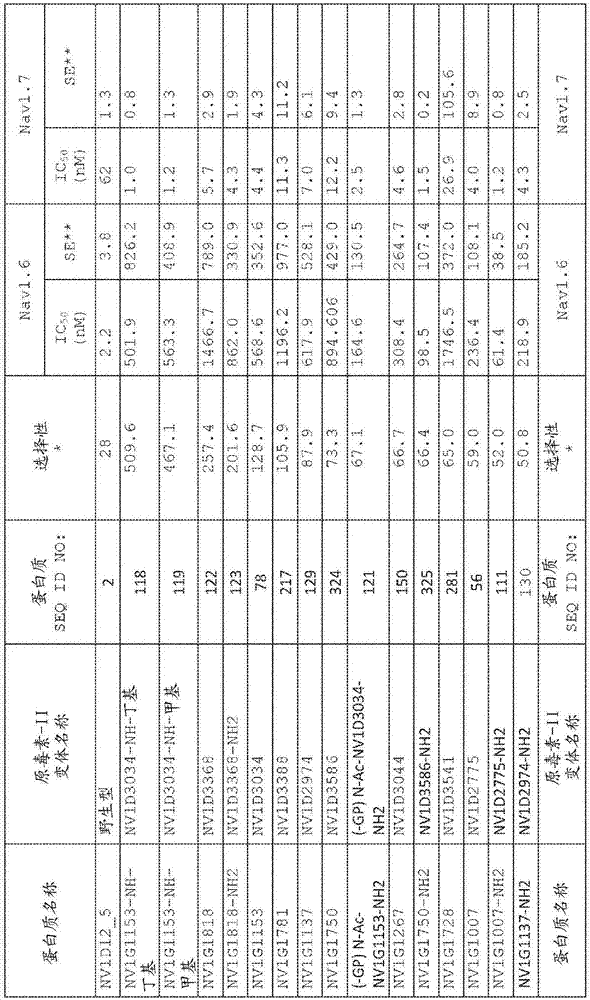
:电压门控钠通道(vgsc)存在于所有可兴奋细胞中,包括心肌细胞、骨骼肌细胞和中枢神经元和周围神经元。在神经元细胞中,钠通道负责放大阈下去极化并引起动作电位的快速上升。同样,钠通道对于神经系统中电信号的产生和传播至关重要。人们认为钠通道功能异常会引起多种医学疾病(hübner和jentsch,hummolgenet11:2435-45,2002),包括癫痫(yogeeswari等人,currdrugtargets5:589-602,2004)、心律失常(tfelt-hansen等人,jcardiovascelectrophysiol21:107-15,2010)、肌强直(cannon和bean,jclininvest120:80-3,2010)和疼痛(cregg等人,jphysiol588:1897-904,2010)。钠通道通常是各种亚基的复合物,基本的亚基是成孔α-亚基,单独的成孔α-亚基即足以发挥功能。人体内存在电压门控钠通道α亚基家族的九个已知成员nav1.1–nav1.9。nav1.x亚家族在药理学上可再分为两组,即河豚毒素(ttx)-敏感型和ttx-不敏感型。nav1.7(也称为pn1或hne)由scn9a基因编码,属于ttx-敏感型,主要在外周交感神经元和感觉神经元中表达。nav1.7积聚在神经纤维末端,使小规模阈下去极化放大,并作为调节兴奋性的阈值通道。nav1.7功能与各种疼痛状态有关,包括急性、炎性和/或神经性疼痛。在人体内,已发现nav1.7的功能获得性突变与原发性红斑肢痛症(pe)(该疾病的特征在于肢体灼痛和炎症)(yang等人,jmedgenet41:171-4,2004)和阵发性剧痛症(pepd)(fertleman等人,neuron52:767-74,2006)相关。与此观察一致的是,非选择性钠通道阻滞剂利多卡因、美西律和卡马西平可缓解这些疼痛疾病的症状(legroux-crespel等人,anndermatolvenereol130:429-33,2003;fertleman等人,neuron52:767-74,2006)。在人体内,nav1.7的功能丧失性突变导致罕见的常染色体隐性疾病先天性无痛症(cip),其特征在于完全缺失对疼痛刺激的感知或对疼痛刺激不敏感(cox等人,nature444:894-8,2006;goldberg等人,clingenet71:311-9,2007;ahmad等人,hummolgenet16:2114-21,2007)。scn9a的编码区中的单核苷酸多态性与伤害感受器兴奋性和疼痛敏感性增加相关。例如,导致人nav1.7中的r1150w置换的多态性rs6746030与骨关节炎疼痛、腰椎间盘切除术疼痛、幻肢疼痛和胰腺炎疼痛相关(reimann等人,procnatlacadsciusa107:5148-53,2010)。表达r1150w突变型nav1.7的drg神经元响应于去极化而表现出放电频率增加(estacion等人,annneurol66:862-6,2009)。伤残形式的纤维肌痛与scn9a钠通道多态性rs6754031相关,表明一些严重纤维肌痛患者可具有背根神经节钠通道病(vargas-alarcon等人,bmcmusculoskeletdisord13:23,2012)。在小鼠中,伤害感受性神经元的scn9a基因缺失导致机械性疼痛阈值和热疼痛阈值降低,以及炎性疼痛响应的减少或消失(nassar等人,procnatlacadsciusa101:12706-11,2004)。去除所有感觉神经元中的scn9a消除了机械性疼痛、炎性疼痛以及对热的反射缩回响应。去除感觉神经元和交感神经元中的scn9a消除了机械性疼痛、热疼痛和神经性疼痛,并且重演了在具有nav1.7功能丧失性突变的人群中发现的无痛表型(minett等人,natcommun3:791,2012)。因此,nav1.7抑制剂或阻滞剂可用于治疗与各种疾病相关的一系列疼痛。已知蜘蛛毒包含许多钠通道阻滞肽,包括虎纹捕鸟蛛毒素-iv(hwtx-iv)(peng等人,jbiolchem277:47564-71,2002)、原毒素-i、原毒素-ii(middleton等人,biochemistry41:14734-47,2002)和智利火鸟蛛毒素–iii(phrixotoxin–iii)(bosmans等人,molpharmacol69:419-29,2006)。需要鉴定另外的nav1.7阻滞剂以治疗一系列疼痛适应症。具体地,需要对其它电压门控钠通道同种型上的nav1.7具有选择性的新型nav1.7阻滞剂。技术实现要素:本发明的一个实施方案为分离的原毒素-ii变体,其中该原毒素-ii变体抑制人nav1.7的活性,其ic50值为约1×10-7m或更小、约1×10-8m或更小、约1×10-9m或更小、约1×10-10m或更小、约1×10-11m或更小、或约1×10-12m或更小,其中ic50值是在稳定表达人nav1.7的hek293细胞中存在25×10-6m3-藜芦酰藜芦瑟文的情况下使用荧光共振能量转移(fret)使用tetra膜去极化测定法而测量,其中原毒素-ii变体具有w7q和/或w30l置换。本发明的另一个实施方案为分离的原毒素-ii变体,其包含seqidno:30、40、44、52、56、56、59、65、78、109、110、111、114、117、118、119、120、121、122、123、124、125、126、127、128、129、130、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、150、151、152、153、154、155、156、157、158、159、162、165、166、167、168、169、170、171、172、173、174、175、177、178、179、180、182、183、184、185、186、189、190、193、195、197、199、206、207、208、209、210、211、212、213、214、215、216、217、218、224、226、227、231、232、243、244、245、247、249、252、255、258、261、263、264、265、266、269、270、271、272、273、274、275、276、277、278、279、280、281、282、283、284、285、286、287、288、289、290、291、292、293、294、295、296、297、298、299、300、301、302、303、304、305、306、307、308、309、310、311、312、313、314、315、316、317、318、319、320、321、322、323、324、325、326、332、334、335、336、337、339、340、341、342、346、351、358、359、364、366、367、368、369、370、371、408、409、410、411、412、413、414、415、416、417、418、419、420、421、422、423、424、425、426、427、428、429、430或431的氨基酸序列。本发明的另一个实施方案为分离的原毒素-ii变体,其包含与seqidno:422的氨基酸序列(gpycqkwmqtcdserkccegmvcrlwckkkll-cooh)具有90%、91%、92%、93%、94%、95%、96%、97%、98%或99%同一性的氨基酸序列;其中当残基编号根据seqidno:1而定时,所述氨基酸序列在位置7处具有q并且在位置30处具有l;并且多肽抑制人nav1.7的活性,其ic50值为约30×10-9m或更小,其中ic50值是在稳定表达人nav1.7的hek293细胞中存在25×10-6m3-藜芦酰藜芦瑟文的情况下使用荧光共振能量转移(fret)使用tetra膜去极化测定法而测量。本发明的另一个实施方案为分离的融合蛋白,其包含缀合至半衰期延长部分的本发明的原毒素-ii变体。本发明的另一个实施方案为编码本发明的原毒素-ii变体的分离的多核苷酸。本发明的另一个实施方案为包含本发明的分离的多核苷酸的载体。本发明的另一个实施方案为包含本发明的载体的宿主细胞。本发明的另一个实施方案为制备本发明的分离的原毒素-ii变体的方法,该方法包括培养本发明的宿主细胞以及回收由宿主细胞产生的原毒素-ii变体。本发明的另一个实施方案为药物组合物,其包含本发明的分离的原毒素-ii变体和融合蛋白以及药学上可接受的赋形剂。本发明的另一个实施方案为治疗受治疗者的nav1.7介导的疼痛的方法,该方法包括向对其有需要的受治疗者施用有效量的本发明的原毒素-ii变体或融合蛋白以治疗疼痛。附图说明图1示出抑制nav1.7的原毒素-ii变体的属氨基酸序列,在fliprtetra测定法中nav1.7的ic50值为30nm或更小。残基编号根据seqidno:1的野生型原毒素-ii而定。属seqidno:403。图2示出在qpatch测定法中nav1.7和nav1.6抑制的nav1.7的ic50值,和由qpatch测定法中获得的ic50(nav1.6)/ic50(nav1.7)比率计算的每个变体的选择性。se:标准误差。图3示出抑制nav1.7的原毒素-ii变体的序列和属序列,在fliprtetra测定法中nav1.7的ic50值为30nm或更小,并且为对nav1.6的选择性的超过30倍。每个变体的选择性由qpatch测定法中获得的ic50(nav1.6)/ic509av1.7)比率计算。残基编号根据seqidno:1的野生型原毒素-ii而定。图4a示出nv1d3034(nv1d3034-oh)(seqidno:78)对cfa-诱导的热痛觉过敏的功效,通过在hargreaves测试中测定cfa注射之前(cfa前)和之后(0)以及肽施用1天后(1)的退缩反应潜伏期来评估该功效。采用双因素方差分析,然后进行bonferroni多重比较,相较于pbs组***p<0.001。图4b示出nv1d3034(nv1d3034-oh)(seqidno:78)在cfa-诱导的热痛觉过敏中的功效,该功效表示为肽施用后第1天每个剂量的mpe(最大可能效应)百分比(mpe%)。采用单因素方差分析,然后进行bonferroni多重比较,相较于pbs组*p<0.05。图5a示出nv1d3368(nv1d3368-oh)(seqidno:198)对cfa-诱导的热痛觉过敏的功效,通过在hargreaves测试中测定cfa注射之前(cfa前)和之后(0)以及肽施用1天后(1)的退缩反应潜伏期来评估该功效。采用双因素方差分析,然后进行bonferroni多重比较,相较于pbs组**p<0.01并且****p<0.0001。图5b示出nv1d3368(nv1d3368-oh)(seqidno:198)在cfa-诱导的热痛觉过敏中的功效,该功效表示为肽施用后第1天每个剂量的mpe百分比(mpe%)。采用单因素方差分析,然后进行bonferroni多重比较,相较于pbs组*p<0.05并且**p<0.01。图6a示出nv1d2775-oh(seqidno:56)对cfa-诱导的热痛觉过敏的功效,通过在hargreaves测试中测定cfa注射之前(cfa前)和之后(0)以及肽施用1天后(1)的退缩反应潜伏期来评估该功效。采用双因素方差分析,然后进行bonferroni多重比较,相较于pbs组****p<0.0001。图6b示出nv1d2775-oh(seqidno:56)在cfa-诱导的热痛觉过敏中的功效,该功效表示为肽施用后第1天每个剂量的mpe百分比(mpe%)。采用单因素方差分析,然后进行bonferroni多重比较,相较于pbs组***p<0.001并且****p<0.0001。图6c示出nv1d2775-oh(seqidno:56)对cfa-诱导的触觉异常痛敏的功效。cfa注射之前(cfa前)和之后(0)以及肽施用1天后(1)的后足的触觉阈值。采用双因素方差分析,然后进行bonferroni多重比较,相较于pbs组****p<0.0001。图6d示出nv1d2775-oh(seqidno:56)对cfa-诱导的触觉异常痛敏的功效,该功效表示为肽施用后第1天的mpe百分比(mpe%)。采用单因素方差分析,然后进行bonferroni多重比较,相较于pbs组***p<0.001。图7a示出了cfa模型中nv1d2775-oh介导的热痛觉过敏逆转的时间进程,通过在hargreaves测试中测量cfa注射之前和之后以及肽施用后的各个时间点的退缩反应潜伏期来评估该逆转。采用双因素方差分析,然后进行bonferroni多重比较,相较于pbs组**p<0.01。阴影区域表示化合物递送周期(0-24hr)。图7b示出了cfa模型中nv1d2775-oh介导的触觉异常痛敏逆转的时间进程,通过测量cfa注射之前和之后以及肽施用后的各个时间点的触觉阈值来评估该逆转。采用双因素方差分析,然后进行bonferroni多重比较,相较于pbs组**p<0.01。阴影区域表示化合物递送周期(0-24hr)。图8示出在热板测试中nv1d2775-oh产生了显著的痛觉丧失。在泵植入之前和之后、在50℃和55℃下评估热退缩潜伏期。在对照pbs组中泵植入对潜伏期无影响。在泵植入一天后,与pbs组相比,由nv1d2775-oh治疗的小鼠表现出潜伏期延长。采用单因素方差分析,然后进行bonferroni多重比较,相较于pbs组*p<0.05并且****p<0.0001。图9示出nv1d2775-oh预处理保护了动物免受角叉菜胶诱导的热痛觉过敏。在泵植入之前和泵植入之后的第1天、足底注射角叉菜胶之前测量退缩潜伏期。在注射角叉菜胶之后2小时、3小时和4小时再次测量潜伏期。图10示出野生型原毒素-ii的nmr结构的表面示意图。左侧所示的疏水面包括残基w5、m6、w7、l23和w24。选择性面在右侧示出,并包括残基s11、e12、k14、e17、g18、l29和w30。残基编号根据seqidno:1而定。图11a示出原毒素-ii变体63955918(seqidno:422)在甩尾测试中单次鞘内(it)施用之后的功效。在肽给药后的指定时间处测量对热刺激的缩尾潜伏期。图11b示出原毒素-ii变体63955918(seqidno:422)在甩尾测试中的功效,该功效表示为单次鞘内(it)施用之后前120min内的曲线下面积百分比(auc%)。采用单因素方差分析,然后进行bonferroni多重比较,相较于pbs组***p<0.001并且****p<0.0001。图11c示出原毒素-ii变体63955918(seqidno:422)在热板测试(52.5℃)中单次鞘内(it)施用之后的功效。在肽给药后的指定时间处测量热板上的伤害性反应的潜伏期。图11d示出原毒素-ii变体63955918(seqidno:422)在热板试验中的功效,以在单次鞘内(it)给药后的前120分钟内曲线下面积百分比(auc%)表示。采用单因素方差分析,然后进行bonferroni多重比较,相较于pbs组***p<0.001并且****p<0.0001。图11e示出原毒素-ii变体63955918(seqidno:422)在福尔马林测试中的功效。将福尔马林注入大鼠后足诱导双相退缩行为。通过自动装置测量第i阶段(福尔马林后0-10min)和第ii阶段(福尔马林后11-60min)的总退缩数。由于小组规模,没有对e)进行统计。图12a示出nv1d2775-oh在甩尾测试中单次鞘内(it)施用之后的功效。对热刺激的缩尾潜伏期在肽施用后指定的时间进行测量。图12b示出nv1d2775-oh在甩尾测试中的功效,该功效表示为单次鞘内(it)施用之后前120min内的曲线下面积百分比(auc%)。采用单因素方差分析,然后进行bonferroni多重比较,相较于pbs组*p<0.05并且**p<0.01。图12c示出nv1d2775-oh在热板测试(52.5℃)中单次鞘内(it)施用之后的功效。热板上感受伤害响应的潜伏期在肽施用后指定的时间进行测量。图12d示出nv1d2775-oh在热板测试中的功效,该功效表示为单次鞘内(it)施用之后前120min内的曲线下面积百分比(auc%)。采用单因素方差分析,然后进行bonferroni多重比较,相较于pbs组**p<0.01并且****p<0.0001。图12e示出nv1d2775-oh在福尔马林试验中的功效。将福尔马林注入大鼠后足诱导双相退缩行为。通过自动装置测量第i阶段(福尔马林后0-10min)和第ii阶段(福尔马林后11-60min)的总退缩数。采用单因素方差分析,然后进行bonferroni多重比较,在第i阶段相较于pbs组**p<0.01,在第ii阶段相较于pbs组*p<0.05。图13a示出nv1d3034-oh在甩尾测试中单次鞘内(it)施用之后的功效。对热刺激的缩尾潜伏期在肽施用后指定的时间进行测量。图13b示出nv1d3034-oh在甩尾测试中的功效,该功效表示为单次鞘内(it)施用之后前120min内的曲线下面积百分比(auc%)。在t检验中,相较于pbs组***p<0.005。图13c示出nv1d3034-oh在热板测试(52.5℃)中单次鞘内(it)施用之后的功效。热板上感受伤害响应的潜伏期在肽施用后指定的时间进行测量。图13d示出nv1d3034-oh在热板测试中的功效,该功效表示为单次鞘内(it)施用之后前120min内的曲线下面积百分比(auc%)。t检验中,相较于pbs组**p<0.01。图13e示出nv1d3034-oh在福尔马林测试中的功效。将福尔马林注入大鼠后足诱导双相退缩行为。通过自动装置测量第i阶段(福尔马林后0-10min)和第ii阶段(福尔马林后11-60min)的总退缩数。t检验中,在第i阶段相较于pbs组*p<0.05,在第ii阶段相较于pbs组**p<0.01。具体实施方式本说明书中所引用的所有出版物,包括但不限于专利和专利申请均以引用方式并入本文,如同在本文中完整给出。说明书和权利要求书中所用的单数形式“一个”、“一种”和“所述”包括复数指代,除非上下文清楚表明并非如此。除非另有定义,否则本文使用的所有技术和科学术语的含义与本发明所属领域的普通技术人员通常所理解的含义相同。虽然本文描述了示例性的组合物和方法,但类似或等同于本文所述的组合物和方法的任何组合物和方法都可用于本发明的实践或测试。术语“多肽”是指包含至少两个由肽键连接形成多肽的氨基酸残基的分子。少于50个氨基酸的小多肽可以称作“肽”。多肽也可被称作“蛋白质”。术语“多核苷酸”是指包含由糖-磷酸主链或其它等同的共价化学方式所共价连接的核苷酸链的分子。双链dna和单链dna以及双链rna和单链rna是多核苷酸的典型示例。术语“互补序列”是指反向平行于第一分离的多核苷酸序列并包含与第一多核苷酸序列中的核苷酸互补的核苷酸的第二分离的多核苷酸序列。术语“载体”是指能够在生物系统内复制或可以在此类系统间移动的非天然多核苷酸。载体多核苷酸通常包含编码目标蛋白质的cdna以及另外的元件,诸如复制起点、聚腺苷酸化信号或选择标记,其功能是促进这些多核苷酸在生物系统中的复制或保持。此类生物系统的示例可包括细胞、病毒、动物、植物和用能够复制载体的生物组分重构的生物系统。包含载体的多核苷酸可为dna或rna分子或这些分子的杂合分子。术语“表达载体”是指可用于在生物系统或重构生物系统中指导由存在于表达载体中的多核苷酸序列所编码的多肽的翻译的载体。如本文所用,术语“变体”是指不同于seqidno:1的野生型原毒素-ii多肽的多肽,或不同于编码具有seqidno:107的序列的野生型原毒素-ii的多核苷酸的多核苷酸,不同之处在于一个或多个修饰,例如核苷酸或氨基酸的置换、插入或缺失。在整个说明书中,原毒素-ii变体中被置换的残基的编号对应于其在seqidno:1的野生型原毒素-ii的位置。例如,说明书中的“y1a”是指对应于seqidno:1的野生型原毒素-ii中位置1的残基位置处的酪氨酸被丙氨酸置换。“互补dna”或“cdna”是指熟知的合成多核苷酸,其序列元件的排列与天然的成熟mrna种类中存在的序列元件排列相同,该基因组dna中存在的连续的外显子,而居间内含子被移除。编码起始甲硫氨酸的密码子可存在或可不存在于cdna中。例如,可通过逆转录或合成基因组件合成cdna。如本文所用,术语“合成”或“非天然”是指非天然存在的多核苷酸或多肽分子。如本文所用,“nav1.7”(也被称为hne或pn1)或“hnav1.7”是指熟知的人钠通道蛋白9型亚基α,其具有genbank登录号np_002968.1和seqidno:79中所示的序列。如本文所用,术语“野生型原毒素-ii”或“野生型protx-ii”是指狼蛛(智利绿丝绒狼蛛(thrixopelmapruriens)(秘鲁绿丝绒狼蛛))毒素肽,其具有氨基酸序列ycqkwmwtcdserkccegmvcrlwckkklw-cooh(seqidno:1),如middleton等人在biochemistry41(50):14734-47,2002中所述。如本文所用,术语“重组原毒素-ii”或“重组protx-ii”是指表达原毒素-ii融合蛋白并随后对其进行切割而获得的重组原毒素-ii,其具有seqidno:2中所示的gpycqkwmwtcdserkccegmvcrlwckkklw-oh序列。与野生型原毒素-ii相比,重组原毒素-ii包含两个氨基酸n-末端延伸(残基g和p)。如本文所用,“阻滞人nav1.7的活性”或“抑制人nav1.7的活性”是指本发明的原毒素-ii变体减少藜芦定(3-藜芦酰藜芦瑟文)诱导的膜去极化的能力,在使用荧光共振能量转移(fret)的tetra膜去极化分析中ic50值为约1×10-7m或更小,其中藜芦定诱导的去极化以fret信号减少测量,该测量使用disbac2(3)([双-(1,3-二乙基硫代巴比妥酸)三次甲基氧杂菁])作为受体,并且pts18(8-十八烷基氧芘-1,3,6-三磺酸三钠)作为供体,通过在390nm-420nm下激发供体,并测量稳定表达人nav1.7的细胞系中515nm-575nm下的fret来进行。如本文所用,“tetra膜去极化测定法”是实施例3所述的分析法。如本文所用,术语“基本上相同”是指被比较的两种原毒素-ii变体氨基酸序列是相同的或具有“非显著性差异”。非显著性差异是原毒素-ii变体氨基酸序列中1、2、3、4、5、6或7个氨基酸发生置换,而这种置换不对肽的性质造成不利影响。与本文所公开的原毒素-ii变体基本上相同的氨基酸序列在本申请范围内。在一些实施方案中,序列同一性可为约80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或更高。百分比同一性可以例如通过使用vectorntiv.9.0.0(invitrogen,carslbad,ca)的alignx模块的默认设置的逐对比对而确定。本发明的蛋白质序列可以用作查询序列以针对公共或专利数据库进行检索,例如以鉴定相关序列。用于进行此类检索的示例性程序是使用默认设置的xblast或blastp程序(http_//www_ncbi_nlm/nih_gov)或genomequesttm(genomequest,westborough,ma)软件包。本文使用如表1中所示的常规单字母和三字母氨基酸代码。表1.氨基酸三字母代码单字母代码丙氨酸alaa精氨酸argr天冬酰胺asnn天冬氨酸aspd半胱氨酸cysc谷氨酸glue谷氨酰胺glnq甘氨酸glyg组氨酸hish异亮氨酸ilei亮氨酸leul赖氨酸lysk甲硫氨酸metm苯丙氨酸phef脯氨酸prop丝氨酸sers苏氨酸thrt色氨酸trpw酪氨酸tyry缬氨酸valv本发明提供了抑制人nav1.7的活性的分离的原毒素-ii(protx-ii)变体多肽、编码该多肽的多核苷酸、载体、宿主细胞以及本发明的多核苷酸和多肽的使用方法。本发明的多肽抑制nav1.7活化引起的去极化,因此可用于治疗与疼痛相关联的各种病症以及与感觉神经元或交感神经元功能异常相关联的病症。本发明的变体是nav1.7的有效抑制剂。本发明至少部分基于如下发现:置换原毒素-ii中的某些残基增强选择性、提高合成收率和/或均质性,而不对所生成的原毒素-ii变体的效力造成不利影响,特别是w7和m19的置换,以及加上残基y1和s11的置换,再加上残基e12、r22的置换(残基编号根据seqidno:1而定)。例如,位置w7和w30的置换增强了原毒素-ii变体折叠并提高了产率。位置s11、e12、k14、e17、g18、l29和w30的置换改善了所得的原毒素-ii变体对nav1.7的选择性。本发明的一个实施方案为分离的原毒素-ii变体,其中该原毒素-ii变体抑制人nav1.7的活性,其ic50值为约1×10-7m或更小、约1×10-8m或更小、约1×10-9m或更小、约1×10-10m或更小、约1×10-11m或更小、或约1×10-12m或更小,其中ic50值是在稳定表达人nav1.7的hek293细胞中存在25×10-6m3-藜芦酰藜芦瑟文的情况下使用荧光共振能量转移(fret)使用tetra膜去极化测定法而测量。本发明的一个另实施方案为分离的原毒素-ii变体,其中该原毒素-ii变体抑制人nav1.7的活性,其ic50值为约1×10-7m或更小、约1×10-8m或更小、约1×10-9m或更小、约1×10-10m或更小、约1×10-11m或更小、或约1×10-12m或更小,其中ic50值是在稳定表达人nav1.7的hek293细胞中存在25×10-6m3-藜芦酰藜芦瑟文的情况下使用荧光共振能量转移(fret)使用tetra膜去极化测定法而测量,其中原毒素-ii变体具有w7q和w30l置换。本发明的另一个实施方案为分离的原毒素-ii变体,其包含序列x1x2x3cx4x5wx6qx7cx8x9x10x11x12ccx13x14fx15cx16lwcx17kkll(seqidno:432),其中x1为g、p、a或缺失;x2为p、a或缺失;x3为s、q、a、r或y;x4为q、r、k、a或s;x5为k、s、q或r;x6为m或f;x7为t、s、r、k或q;x8为d或t;x9为s、a或r;x10为e、r、n、k、t或q;x11为r或k;x12为k、q、s或a;x13为e、q或d;x14为g或q;x15为v或s;x16为r或t;并且x17为k或r;该分离的原毒素-ii变体任选地具有n-末端延伸或c-末端延伸,其中多肽抑制人nav1.7的活性,其ic50值为约1×10-7m或更小,其中ic50值是在稳定表达人nav1.7的hek293细胞中存在25×10-6m3-藜芦酰藜芦瑟文的情况下使用荧光共振能量转移(fret)使用tetra膜去极化测定法而测量。原毒素-ii位置w7q和w30l处的置换改善所得原毒素-ii变体的再折叠和收率。在一些实施方案中,n-末端延伸包含seqidno:372、373、374、375、376、377、378、379、380、381、382、383、384或385的氨基酸序列。在一些实施方案中,c-末端延伸包含seqidno:374、386、387、388、389、390、391、392、393、394、395、396或397的氨基酸序列。在一些实施方案中,n-末端延伸和/或c-末端延伸通过接头缀合至原毒素-ii变体。在一些实施方案中,接头包含seqidno:383、392、398、399、400、401或402的氨基酸序列。在一些实施方案中,n-末端延伸由seqidno:372、373、374、375、376、377、378、379、380、381、382、383、384或385的氨基酸序列构成。在一些实施方案中,c-末端延伸由seqidno:374、386、387、388、389、390、391、392、393、394、395、396或397的氨基酸序列构成。在一些实施方案中,接头由seqidno:383、392、398、399、400、401或402的氨基酸序列构成。本发明的另一个实施方案为分离的原毒素-ii变体,其包含序列x1x2x3cx4x5wx6qx7cx8x9x10x11x12ccx13x14fx15cx16lwcx17kklw(seqidno:403),其中x1为g、p、a或缺失;x2为p、a或缺失;x3为s、q、a、r或y;x4为q、r、k、a或s;x5为k、s、q或r;x6为m或f;x7为t、s、r、k或q;x8为d或t;x9为s、a或r;x10为e、r、n、k、t或q;x11为r或k;x12为k、q、s或a;x13为e、q或d;x14为g或q;x15为v或s;x16为r或t;并且x17为k或r;该分离的原毒素-ii变体任选地具有n-末端延伸或c-末端延伸,其中多肽抑制人nav1.7的活性,其ic50值为约1×10-7m或更小,其中ic50值是在稳定表达人nav1.7的hek293细胞中存在25×10-6m3-藜芦酰藜芦瑟文的情况下使用荧光共振能量转移(fret)使用tetra膜去极化测定法而测量。本发明的原毒素-ii变体是有效的nav1.7抑制剂。在藜芦定诱导的去极化抑制测定法中,重组原毒素-ii(seqidno:2)对于人nav1.7具有约4×10-9m的ic50值,该测定法使用tetra仪器(moleculardevices)通过实施例3中详述的实验测量稳定表达nav1.7的细胞中fret(荧光共振能量转移)的减少。当上述测定法和实验3中的ic50值为约30×10-9m或更小,即在重组原毒素-ii的ic50值的10倍以内时,原毒素-ii变体是“有效的”nav1.7抑制剂。为清楚起见,30×10-9m的ic50相当于3.0×10-8m的ic50。本发明的原毒素-ii变体多肽可以通过化学合成制备,诸如在自动肽合成仪上进行的固相肽合成。另选地,本发明的多肽可通过使用无细胞表达系统(诸如基于网织红细胞溶解物的表达系统)或通过重组表达系统从编码多肽的多核苷酸获得。本领域的技术人员将认识到其它用于获得本发明多肽的技术。在示例性方法中,通过如下方式生成本发明的原毒素-ii变体:将这些变体表达为人血清白蛋白(hsa)融合蛋白,该融合蛋白利用富含甘氨酸的接头诸如(ggggs)4(seqidno:80)或(ggggs)6(seqidno:81),该富含甘氨酸的接头偶联到可被蛋白酶切割的接头诸如hrv3c蛋白酶(人鼻病毒重组型143c蛋白酶)的识别序列levlfqgp(hrv3c接头)(seqidno:82),并且用hrv3c蛋白酶切割所表达的融合蛋白,以释放重组原毒素-ii变体肽。六聚组氨酸(seqidno:108)或其它标签可用于促进采用熟知的方法进行纯化。本发明的原毒素-ii变体可使用本文所述的方法纯化。在一个示例性方法中,本发明的原毒素-ii变体表达为hsa融合蛋白,并被hrv3c蛋白酶切割,可使用本文所述的固相萃取(spe)法将该变体纯化。通常在核酸水平实现任选地具有n-末端延伸和/或c-末端延伸的原毒素-ii变体的生成和原毒素-ii变体融合蛋白的生成。可使用根据美国专利6,521,427和6,670,127所述方法的化学基因合成法利用简并寡核苷酸生成所需的变体来合成多核苷酸,或通过标准pcr克隆和诱变来合成多核苷酸。变体库可通过标准克隆技术生成,以将编码原毒素-ii变体的多核苷酸克隆到载体中用于表达。与seqidno:1的野生型原毒素-ii相比,原毒素-ii变体可包含例如由克隆和/或表达方案产生的另外的n-末端和/或c-末端氨基酸。例如,将变体表达为hsa-接头-hrv3c可切割肽-原毒素-ii变体融合蛋白,然后从hsa切割可使每个原毒素-ii变体的n-末端与另外的两个残基诸如g和p结合。使用本文所述的方法测试本发明的原毒素-ii变体抑制人nav1.7的能力。示例性测定法为藜芦定诱导的去极化抑制测定法,该测定法测量稳定表达nav1.7的细胞中fret(荧光共振能量转移)的减少。另一种示例性测定法采用电生理学记录以使用熟知的膜片钳技术和本文所述的技术测量nav1.7介导的电流的变化。本发明的另一个实施方案为分离的原毒素-ii变体,其包含seqidno:3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35,36,37,38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、109、110、111、112、113、114、115、116、117、118、119、121、122、123、124、125、126、127、128、129、130、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、150、151、152、153、154、155、156、157、158、159、160、161、162、163、164、165、166、167、168、169、170、171、172、173、174、175、176、177、178、179、180、181、182、183、184、185、186、187、188、189、190、191、192、193、194、195、196、197、198、199、200、201、202、203、204、205、206、207、208、209、210、211、212、213、214、215、216、217、218、219、220、221、222、223、224、225、226、227、228、229、230、231、232、233、234、235、236、237、238、239、240、241、242、243、244、245、246、247、248、249、250、251、252、253、254、256、257、258、259、260、261、262、263、264、265、266、267、268、269、270、271、272、273、274、275、276、277、278、279、280、281、282、283、284、285、286、287、288、289、290、291、292、293、294、295、296、297、298、299、300、301、302、303、304、305、306、307、308、309、310、311、312、313、314、315、316、317、318、319、320、321、322、323、324、325、326、327、328、329、330、331、332、333、334、335、336、337、338、339、340、341、342、343、344、345、346、347、348、349、350、351、352、353、354、355、35、357、358、359、360、361、362、363、364、365、366、367、368369、370、371、408、409、410、411、412、413、414、415、416、417、418、419、420、421、422、423、424、425、426、427、428、429、430或431的氨基酸序列。本发明的原毒素-ii变体可抑制人nav1.7,其ic50值为约1×10-7m或更小、约1×10-8m、约1×10-9或更小、约1×10-10m或更小、约1×10-11m或更小,或约1×10-12m或更小。展示出ic50值的范围的示例性变体是具有seqidno:30、40、44、52、56、56、59、65、78、109、110、111、117、118、119、120、121、122、123、124、125、126、127、128、129、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、150、151、152、153、154、155、156、157、158、159、162、165、166、167、168、169、170、171、172、173、174、175、177、178、179、180、182、183、184、185、186、189、190、193、195、197、199、206、207、208、209、210、211、212、213、214、215、216、217、218、224、226、227、231、232、243、244、245、247、249、252、255、258、261、263、264、265、266、269、270、271、272、273、274、275、276、277、278、279、280、281、282、283、284、285、286、287、288、289、290、291、292、293、294、295、296、297、298、299、300、301、302、303、304、305、306、307、308、309、310、311、312、313、314、315、316、317、318、319、320、321、322、323、324、325、326、332、334、335、336、337、339、340、341、342、346、351、358、359、364、366、367、368、408、409、410、411、412、413、414、415、416、417、418、419、420、421、422、423、424、425、426、427、428、429、430或431中所示的氨基酸序列的变体。表2、表3和表13示出所选原毒素-ii变体的序列。表2.表3.在一些实施方案中,分离的原毒素-ii变体抑制人nav1.7的活性,其ic50值为约3×10-8m或更小。在一些实施方案中,分离的原毒素-ii变体抑制人nav1.7的活性,其ic50值介于约3×10-8m至约1×10-9m之间。本发明的另一个实施方案为分离的原毒素-ii变体,其包含氨基酸序列gpqcx1x2wx3qx4cx5x6x7x8x9ccx10x11fx12cx13lwcx14kkll(seqidno:433),其中x1为q、r、k、a或s;x2为k、s、q或r;x3为m或f;x4为t、s、r、k或q;x5为d或t;x6为s、a或r;x7为e、r、n、k、t或q;x8为r或k;x9为k、q、s或a;x10为e、q或d;x11为g或q;x12为v或s;x13为r或t;并且x14为k或r。抑制人nav1.7的活性且具有约30×10-9m或更小的ic50值的示例性原毒素-ii变体为包含seqidno:56、78、111、114、117、118、119、122、123、129、130、131、132、133、134、135、136、138、139、140、141、142、145、146、147、149、150、151、152、153、154、156、158、159、165、172、173、175、177、178、183、184、185、186、189、190、193、197、199、207、210、211、216、217、224、266、273、282、335、408、409、410、422、424、425、426、427和428的氨基酸序列的变体。在一些实施方案中,分离的原毒素-ii变体选择性地抑制人nav1.7。与重组原毒素-ii(seqidno:2)相比时,本发明的原毒素-ii变体可对nav1.7具有更大选择性。在qpatch电生理学测定法中,重组原毒素-ii对于nav1.7的ic50为约2.2×10-9m,并且对于nav1.6的ic50为约62×10-9m,因此对于nav1.6的ic50是对于nav1.7的ic50的约28倍。本文所用的“选择性”或“选择的”或“选择性更大”或“选择性地阻滞”或“选择性地抑制”是指原毒素-ii变体对于nav1.6的ic50与对于nav1.7的的ic50的比率(ic50(nav1.6)/ic50(nav1.7))等于或大于约30。可采用针对nav1.7所述的类似的方法在qpatch电生理学测定法中使用稳定表达nav1.6的细胞系测定针对nav1.6的ic50。原毒素-ii中可被诱变而提高选择性的残基位置包括残基7、11、12、14、17、18和19,并且任选地残基1、20、22和26,(残基编号根据seqidno:1而定)。提高选择性的示例性置换为y1q、w7q、s11r、s11a、e12t、m19f、v20s、r22t和k26r。选择性提高的示例性原毒素-ii变体为seqidno:56、59、65、78、111、114、117、118、119、121、122、123、129、130、133、150、190、217、281、324、325或326的变体。本发明的另一个实施方案为分离的原毒素-ii变体,其包含序列gpx1cqkwmqx2cdx3x4rkccx5gfx6cx7lwcx8kklw(seqidno:405);其中x1为y、q、a、s或r;x2为t或s;x3为s、r或a;x4为e、t或n;x5为e或q;x6为v或s;x7为r或t;并且x8为k或r;其中原毒素-ii变体抑制人nav1.7的活性,其ic50值为约3×10-8m或更小,并且选择性地抑制人nav1.7。在一些实施方案中,分离的原毒素-ii变体包含序列gpqcqkwmqx1cdx2x3rkccx4gfx5cx6lwcx8kklw(seqidno:406);其中x1为t或s;x2为s、r或a;x3为e、t或n;x4为e或q;x5为v或s;x6为r或t;并且x7为k或r。另一个实施方案为分离的原毒素-ii变体,其包含与seqidno:422的氨基酸序列(gpycqkwmqtcdserkccegmvcrlwckkkll-cooh)具有90%、91%、92%、93%、94%、95%、96%、97%、98%或99%同一性的氨基酸序列;其中当残基编号根据seqidno:1而定时,该氨基酸序列在位置7处具有q并且在位置30处具有l;并且多肽抑制人nav1.7的活性,其ic50值为约30×10-9m或更小,其中ic50值是在稳定表达人nav1.7的hek293细胞中存在25×10-6m3-藜芦酰藜芦瑟文的情况下使用荧光共振能量转移(fret)使用tetra膜去极化测定法而测量。当与野生型原毒素-ii相比时,具有置换w7q和w30l的原毒素-ii变体具有改善的折叠、产率和选择性。另一个实施方案为分离的原毒素-ii变体,其包含与seqidno:78的氨基酸序列(gpqcqkwmqtcdrerkccegfvctlwcrkklw-cooh)具有90%、91%、92%、93%、94%、95%、96%、97%、98%或99%同一性的氨基酸序列;其中当残基编号根据seqidno:1而定时,氨基酸序列在位置1处具有q,在位置7处具有q,并且在位置19处具有f;多肽抑制人nav1.7的活性,其ic50值为约30×10-9m或更小,其中ic50值是在稳定表达人nav1.7的hek293细胞中存在25×10-6m3-藜芦酰藜芦瑟文的情况下使用荧光共振能量转移(fret)使用tetra膜去极化测定法而测量;并且该多肽选择性地抑制nav1.7。在一些实施方案中,分离的原毒素-ii变体具有游离的c-末端羧酸、酰胺、甲基酰胺或丁基酰胺基团,这些基团通过常规合成方法生成。本发明的另一个实施方案为分离的融合蛋白,其包含seqidno:3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35,36,37,38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、109、110、111、112、113、114、115、116、117、118、119、121、122、123、124、125、126、127、128、129、130、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、150、151、152、153、154、155、156、157、158、159、160、161、162、163、164、165、166、167、168、169、170、171、172、173、174、175、176、177、178、179、180、181、182、183、184、185、186、187、188、189、190、191、192、193、194、195、196、197、198、199、200、201、202、203、204、205、206、207、208、209、210、211、212、213、214、215、216、217、218、219、220、221、222、223、224、225、226、227、228、229、230、231、232、233、234、235、236、237、238、239、240、241、242、243、244、245、246、247、248、249、250、251、252、253、254、256、257、258、259、260、261、262、263、264、265、266、267、268、269、270、271、272、273、274、275、276、277、278、279、280、281、282、283、284、285、286、287、288、289、290、291、292、293、294、295、296、297、298、299、300、301、302、303、304、305、306、307、308、309、310、311、312、313、314、315、316、317、318、319、320、321、322、323、324、325、326、327、328、329、330、331、332、333、334、335、336、337、338、339、340、341、342、343、344、345、346、347、348、349、350、351、352、353、354、355、35、357、358、359、360、361、362、363、364、365、366、367、368369、370、371、408、409、410、411、412、413、414、415、416、417、418、419、420、421、422、423、424、425、426、427、428、429、430或431的原毒素-ii变体。此类第二多肽可以是熟知的前导序列或分泌信号序列,或合成序列例如从克隆步骤获得的合成序列,或标签诸如六聚组氨酸标签(seqidno:108)。此类第二多肽可以是半衰期延长部分。在一个实施方案中,分离的融合蛋白包括缀合至半衰期延长部分的本发明的原毒素-ii变体。可使用的示例性半衰期延长部分包括熟知的人血清白蛋白、甲状腺素运载蛋白(ttr)、甲状腺素结合球蛋白(tgb)、白蛋白结合域或fc或其片段。也可使用生物学上合适的聚合物或共聚物,例如乙二醇或聚乙二醇(peg)分子,诸如peg5000或peg20000、葡聚糖、聚赖氨酸;不同链长的脂肪酸和脂肪酸酯,例如月桂酸酯、肉豆蔻酸酯、硬脂酸脂、花生酸酯、山嵛酸酯、油酸酯、花生四烯酸酯、辛二酸、十四烷二酸、十八烷二酸、二十二烷二酸等;辛烷或碳水化合物(葡聚糖、纤维素、寡糖或多糖)。这些部分可与原毒素-ii变体多肽直接融合并且可通过标准的克隆和表达技术生成。另选地,熟知的化学偶联方法可用于将这些部分连接至本发明的重组制备的原毒素-ii变体。在另一个实施方案中,本发明的融合蛋白的半衰期延长部分为人血清白蛋白、白蛋白结合域(abd)或聚乙二醇(peg)。在另一个实施方案中,半衰期延长部分通过接头缀合至原毒素-ii变体。合适的接头是众所周知的,并且包括具有seqidno:80或81所示的序列的接头。包含本发明的原毒素-ii变体的示例性融合蛋白是具有seqidno:83、85、87、89、91、93、95、97、99、101或103的多肽序列的那些融合蛋白。可通过多种熟知的测定法来比较引入另外部分的本发明的原毒素-ii变体的官能度。例如,偶联到peg的原毒素-ii变体的药代动力学特性可在熟知的体内模型中进行评估。另外的原毒素-ii变体和原毒素-ii变体融合蛋白在本发明的范围内。与亲本肽相比,只要所得的变体或融合蛋白保持类似的特性,便可对本发明的原毒素-ii变体进行另外的置换。示例性修饰为例如将使得原毒素-ii变体具有与亲本分子类似的特性的保守置换。保守置换是在其侧链中相关的氨基酸家族内发生的那些。基因编码的氨基酸可被分为四类:(1)酸性(天冬氨酸、谷氨酸);(2)碱性(赖氨酸、精氨酸、组氨酸);(3)非极性(丙氨酸、缬氨酸、亮氨酸、异亮氨酸、脯氨酸、苯丙氨酸、甲硫氨酸、色氨酸);以及(4)非荷电极性(甘氨酸、天冬酰胺、谷氨酰胺、半胱氨酸、丝氨酸、苏氨酸、酪氨酸)。苯丙氨酸、色氨酸和酪氨酸有时被共同分类为芳族氨基酸。另选地,氨基酸所有组成成分可以分组为:(1)酸性(天冬氨酸、谷氨酸);(2)碱性(赖氨酸、精氨酸、组氨酸)、(3)脂族(甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸、丝氨酸、苏氨酸),其中丝氨酸和苏氨酸任选地分别分组为含羟基脂族的;(4)芳族(苯丙氨酸、酪氨酸、色氨酸);(5)酰胺(天冬酰胺、谷氨酰胺);和(6)含硫的(半胱氨酸和甲硫氨酸)(stryer(编),biochemistry,第2版,whfreemanandco.,1981)。可对原毒素-ii变体进行非保守置换,该非保守置换涉及不同类别氨基酸之间的氨基酸残基置换,以改善原毒素-ii变体和原毒素-ii变体融合蛋白的性质。可以使用本文描述的测定法,通过评估修饰的多肽或片段以类似于未经修饰的多肽或片段的方式产生应答的能力,容易地测定多肽或其片段的氨基酸序列中的变化是否导致功能同源物。其中发生了多于一个替换的肽、多肽或蛋白质可以容易地以相同方式进行测试。本发明的另一个实施方案为分离的合成多核苷酸,其包含编码本发明的原毒素-ii变体的多核苷酸。本文公开了某些示例性的合成多核苷酸,然而,在给定的表达系统中给定遗传密码的简并性或密码子偏好,编码本发明的原毒素-ii变体和原毒素-ii变体融合蛋白的其它合成多核苷酸也在本发明的范围内。示例性的合成多核苷酸为例如seqidno:84、86、88、90、92、94、96、98、100、102和104所示的多核苷酸序列,该多核苷酸序列编码本发明的原毒素-ii变体融合蛋白。本领域的技术人员能够容易地鉴定编码原毒素-ii变体本身的融合蛋白中的多核苷酸片段。编码本发明的原毒素-ii变体或融合蛋白的合成多核苷酸序列可有效连接至一个或多个调控元件,诸如允许核苷酸序列在预期宿主细胞中表达的启动子和增强子。该合成多核苷酸可以是cdna。本发明的多核苷酸可以通过化学合成产生,诸如在自动化多核苷酸合成仪上进行的固相多核苷酸合成。另选地,本发明的多核苷酸可以通过其它技术产生,诸如基于pcr的复制、基于载体的复制或基于限制性内切酶的dna操作技术。用于制备或获得具有已知序列的多核苷酸的技术是已知的。本发明的多核苷酸还可以包含至少一个非编码序列,诸如转录但不翻译的序列、终止信号、核糖体结合位点、mrna稳定序列、内含子和聚腺苷酸化信号。多核苷酸序列还可以包含编码另外的氨基酸的另外的序列。这些另外的多核苷酸序列可以(例如)编码标记或熟知的标签序列,诸如六聚组氨酸(seqidno:108)或ha标签,这些标签有利于融合多肽的纯化。本发明的另一个实施方案为包含本发明的多核苷酸的载体。此类载体可以是质粒载体、病毒载体、用于杆状病毒表达的载体、基于转座子的载体或任何其它适于通过任何手段将本发明的多核苷酸引入给定生物体或遗传背景的载体。例如,编码本发明的原毒素-ii变体或原毒素-ii变体融合蛋白的多核苷酸被插入表达载体中,并且可操作地连接至表达载体中的控制序列以确保有效表达,诸如信号序列、启动子(例如,天然关联的或异源的启动子)、增强子元件和转录终止序列,并且该多核苷酸被选择为与所选的表达本发明的原毒素-ii变体或原毒素-ii变体融合蛋白的宿主细胞相容。一旦载体掺入到适当的宿主中,该宿主即保持在适于高水平表达由所掺入的多核苷酸编码的蛋白质的条件下。合适的表达载体通常可作为附加体或作为宿主染色体dna的组成部分在宿主生物中复制。通常,表达载体包含选择标记,诸如氨苄青霉素抗性标记、潮霉素抗性标记、四环素抗性标记、卡那霉素抗性标记或新霉素抗性标记,以允许检测转染了所需dna序列的那些细胞。合适的启动子和增强子元件在本领域中是已知的。对于在细菌细胞中表达的情况,合适的启动子包括但不限于lacl、lacz、t3、t7、gpt、λp和trc。对于在真核细胞中表达的情况,合适的启动子包括但不限于轻链和/或重链免疫球蛋白基因启动子和增强子元件、细胞巨化病毒立即早期启动子、单纯疱疹病毒胸苷激酶启动子、早期和晚期sv40启动子、逆转录病毒的长末端重复序列中存在的启动子、小鼠金属硫蛋白-i启动子、以及本领域已知的各种组织特异性启动子。对于在酵母细胞中表达的情况,合适的启动子是组成型启动子诸如adh1pgk1、eno或pyk1启动子等等,或者是可调节的启动子诸如gal1或gal10启动子。选择适当的载体和启动子在本领域普通技术人员的水平范围内。许多合适的载体和启动子是本领域技术人员已知的;它们中的很多可商购获得以用于制备重组构建体。以下载体以举例的方式提供。细菌:pbs、phagescript、psix174、pbluescriptsk、pbsks、pnh8a、pnh16a、pnh18a、pnh46a(stratagene,lajolla,calif.,usa);ptrc99a、pkk223-3、pkk233-3、pdr540和prit5(pharmacia,uppsala,sweden)。真核生物:pwlneo、psv2cat、pog44、pxr1、psg(stratagene)、psvk3、pbpv、pmsg和psvl(pharmacia)。用于表达原毒素-ii变体或原毒素-ii变体融合蛋白的示例性载体为具有以下组成部分的载体:氨苄青霉素抗性选择标记、cmv启动子、cmv内含子、信号肽、新霉素抗性、f1复制起点、sv40聚腺苷酸化信号和编码本发明的原毒素-ii变体或原毒素-ii变体融合蛋白的cdna。本发明的另一个实施方案为包含本发明的载体的宿主细胞。术语“宿主细胞”是指已引入载体的细胞。应该理解,术语宿主细胞不仅旨在指特定的主体细胞,还指这种细胞的子代。因为由于突变或者由于环境影响,在后代中可发生某些修饰,因此这种子代可与母体细胞不同,但仍包括在本文所用的术语“宿主细胞”的范围内。此类宿主细胞可以是真核细胞、原核细胞、植物细胞或古菌细胞。原核宿主细胞的示例是大肠杆菌(escherichiacoli)、杆菌属(bacilli)诸如枯草芽孢杆菌(bacillussubtilis)和其它肠杆菌科(enterobacteriaceae)诸如沙门氏菌(salmonella)、沙雷氏菌(serratia)以及各种假单胞菌属(pseudomonas)物种。其它微生物诸如酵母也可用于表达。合适的酵母宿主细胞的示例是酵母属(saccharomyces)(例如,酿酒酵母(s.cerevisiae))和毕赤酵母属(pichia)。示例性真核细胞可以是哺乳动物、昆虫、禽类或其它动物来源。哺乳动物真核细胞包括无限增殖化细胞系,诸如杂交瘤或骨髓瘤细胞系,诸如sp2/0(美国典型培养物保藏中心(atcc),manassas,va,crl-1581)、ns0(欧洲细胞培养物保藏中心(ecacc),salisbury,wiltshire,uk,ecaccno.85110503)、fo(atcccrl-1646)和ag653(atcccrl-1580)鼠细胞系。一种示例性人骨髓瘤细胞系是u266(attccrl-tib-196)。其它可用的细胞系包括衍生自中国仓鼠卵巢(cho)细胞的那些细胞系,诸如cho-k1sv(lonzabiologics,walkersville,md)、cho-k1(atcccrl-61)或dg44。多核苷酸(诸如载体)向宿主细胞中的引入可通过本领域技术人员熟知的方法实现。示例性方法为磷酸钙转染、deae-葡聚糖介导的转染、显微注射、阳离子脂质介导的转染和电穿孔。本发明的另一个实施方案为用于制备本发明的原毒素-ii变体的方法,该方法包括以下步骤:提供本发明的宿主细胞;以及在足以表达至少一种本发明的原毒素-ii变体的条件下培养该宿主细胞。宿主细胞可以在任何适用于保持或增殖给定类型的宿主细胞并足以表达多肽的条件下培养。足以表达多肽的培养条件、培养基和相关方法是本领域熟知的。例如,许多哺乳类细胞类型可以在37℃下用适当缓冲的dmem培养基进行有氧培养,而细菌、酵母和其它细胞类型可以在37℃和适当的大气条件下在lb培养基中进行培养。在本发明的方法中,原毒素-ii变体的表达可使用各种熟知的方法来确认。例如,可采用诸如sds-page或hplc使用检测试剂确认多肽的表达。本发明的另一方面是调节生物组织中nav1.7活性的方法,该方法包括:使表达nav1.7的生物组织与nav1.7调节量的本发明的原毒素-ii变体接触。治疗方法本发明的原毒素-ii变体可用于期望的任何治疗,在这些治疗中,期望该变体治疗、减轻或缓解疼痛的症状或与感觉神经元或交感神经元功能异常相关的其它疾病。采用本发明的原毒素-ii变体治疗的疼痛可以是任何类型的疼痛,诸如慢性疼痛、急性疼痛、神经性疼痛、伤害性疼痛、内脏疼痛、背部疼痛、与炎性病症相关联的疼痛、术后疼痛、热疼痛,或与疾病和退变相关联的疼痛。用本发明的原毒素-ii变体治疗的疼痛可以是nav1.7介导的疼痛。如本文所用,nav1.7介导的疼痛是指至少部分地由于nav1.7通道活性增加而导致的疼痛。本发明的方法可用于治疗属于任何分类的动物患者。此类动物的示例包括哺乳动物,诸如人、啮齿动物、犬类、猫类和农畜。疼痛和/或nav1.7介导的疼痛可能由于一种或多种原因引起,诸如周围神经病变,中枢神经病变,神经压迫或神经压迫综合征诸如腕管综合征,踝管综合征,尺神经卡陷,压迫性神经根病,腰椎椎管狭窄,坐骨神经压迫,脊髓根部压迫,肋间神经痛,压迫性神经根病和根性下背痛,脊髓根部病变,神经炎,自身免疫性疾病,全身性炎症,慢性炎症疾病,关节炎,风湿性疾病,狼疮,骨关节炎,一般胃肠疾病,结肠炎,胃溃疡,十二指肠溃疡,炎症性肠病,肠易激综合征,与腹泻相关的疼痛,炎症性眼病,炎症性或不稳定性膀胱病,牛皮癣,具有炎症成分的皮肤病症,晒伤,心炎,皮炎,肌炎,神经炎,胶原血管疾病,炎症性疼痛及相关性痛觉过敏和异常性疼痛,神经性疼痛及相关的痛觉过敏和异常性疼痛,多发性硬化症,脱髓鞘疾病,糖尿病,糖尿病性神经病疼痛,灼痛,截肢或脓肿引起的疼痛,幻肢疼痛,骨折疼痛,骨损伤,直接创伤,hiv感染,获得性免疫缺陷综合征(“aids”),天花感染,疱疹感染,暴露于毒素或其它异物颗粒或分子,浸润性癌症,癌症,化疗,放疗,激素治疗,烧伤,先天性缺陷,牙痛,痛风引起的疼痛,纤维肌痛,脑炎,慢性酒精中毒,甲状腺功能减退,尿毒症和维生素缺乏,三叉神经痛,中风,丘脑痛综合征,一般头痛,偏头痛,丛集性头痛,紧张性头痛,混合血管和非血管性综合征,交感神经保持疼痛,传入神经阻滞综合征,哮喘,上皮组织损伤或功能障碍,呼吸道、泌尿生殖道、胃肠道或血管区域的内脏运动紊乱,创伤,烧伤,过敏性皮肤反应,瘙痒症,血管舒缩或过敏性鼻炎,或支气管疾病,痛经,劳动和分娩期间的疼痛,消化不良,胃食管反流,胰腺炎和内脏痛。可由本发明的原毒素-ii变体缓解的与感觉神经元或交感神经元功能异常相关的其它疾病包括瘙痒、咳嗽和哮喘。小鼠的scn9a基因的完全缺失导致小鼠对组胺诱导的瘙痒完全不敏感(gingras等人,americanpainsocietymeetingabstract2013和美国专利公布2012/0185956)。该发现表明,nav1.7肽阻滞剂可用于治疗可由各种来源引起的瘙痒,诸如皮肤病或炎性病症;或诸如肾或肝胆疾病、免疫疾病、药物反应和未知/原发病症的炎性病症,包括皮炎、牛皮癣、湿疹或昆虫螫咬。nav1.7也在支配气道的感觉神经中表达(muroi等人,jphysiol.2011年12月1日,589(pt23):5663-76;muroi等人,amjphysiolregulintegrcompphysiol.2013年4月10日),这表明nav1.7肽阻滞剂可有利于治疗咳嗽如急性或慢性咳嗽、或胃食管逆流性疾病的刺激导致的咳嗽以及气道的炎性病症诸如哮喘和过敏相关的免疫应答、支气管痉挛、慢性阻塞性肺疾病、慢性支气管炎、气肿和呃逆(打嗝、呃逆)。在豚鼠体内使用shrna使结状神经节中的nav1.7沉默几乎消除了机械探测导致的咳嗽逆流(muroi等人,amjphysiolregulintegrcompphysiol,2013年4月10日)。本发明的一个方面是缓解或治疗受治疗者的瘙痒、咳嗽或哮喘的方法,该方法通过向对其有需要的受治疗者施用治疗有效量的本发明的原毒素-ii变体持续足以缓解瘙痒、咳嗽或哮喘的时间来进行。本发明的另一个方面是缓解或治疗受治疗者的nav1.7介导的瘙痒、nav1.7介导的咳嗽或nav1.7介导的哮喘的方法,该方法通过向对其有需要的受治疗者施用治疗有效量的本发明的原毒素-ii变体持续足以缓解瘙痒、咳嗽或哮喘的时间来进行。如本文所用,nav1.7介导的瘙痒是指至少部分地由于nav1.7通道活性增加而导致的瘙痒。如本文所用,nav1.7介导的咳嗽是指至少部分地由于nav1.7通道活性增加而导致的咳嗽。如本文所用,nav1.7介导的哮喘是指至少部分地由于nav1.7通道活性增加而导致的哮喘。可使用以下模型来测试本发明的原毒素-ii变体在减轻或缓解疼痛和/或nav1.7介导的疼痛方面的作用:本文所述的动物模型,和诸如大鼠脊神经结扎(snl)神经性疼痛的模型、角叉菜胶诱导的异常痛敏模型、freund完全佐剂(cfa)诱导的异常痛敏模型、热伤模型、福尔马林模型和bennett模型的模型,以及美国专利申请2011/0124711和美国专利7,998,980中所述的模型。角叉菜胶诱导的异常痛敏和cfa诱导的异常痛敏是炎性疼痛的模型。bennett模型提供了慢性疼痛的动物模型,包括术后疼痛、复杂性局部疼痛综合征以及反射交感性营养不良。上述动物模型中的任一模型均可用于评估本发明的原毒素-ii变体抑制剂在治疗疼痛和/或nav1.7介导的疼痛方面的功效。将本发明的原毒素-ii变体的功效与未治疗组或空白对照组进行比较。除此之外或另选地,可与一种或多种已知的疼痛缓解药物相比较来评估功效。本发明提供使用本发明的原毒素-ii变体治疗nav1.7介导的疼痛的方法。在本发明人的待审专利申请(美国专利申请号61/781,276)中已发现,施用nav1.7封闭肽对于治疗和/或缓解各种疼痛动物模型的疼痛是有效的,这与文献中所公开和提出的内容相反。虽然已证实nav1.7肽抑制剂在过表达nav1.7的体外细胞培养物模型,或在分离的神经元(其中血-神经屏障通过脱鞘或高渗盐水注射来跨过)中对nav1.7有效和/或具有选择性,但目前尚未证明nav1.7肽抑制剂在动物体内疼痛模型中有效,据报道该模型中肽不能跨过血-神经屏障引起功效的缺乏。多个出版物描述了nav1.7封闭肽在疼痛动物模型或分离的神经中缺乏功效。例如在hackel等人,procnatlacadsci109:e2018-27,2012中描述了除非神经周屏障失能,否则protx-ii在分离的神经中不能抑制动作电位的产生,神经周屏障在该模型中提供扩散屏障。据发现,protx-ii在急性和炎性疼痛的啮齿动物模型中无效;这合理地解释了protx-ii不能跨过血-神经屏障的现象(schmalhofer等人,molpharmacol74:1476–1484,2008)。据发现,nav1.7肽毒素阻滞剂的口服生物利用率低,并且它们难以递送到神经末梢,这说明它们作为治疗剂的用途仍然有限(dib-hajj等人,naturerevneuroscience14,49-62,2013)。nav1.7表达于周围神经系统中,例如表达于伤害感受性背根神经节(drg)中,最明显地表达于伤害感受性小直径drg神经元中,尤其是表达于皮肤中的周围末端中,但很少表达于大脑中。nav1.7的分布(例如感觉末梢)和生理机能使其倾向于在疼痛刺激的传输中发挥主要作用。本发明的一个实施方案为治疗nav1.7介导的疼痛的方法,该方法通过向对其有需要的受治疗者施用治疗有效量的本发明的原毒素-ii变体持续足以治疗nav1.7介导的疼痛的时间来进行。本发明的原毒素-ii变体nav1.7可用于期望的任何疗法,在这些疗法中,期望治疗nav1.7介导的疼痛,或与感觉神经元或交感神经元功能异常相关的其它疾病。疼痛的“治疗”是指包括部分或完全防止、阻止、抑制、减轻或延缓疼痛的感觉。在一些实施方案中,nav1.7介导的疼痛为慢性疼痛,急性疼痛,神经性疼痛,伤害性疼痛,内脏疼痛,背部疼痛,术后疼痛,热疼痛,幻肢疼痛,或者与下列相关的疼痛:炎性病症、原发性红斑肢痛症(pe)、阵发性剧痛症(pepd)、骨关节炎、类风湿性关节炎、腰椎间盘切除术、胰腺炎、纤维肌痛、疼痛性糖尿病性神经病变(pdn)、疱疹后神经病变(phn)、三叉神经痛(tn)、脊髓损伤或多发性硬化症,或与疾病和退变相关的疼痛。神经性疼痛包括例如疼痛性糖尿病性神经病变(pdn)、疱疹后神经病变(phn)或三叉神经痛(tn)。神经性疼痛的其它原因包括脊髓损伤、多发性硬化症、幻肢疼痛、中风后疼痛和hiv相关的疼痛。病症诸如慢性背部疼痛、骨关节炎和癌症也可导致产生神经相关的疼痛,因此可能适于使用本发明的原毒素-ii变体进行治疗。在另一个实施方案中,nav1.7介导的疼痛与原发性红斑肢痛症(pe)、阵发性剧痛症(pepd)、骨关节炎、类风湿性关节炎、腰椎间盘切除术、胰腺炎或纤维肌痛相关。在本发明的方法中,本发明的原毒素-ii变体可缀合至第二多肽以形成融合蛋白。此类融合蛋白为例如熟知的fc融合体,或融合至人血清白蛋白以延长肽抑制剂半衰期的融合体。缀合可以通过接头诸如富含甘氨酸-丝氨酸的接头直接缀合。此类接头是本领域所熟知的。可在使用熟知的方法和本文所述的那些方法治疗或减轻疼痛的过程中对包含另外部分的本发明的原毒素-ii变体的nav1.7阻滞能力和功效进行比较。可用本发明的原毒素-ii变体治疗的感觉神经元或交感神经元功能异常的其它疾病包括哮喘、咳嗽、胃灼热、瘙痒、皮炎、不稳定性膀胱和雷诺氏病。药物组合物本发明的原毒素-ii变体可在药学上可接受的媒介物或载体中配制。本发明的一个实施方案为药物组合物,其包含本发明的分离的原毒素-ii变体以及药学上可接受的赋形剂。合适的媒介物或载体可以是注射用水、生理盐水溶液或人工脑脊液,可能补充有在非肠道给药的组合物中常见的其它材料。另外的示例性媒介物是中性缓冲盐水或混合有血清白蛋白的盐水。这些溶液是无菌的,并且基本上不含颗粒物,并且可通过常规熟知的杀菌技术(例如,过滤)进行杀菌。组合物可包含接近生理条件所需的药学上可接受的赋形剂,诸如ph调节剂和缓冲剂、稳定剂、增稠剂、润滑剂和染色剂等。合适的媒介物及其制剂和封装在例如remington:thescienceandpracticeofpharmacy(第21版,troy,d.编,lippincottwilliams&wilkins,baltimore,md(2005)第40和41章)中有所描述。在本发明的方法中,本发明的原毒素-ii变体可外周施用。“外周施用”或“外周施用的”是指将试剂引入受治疗者中枢神经系统的外部。外周施用涵盖除直接施用到脊或大脑之外的任何施用途径。外周施用可为局部或全身的。局部施用可用于将治疗剂浓缩至作用位点,诸如局部施用于关节、脊髓、外科伤口、损伤/创伤部位、外周神经纤维、各种器官(gi、泌尿生殖器官等)或发炎组织。根据组合物的性质,全身施用使药物组合物基本上递送至受治疗者的整个外周神经系统,也可递送至中枢神经系统。外周施用途径涵盖但不限于局部施用、静脉注射或其它注射方式,以及植入微型泵或其它缓释装置或制剂。本发明的药物组合物包括涉及本发明的原毒素-ii变体的缓释或控释制剂的制剂。这些制剂可通过使用以下装置而获得:例如可注射微球体、可生物消化颗粒、微乳液、纳米粒子、毫微囊、粗乳剂、聚合化合物(诸如聚酯、聚氨基酸、水凝胶、聚(乳酸)、聚乙醇酸或乙烯基乙酸亚乙酯共聚物)、微珠或脂质体、透明质酸或可植入的药物递送装置。本发明的原毒素-ii变体可被制备成用于肠胃外(皮下、肌内或静脉内)、大脑内(实质内)、脑室内、肌内、眼内、动脉内、门脉内或病灶内途径;通过缓释系统或通过植入装置、或任何其它施用方式,特别是以液体溶液或悬液的形式;用于颊或舌下施用,例如以片剂或胶囊的形式;或者鼻内施用,例如以粉末、滴鼻剂或气雾剂或某些试剂的形式;经皮施用,以凝胶、软膏、洗剂、霜剂或扑粉剂、悬液或贴剂递送系统的形式,其具有改变皮肤结构或增大透皮贴剂中药物浓度的化学增强剂、或者能够将包含蛋白质或肽的制剂施用到皮肤上的试剂(国际专利公布wo98/53847),或用于以电场产生瞬时转运通道诸如电穿孔的应用,或用于增加带电药物通过皮肤的移动性诸如离子电渗,或用于超声诸如透皮吸收超声波的应用(美国专利4,309,989和4,767,402)。组合物也可通过植入吸收或包封有所需分子的膜、海绵或其它适当材料而局部施用。在某些实施方案中,当使用植入装置时,可将该装置植入任何合适的组织或器官,并且可通过扩散、定时释放丸剂或连续施用来递送所需分子。根据所选的具体施用模式,此类药物制剂中的本发明的原毒素-ii变体或其它nav1.7肽抑制剂的浓度可广泛的变化,例如约0.1重量%、0.2重量%、0.3重量%、0.4重量%、0.5重量%、0.6重量%、0.7重量%、0.8重量%、0.9重量%、1.0重量%、1.1重量%、1.2重量%、1.3重量%、1.4重量%、1.5重量%、1.6重量%、1.7重量%、1.8重量%、1.9重量%、2重量%,或在2重量%至5重量%之间,至多达15重量%、20重量%、30重量%、40重量%、50重量%、60重量%或70重量%,并且主要基于流体体积、粘度和其它因素进行选择。可将本发明的原毒素-ii变体冻干用于贮存,并在使用前在合适的媒介物中复原。已证实该技术对常规的蛋白质制备物是有效的。冻干和复原技术是本领域所熟知的。本发明的示例性药物组合物可包含ph为约7.0-8.5的tris缓冲液,或ph为约ph4.0-5.5的乙酸缓冲液,并且还可包括山梨糖醇、蔗糖、tween-20和/或其合适的替代品。本领域的技术人员可以很容易地确定适当的治疗有效剂量。有效剂量是指足以产生所需结果,即部分或完全防止、阻止、抑制、减轻或延缓与任何疼痛医学病症相关的疼痛感觉的量或剂量。有效量可根据具体媒介物和所选的本发明的原毒素-ii变体而发生变化,并且也取决于多种因素和与待治疗的受治疗者相关的病症以及疼痛的严重性。例如,诸如待施用本发明的药物组合物的受治疗者的年龄、体重和健康状态等因素,以及临床前动物工作获得的剂量响应曲线和毒性数据可在考虑范围内。如果必要,可将确定的剂量在治疗期间以适当的时间间隔加以重复,该时间间隔是由医师或相关领域的其他技术人员(如,护士、兽医或兽医技师)适当选择的。给定试剂的有效量或治疗有效量的测定在本领域的技术人员的能力范围之内。因此,用于肌内注射的本发明的药物组合物可制备为含有1ml无菌缓冲水,以及约1ng至约100mg、约50ng至约30mg、或约5mg至约25mg的本发明的原毒素-ii变体。类似地,用于静脉内输注的本发明的药物组合物可制成为含有约250ml无菌林格氏溶液,以及约1mg至约30mg、或约5mg至约25mg的本发明的原毒素-ii变体。用于制备非肠道施用的组合物的实际方法是熟知的,并且更详细地描述于例如“remington'spharmaceuticalscience”,第15版,mackpublishingcompany,easton,pa中。本发明的其它实施方案下文根据本文别处的公开内容陈述了本发明的某些其它实施方案。上文陈述为与本文所公开的发明相关的本发明的实施方案的特征还涉及这些其它带有编号的实施方案中的每一者。1)一种分离的原毒素-ii变体,该分离的原毒素-ii变体包含序列x1x2x3cx4x5wx6qx7cx8x9x10x11x12ccx13x14fx15cx16lwcx17kklw(seqidno:403),其中x1为g、p、a或缺失;x2为p、a或缺失;x3为s、q、a、r或y;x4为q、r、k、a或s;x5为k、s、q或r;x6为m或f;x7为t、s、r、k或q;x8为d或t;x9为s、a或r;x10为e、r、n、k、t或q;x11为r或k;x12为k、q、s或a;x13为e、q或d;x14为g或q;x15为v或s;x16为r或t;并且x17为k或r;该分离的原毒素-ii变体任选地具有n-末端延伸或c-末端延伸,其中多肽抑制人nav1.7的活性,其ic50值为约1×10-7m或更小,其中ic50值是在稳定表达人nav1.7的hek293细胞中存在25×10-6m3-藜芦酰藜芦瑟文的情况下使用荧光共振能量转移(fret)使用tetra膜去极化测定法而测量。2)根据权利要求1所述的原毒素-ii变体,其中n-末端延伸包含seqidno:372、373、374、375、376、377、378、379、380、381、382、383、384或385的氨基酸序列。3)根据权利要求1或2所述的原毒素-ii变体,其中所述c-末端延伸包含seqidno:374、386、387、388、389、390、391、392、393、394、395、396或397的氨基酸序列。4)根据权利要求2或3所述的原毒素-ii变体,其中所述n-末端延伸和/或所述c-末端延伸通过接头缀合至所述原毒素-ii变体。5)根据权利要求4所述的原毒素-ii变体,其中接头包含seqidno:383、392、398、399、400、401或402的氨基酸序列。6)根据权利要求1-5中任一项所述的分离的原毒素-ii变体,该分离的原毒素-ii变体包含seqidno:30、40、44、52、56、56、59、65、78、109、110、111、114、117、118、119、120、121、122、123、124、125、126、127、128、129、130、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、150、151、152、153、154、155、156、157、158、159、162、165、166、167、168、169、170、171、172、173、174、175、177、178、179、180、182、183、184、185、186、189、190、193、195、197、199、206、207、208、209、210、211、212、213、214、215、216、217、218、224、226、227、231、232、243、244、245、247、249、252、255、258、261、263、264、265、266、269、270、271、272、273、274、275、276、277、278、279、280、281、282、283、284、285、286、287、288、289、290、291、292、293、294、295、296、297、298、299、300、301、302、303、304、305、306、307、308、309、310、311、312、313、314、315、316、317、318、319、320、321、322、323、324、325、326、332、334、335、336、337、339、340、341、342、346、351、358、359、364、366、367或368的氨基酸序列。7)根据权利要求1-6中任一项所述的分离的原毒素-ii变体,该分离的原毒素-ii变体抑制人nav1.7的活性,其ic50值为约3×10-8m或更小。8)根据权利要求7所述的分离的原毒素-ii变体,该分离的原毒素-ii变体抑制人nav1.7的活性,其ic50值介于约3×10-8m至约1×10-9m之间。9)根据权利要求7或8所述的分离的原毒素-ii变体,该分离的原毒素-ii变体包含氨基酸序列gpqcx1x2wx3qx4cx5x6x7x8x9ccx10x11fx12cx13lwcx14kklw(seqidno:404);其中x1为q、r、k、a或s;x2为k、s、q或r;x3为m或f;x4为t、s、r、k或q;x5为d或t;x6为s、a或r;x7为e、r、n、k、t或q;x8为r或k;x9为k、q、s或a;x10为e、q或d;x11为g或q;x12为v或s;x13为r或t;并且x14为k或r。10)根据权利要求9所述的分离的原毒素-ii变体,该分离的原毒素-ii变体包含seqidno:56、78、111、114、117、118、119、122、123、129、130、131、132、133、134、135、136、138、139、140、141、142、145、146、147、149、150、151、152、153、154、156、158、159、165、172、173、175、177、178、183、184、185、186、189、190、193、197、199、207、210、211、216、217、224、266、273、282或335的氨基酸序列。11)根据权利要求1-10中任一项所述的分离的原毒素-ii变体,其中变体选择性地抑制人nav1.7。12)根据权利要求11所述的分离的原毒素-ii变体,该分离的原毒素-ii变体包含序列gpx1cqkwmqx2cdx3x4rkccx5gfx6cx7lwcx8kklw(seqidno:405);其中x1为y、q、a、s或r;x2为t或s;x3为s、r或a;x4为e、t或n;x5为e或q;x6为v或s;x7为r或t;并且x8为k或r。13)根据权利要求12所述的分离的原毒素-ii变体,该分离的原毒素-ii变体包含seqidno:56、59、65、78、111、114、117、118、119、121、122、123、129、130、133、150、190、217、281、324、325或326的氨基酸序列。14)根据权利要求12所述的分离的原毒素-ii变体,该分离的原毒素-ii变体包含序列gpqcqkwmqx1cdx2x3rkccx4gfx5cx6lwcx8kklw(seqidno:406);其中x1为t或s;x2为s、r或a;x3为e、t或n;x4为e或q;x5为v或s;x6为r或t;并且x7为k或r。15)一种分离的原毒素-ii变体,该分离的原毒素-ii变体包含与seqidno:78的氨基酸序列(gpqcqkwmqtcdrerkccegfvctlwcrkklw-cooh)具有90%、91%、92%、93%、94%、95%、96%、97%、98%或99%同一性的氨基酸序列,其中a)当残基编号根据seqidno:1而定时,氨基酸序列在位置1处具有q,在位置7处具有q,并且在位置19处具有f;b)多肽抑制人nav1.7的活性,其ic50值为约30×10-9m或更小,其中ic50值是在稳定表达人nav1.7的hek293细胞中存在25×10-6m3-藜芦酰藜芦瑟文的情况下使用荧光共振能量转移(fret)使用tetra膜去极化测定法而测量;并且c)多肽选择性地抑制nav1.7。16)根据权利要求1-15中任一项所述的分离的原毒素-ii变体,该分离的原毒素-ii变体具有游离的c-末端羧酸、酰胺、甲基酰胺或丁基酰胺基团。17)一种分离的融合蛋白,该分离的融合蛋白包括缀合至半衰期延长部分的根据权利要求1-16中任一项所述的原毒素-ii变体。18)根据权利要求17所述的融合蛋白,其中半衰期延长部分为人血清白蛋白(hsa)、白蛋白结合域(abd)、fc或聚乙二醇(peg)。19)一种分离的多核苷酸,该分离的多核苷酸编码根据权利要求12或15所述的原毒素-ii变体。20)一种载体,该载体包含根据权利要求19所述的分离的多核苷酸。21)一种宿主细胞,该宿主细胞包含根据权利要求20所述的载体。22)一种制备分离的原毒素-ii变体的方法,该方法包括培养根据权利要求21所述的宿主细胞,以及回收由宿主细胞产生的原毒素-ii变体。23)一种药物组合物,该药物组合物包含根据权利要求1、6、12、13或15所述的分离的原毒素-ii变体以及药学上可接受的赋形剂。24)一种治疗受治疗者的nav1.7介导的疼痛的方法,该方法包括向对其有需要的受治疗者施用有效量的根据权利要求1-16中任一项所述的原毒素-ii变体以治疗所述疼痛。25)根据权利要求24所述的方法,其中疼痛为慢性疼痛,急性疼痛,神经性疼痛,伤害性疼痛,内脏疼痛,背部疼痛,术后疼痛,热疼痛,幻肢疼痛,或者与下列相关的疼痛:炎性病症、原发性红斑肢痛症(pe)、阵发性剧痛症(pepd)、骨关节炎、类风湿性关节炎、腰椎间盘切除术、胰腺炎、纤维肌痛、疼痛性糖尿病神经病变(pdn)、疱疹后神经病变(phn)、三叉神经痛(tn)、脊髓损伤或多发性硬化症。26)根据权利要求24所述的方法,其中原毒素-ii变体是外周施用的。27)根据权利要求24所述的方法,其中将原毒素-ii变体局部施用于关节、脊髓、外科伤口、损伤或创伤部位、外周神经纤维、泌尿生殖器官或发炎组织。28)根据权利要求24所述的方法,其中受治疗者是人。29)根据权利要求1-16中任一项所述的原毒素-ii变体,该原毒素-ii变体用于治疗对其有需要的受治疗者的疼痛。30)根据权利要求29所述的所用原毒素-ii变体,其中疼痛为慢性疼痛,急性疼痛,神经性疼痛,伤害性疼痛,内脏疼痛,背部疼痛,术后疼痛,热疼痛,幻肢疼痛,或者与下列相关的疼痛:炎性病症、原发性红斑肢痛症(pe)、阵发性剧痛症(pepd)、骨关节炎、类风湿性关节炎、腰椎间盘切除术、胰腺炎、纤维肌痛、疼痛性糖尿病神经病变(pdn)、疱疹后神经病变(phn)、三叉神经痛(tn)、脊髓损伤或多发性硬化症。31)根据权利要求29或30所述的所用原毒素-ii变体,其中原毒素-ii变体是外周施用的。32)根据权利要求29、30或31所述的所用原毒素-ii变体,其中将原毒素-ii变体局部施用于关节、脊髓、外科伤口、损伤或创伤部位、外周神经纤维、泌尿生殖器官或发炎组织。本发明现将结合以下具体的、非限制性实施例进行描述。实施例1:原毒素-ii变体的设计和制备原毒素-ii单一位置受限的氨基酸扫描文库置换被设计为评估选择性、肽收率和均质性所能改善的程度。原毒素-ii变体被设计为如下形式的可被hrv3c蛋白酶切割的hsa融合蛋白:从n-至c-末端:6×his-hsa-接头-hrv3c可切割肽-原毒素-ii变体(“6×his”被公开为seqidno:108);接头为(ggggsggggsggggsggggs;seqidno:80,hsa的序列为seqidno:106,hrv3c可切割肽的序列为seqidno:82)。每个原毒素-ii变体从hsa切割后均具有来自切割位点的剩余n-末端gp。变体在以下测定法中进行表征:如实施例3tetra膜去极化测定法所述,使用tetra进行膜去极化测定法;以及使用如实施例3所述的qpatch测定法进行全细胞膜片钳实验。组合文库被设计为在生成nav1.7拮抗剂的尝试中测试所选单一位置命中的加合效应,其具有相比天然肽更进一步改善的效力和选择性特征。表达载体的构建使用美国专利us6,521,427中所述的合成基因组件技术生成所设计的原毒素-ii变体基因。使用人高频密码子将所设计的肽变体的氨基酸序列回译为dna序列。每个变体基因的dna序列与包含dna克隆位点的载体dna的一部分一起合成为多个寡核苷酸,这些寡核苷酸中的一些包含简并密码子,并组装成全长dna片段。所组装的dna片段通过pcr扩增,随后将pcr产物克隆为库。将pcr产物库用适当的限制性酶消化,并以使每个毒素变体基因融合至载体中包含的信号肽和融合伴侣6×his-hsa-接头-hrv3c可切割肽(公开为seqidno:108的“6×his”)的方式克隆到设计的表达载体中。使用标准分子生物学技术鉴定每个设计变体的阳性克隆。纯化这些阳性克隆的质粒dna,先确认序列,再使用标准方法表达原毒素-ii肽变体融合蛋白。蛋白质表达将hek293-f细胞保持在293freestyletm培养基(invitrogen目录号12338)中,当细胞浓度介于1.5×106和2.0×106个细胞/ml之间时分瓶。将细胞在37℃和8%co2下培养于湿化培养箱中的悬浮液中,并以125rpm进行摇动。在hek293f细胞稀释至1.0×106个细胞/ml后,使用dna/脂质复合物瞬时转染。为生成复合物,将1.25μgdna/ml的转染物稀释在1.0mloptipro培养基(invitrogen目录号12309)中,并将1.25mlfreestyletmmax转染试剂(invitrogen目录号16447)稀释在1.0mloptipro培养基中。将dna和max转染试剂混合在一起,并在室温下温育10分钟,然后加入细胞中。将转染的细胞在37℃、8%co2的湿化培养箱中放置4天,并以125rpm进行摇动。通过以5,000×g离心10分钟将上清液从细胞分离并滤过0.2μm过滤器(corning;目录号:431153),然后用amiconultra浓缩器10k(目录号:ufc901096)浓缩10倍和50倍,并以3,750×g离心大约10分钟。实施例2:原毒素-ii变体的纯化如实施例1所指出的那样,将原毒素-ii变体表达为hsa融合蛋白,并在进行纯化之前采用hrv3c蛋白酶切割原毒素-ii变体肽。用两种方法测试原毒素-ii变体是否已有效纯化。蛋白质纯化通过rp-hplc纯化原毒素-ii变体使用1mlhistraphp柱(gehealthcare目录号17-5247-01)通过imac从表达上清液纯化分泌蛋白。使用aktaxpress运行色谱法,并使用分级梯度的咪唑将蛋白质从柱洗脱。合并峰值级分并用hrv3c蛋白酶消化过夜(1μg蛋白酶/150μg融合)。使用具有反相phenomenexluna5μmc18(2)柱(目录号00b-4252-p0-ax)的dionexhplc系统进一步纯化切割肽融合库。用0-68%乙腈(0.05%tfa)线性梯度从柱洗脱样品。合并洗脱级分,冻干过夜并在ph7.4的hepes缓冲盐溶液(10mmhepes、137mmnacl、5.4mmkcl、5mm葡萄糖、2mmcacl2、1mmmgcl2)中复原。表4示出了通过rp-hplc纯化的原毒素-ii变体的收率。平均收率为0.01615mg/l。表4.通过固相萃取(spe)纯化原毒素-ii变体使用1mlhistraphp柱(gehealthcare目录号17-5247-01)通过imac从表达上清液纯化分泌蛋白。使用aktaxpress运行色谱法,并使用分级梯度的咪唑将蛋白质从柱洗脱。合并峰值级分并用hrv3c蛋白酶消化过夜(1μg蛋白酶/150μg融合)。将切割的样品上样到50kda分子量截留离心过滤单元(milliporeufc805096),并将切割肽收集在滤液级分中。将肽库上样到96孔固相萃取块(agilentbondelutplexaa3969030)用于进一步纯化、脱盐并浓缩。将块与真空歧管(whatman)结合使用。将肽样品上样,在溶于水的0.05%tfa中洗涤,并且用分级梯度的乙腈和溶于水的0.05%tfa洗脱。然后将洗脱级分冻干过夜,并在ph7.4的hepes缓冲盐溶液(10mmhepes、137mmnacl、5.4mmkcl、5mm葡萄糖、2mmcacl2、1mmmgcl2)中复原。肽在ph7.4的补充hepes缓冲盐溶液(10mmhepes、137mmnacl、5.4mmkcl、5mm葡萄糖、2mmcacl2、1mmmgcl2)中复原,并测量280nm下的吸光度。然后使用每个样品的消光系数计算浓度值。将2μg的每种肽上样到invitrogenbis-trisgel15孔凝胶上,并在非还原性mes缓冲液中跑胶。使用溶于0.05%tfa的线性梯度的4%-80%乙腈和phenomenexlunac18(2)分析柱(目录号00a-4041-b0)在agilent1100hplc上分析样品。使所有肽的浓度归一化,每种肽注入10μl,每个样品总共1.3μg。监测220nm下的吸光度,并使用chromeleon软件进行色谱分析。表5示出了通过spe纯化的原毒素-ii变体的收率(mg)。平均收率为0.05353mg/l。spe纯化过程中因为样品在96孔板中平行处理,而不是在rp-hplc上连续处理,因而具有简单且纯化通量高的优点,并且收率得到了提高。通过spe纯化的变体的收率(mg/l)是通过rp-hplc纯化的变体的收率的平均3倍以上。表5.实施例3:原毒素-ii变体的表征所选原毒素-ii变体以膜去极化和全细胞膜片钳测定法进行表征,以评估它们对于nav1.7的效力和选择性。tetra膜去极化测定法使用fret(荧光共振能量转移)测定法在tetra上测量所生成的肽抑制nav1.7激动剂藜芦定(3-藜芦酰藜芦瑟文;biomol,catalog#na125)诱导的膜去极化的能力,该测定法使用disbac2(3)(invitrogen,k1018)作为电子受体,并使用pts18(8-十八烷基氧芘-1,3,6-三磺酸三钠)(sigma)作为供体,通过在390nm-420nm下激发供体,并测量515nm-575nm下的fret来进行。将稳定表达人nav1.7的hek293细胞在补充有10%胎牛血清、1%青霉素/链霉素、400μg/ml遗传霉素和100μmneaa(所有试剂均购自invitrogen)的dmem/f-12培养基(1:1)中培养。将50μl收获的细胞以25,000个细胞/孔铺到聚赖氨酸涂布的384孔黑色透明底板中。板在室温(rt)下温育15min,然后在37℃下过夜温育。除非另有说明,否则所有温育均在黑暗中进行。次日,用测定缓冲液(137mmnacl、4mmkcl、2mmmgcl2、2mmcacl2、5mm葡萄糖、10mmhepes,ph7.4)将孔洗涤4次,并将孔重悬于25μl测定缓冲液中。通过将染料以1:1(v/v比)的比例悬浮于溶于dmso的10%普兰尼克(pluronic)f127中来制备2倍的pts18染料原液(6μm)。将25μl的2倍pts18原液加入孔中,并在室温下使细胞染色30min,然后用测定缓冲液洗掉染料。将肽以3倍的终浓度悬浮于包含10μmdisbac2(3)和400μmvabsc-1的测定缓冲液中,以抑制背景荧光(sigma,目录号201987)。悬浮肽以25μl/孔的量加入每个孔中,并在室温下温育60分钟。由25μm终浓度的藜芦定(通过加入25μl/孔的75μm3倍原液)诱导去极化,在加入激动剂30-100秒后测量fret染料荧光的平均强度减少。按照惯例,加入藜芦定后每个所测量的肽稀释1.3倍,记录tetra测定法开始时的浓度。构建每个实验系列的合成原毒素-ii(peptideinternational)的浓度响应曲线,并作为对照。通过将信号归一化为阴性对照值(对单独激动剂藜芦定的响应)和阳性对照(在存在10μm丁卡因的情况下对藜芦定的响应)值之间的差值来将每个孔的荧光计数转换为%响应。在测量时,在tetra中打开“空间均匀性校正”(将所有荧光迹线归一化为平均初始起始强度)和“差值偏移值”(从每条迹线减去初始起始强度)。每个数据点表示单个孔的响应。使用origin(microcal)在非线性最小二乘拟合过程中采用所有单个数据点查找hill函数的最佳拟合。从所得的拟合曲线提取ic50值。使用阳性(p)和阴性(n)对照的平均响应根据下式计算%响应:%响应=100*(n-r)/(n-p)。如果该日对照拮抗剂的效力在其历史平均值的±0.5对数单位之内,则该测定板是可接受的。qpatch测定法将稳定表达人nav1.5(seqidno:105)、nav1.7(seqidno:79)或nav1.6(seqidno:407)的hek293细胞在补充有10%胎牛血清、1%青霉素/链霉素、400μg/ml遗传霉素和100μmneaa(所有试剂均购自invitrogen)的dmem/f-12培养基(1:1)中培养。将细胞保持在37℃下和5%co2中,并在达到约50%-90%汇合度时测定。将稳定表达人nav1.6的cho细胞以四环素诱导的方式(seqidno:407)在补充有10%胎牛血清、1%青霉素/链霉素、10μg/ml杀稻瘟菌素和400μg/ml博莱霉素的hamsf12中培养。将细胞保持在37℃下和5%co2中,并在达到约50%-90%汇合度时进行测定。在实验前24-48h用1μg/ml四环素诱导nav1.6表达。在qpatchht(sophion)中进行测试之前,先使用0.05%胰蛋白酶(5min,37℃下)解离细胞,将细胞重悬于cho-s-sfm培养基(lifetechnologies)中,并轻轻研磨以破碎细胞团块。用相同的培养基将细胞密度调整为1-2×106/ml,将细胞转移到qpatchht中的细胞“旅馆”并用于数小时的实验。对于千兆欧姆阻抗封接形成和全细胞膜片钳记录,细胞外溶液包含137mmnacl、5.4mmkcl、1mmmgcl2、2mmcacl2、5mm葡萄糖和10mmhepes,ph=7.4并且同渗容摩=315mosm。细胞内溶液包含135mmcsf、10mmcscl、5mmegta、5mmnacl和10mmhepes,ph=7.3并且同渗容摩=290mosm。测定法所用的电压方案如下。细胞首先从保持电位-75mv(nav1.7)、-60mv(nav1.6)或-105mv(nav1.5)超极化2秒至-120mv,然后去极化5ms至0mv,然后恢复到保持电位。在液体施用期间将该方案每60秒重复一次(参见下文)。未执行上述电压方案时,则将细胞保持于保持电位。在建立全细胞记录模式时,对待记录的细胞总共施用五次细胞外溶液(均包含0.1%牛血清白蛋白(bsa),同时含有或不含测试化合物,但最后一次施用除外,最后一次施用的细胞外溶液包含1μmttx或10mm利多卡因作为阳性对照)。第一次液体施用仅包含对照缓冲液(5μl)。在施用五秒后执行电压方案10次(总共持续10分钟)。接下来的三次液体施用(每次5μl)包含测试化合物(全部三次施用的化合物相同、浓度相同)或对照缓冲液(仅施用于对照细胞)。在每次施用五秒后,再次执行电压方案10次(同样每分钟一次)。最后一次施用包含阳性对照(由三次10μl次级施用构成,每次间隔2秒),五秒后执行相同的电压方案两次,得到基线电流。在25khz下对电流取样,使用八极点贝塞尔(bessle)滤波器在5khz下过滤。串联电阻补偿水平设定为80%。对于每个细胞,在存在阳性对照的情况下,首先从上次迹线的振幅减去前四次液体施用中每次电流迹线在0mv下的峰值电流振幅,然后归一化为第一次(对照缓冲液)施用中的上次迹线的振幅,作为%抑制。为控制电流下降,在存在测试化合物的情况下,将每个细胞的该值(%抑制值)进一步归一化为同一实验中对照细胞(通常5-6个)的平均%抑制值。在最后一次化合物施用中,将上两次此类值的平均值(即,每个浓度的测试化合物的校正%抑制值)作为所测试的特定化合物浓度下每个细胞的%抑制值。对每个化合物浓度下所有测试细胞的%抑制值取平均值,用于浓度响应计算。所有实验均在室温下进行(约22℃)。数据表示为平均值±se。野生型原毒素-ii作为阳性对照包括在每个实验中。只有原毒素-ii的效力在历史平均值的±0.5对数单位之内,数据才是可接受的。表6示出了使用tetra获得的所选原毒素-ii变体的针对nav1.7的ic50值。表6.使用qpatch测试所选原毒素-ii变体对人nav1.5的选择性。表7示出了使用qpatch获得的所选肽针对nav1.7和nav1.5两者的ic50值。表7.实施例4.组合原毒素-ii变体的制备和表征组合文库被设计为在生成nav1.7拮抗剂的尝试中测试所选单个位置命中的加合效应,与天然肽相比,其效力和选择性特征使用多种方法进一步改善。使用a、d、q、r、k和s对所有非半胱氨酸原毒素-ii位置进行限制氨基酸扫描,使这些位置多样化。如实施例1所述的那样,在这些实验中,将原毒素-ii作为单价fc融合蛋白进行表达和测试。从该扫描中鉴定了使所得的变体的效力和/或选择性得以改善的置换y1q、w7q、s11a。还对位置m6和m19进行全氨基酸扫描(除cys和trp之外)。从该扫描中鉴定了使所得的变体的效力和/或选择性得以改善的m19f置换。双向地设计原毒素-ii/虎纹捕鸟蛛毒素-iv单一位置嵌合体。该文库的目的是获得保持野生型原毒素-ii的效力和选择性特征的原毒素-ii变体,并且可实现与虎纹捕鸟蛛毒素-iv相关的有利的再折叠特性。从该扫描中鉴定了置换r22t和e12n。通过使用r、k、t、a、d、e、q和s的限制氨基酸扫描使位置y1多样化,并且通过电荷簇工程化来进一步工程化肽nv1g1153,其中在肽的三维结构(d10/e12、k4/e17、d10/e12/r13)中所有带电残基组是突变的。将n-末端延伸和c-末端延伸引入包括nv1g1153在内的所选肽,目的是在不显著增加所得的肽的分子量的情况下,改善作用位点的肽分布以及改善肽的半衰期。所用的n-末端延伸和c-末端延伸分别示于表8和表9中,并且在oi等人,neuroscienceletters434,266-272,2008;whitney等人,naturebiotechnology201129:4,352-356;sockolosky等人,(2012)109:40,16095–16100中有所描述。也可使用细胞渗透肽hivtat和聚精氨酸。使用各种接头将原毒素-ii变体偶联到n-末端延伸和/或c-末端延伸。所用的接头示于表10中。使用实施例3所述的方法测试每个活动的原毒素-ii变体对nav1.7的效力和选择性。抑制nav1.7且ic50值为200nm或更小的变体的氨基酸序列示于表3中。表11示出与野生型原毒素-ii相比的所选变体中的氨基酸置换,以及fliprtetra测定法中nav1.7抑制的ic50值。表8.表9.表10.表11.如实施例3所述的那样,野生型原毒素-ii抑制nav1.7,在flipr测定法中,其ic50值为约4nm。对保持显著nav1.7效力的变体进行进一步表征。图1示出所生成的抑制nav1.7且具有30nm或更小的ic50值的原毒素-ii变体的序列属。使用qpatch测试所选原毒素-ii变体对nav1.7的抑制以及对人nav1.6的选择性。图2示出使用qpatch获得的所选肽针对nav1.7和nav1.6两者的ic50值。这些肽抑制nav1.7,其ic50值为30nm或更小,并且这些肽对nav1.7的选择性为对nav1.6的选择性的至少30倍。图3示出了如图2所示的肽的氨基酸序列。与野生型原毒素-ii相比,所有这些肽均具有w7q和m19f置换。如实施例1所述表达并纯化原毒素-ii变体,或通过标准固相合成法合成该变体。将重组或合成肽的收率与野生型原毒素的收率进行比较。表12示出所选原毒素-ii变体的收率显著大于原毒素-ii的收率,表明变体的折叠特性改善。固相合成的规模为0.5mmol。表12.实施例5.原毒素-ii变体在体内疼痛模型中有效材料和方法动物本研究使用购自charlesriver并单独饲养的雄性c57bl/6小鼠(24g-26g)。行为测试vonfrey测试:采用vonfreyhairs纤毛机械刺激针根据上-下法评估机械(触觉)阈值(dixon,1980,chaplan等人,1994)。使用7级刺激(vonfrey细丝:0.03、0.07、0.16、0.4、0.6、1、2g;stoelting,wooddale,il)。将纤毛机械刺激针垂直于后足脚底的中央区域(环面之间)放置。施加足够的力使细丝稍微弯曲并保持3秒。根据chaplan的文章,正响应可以是1)急剧退缩或2)在细丝去除时立即退缩。更多详情参见chaplan等人的文章。在进行测试之前使小鼠适应测试室中的丝网30-60分钟。hargreaves测试:使用经改进的hargreaves盒测量热退缩潜伏期(pwl)(hargreaves等人,1988,pain,32:77-88;dirig等人,1997,jneurosci.methods,76:183-191)。该盒由具有升高玻璃板的室构成,该玻璃板保持在恒温(27℃)下。玻璃表面以下的投影灯泡光束产生热伤害感受性刺激。该光束瞄准环面之间的区域(脚底中央)。“开始”按钮将打开光照并开始计时。爪受刺激而产生的运动(诸如突然退缩)将触发开关关闭光照并停止计时。潜伏期以秒显示。如果未发生运动,则灯泡将在20秒后关闭(截止)以防止组织损伤。在pwl测量之前,先使动物习惯玻璃表面30-60分钟。在整个研究中使用恒定安培数,当至少相隔5分钟采集平均超过3至6次读数时,得到的预测试退缩潜伏期在8-12秒之间。mpe%计算:最大可能效应百分比(mpe%)=(t1–t0)/(tc–t0)×100%。t0:第0天的阈值(cfa后,泵植入前);t1:泵植入后第1天的阈值;tc:测试截止(hargreaves测试中为20s,vonfrey测试中为2g)。热板测试:将动物置于4个树脂玻璃壁(高15英寸)围绕的10"×10"金属板上。将板保持在50℃或55℃的温度下。测量响应潜伏期(动物第一次退缩或舔后足、跳跃或发声的时间)并从板上移走动物。在40s(50℃)或20s(55℃)后从板上移走未表现出响应的动物,以防止任何可能的组织损伤。该试验在一天内每15-60分钟重复2-5次。炎性疼痛模型cfa模型:用异氟烷(用4%的浓度诱导并保持在2%)麻醉动物,并使用附接到50μlhamilton注射器的27号针将20μl100%完全freund佐剂(cfa;sigma-aldrich;saintlouis,mo)注入一只后足足底的中央区域。角叉菜胶模型:用异氟烷(用4%的浓度诱导并保持在2%)麻醉动物,使用胰岛素注射器(bd;franklinlakes,newjersey)将25μl溶解于生理盐水的2%λ-角叉菜胶(sigma-aldrich;saintlouis,mo)注入后足足底的中央区域。微型泵的植入根据制造商的指南填充并灌注alzet微渗透微型泵(durectcorporation,1003d型和2001d型)。用异氟烷(用5%的浓度诱导;并保持在2%)麻醉小鼠。剃光小鼠背部,用异丙醇和聚乙烯吡咯烷酮擦拭,并在肩胛骨之间作出小切口。用一对钳子或止血钳展开皮下结缔组织,形成小凹坑。将泵插入凹坑,并使流动调节器指向远离切口的方向。然后使用7mm缝钉缝合皮肤切口,动物在笼窝中得到恢复。数据分析数据表示为平均值±s.e.m。使用prism(graphpadsoftwareinc.,lajolla,ca)进行绘图和统计分析。对于阈值与时间的比较,使用双因素方差分析然后进行bonferroni多重比较测试,显著性水平为p<0.05。对热板和mpe%数据进行单因素方差分析然后进行bonferroni多重比较测试。结果在常用的炎性疼痛模型——cfa模型中研究变体nv1d3034-oh(nv1d3034-cooh)、nv1d3368-oh(nv1d3368-cooh)和nv1d2775-oh(nv1d2775-cooh)的功效。cfa注入后足诱导了爪浮肿(未示出)以及对热刺激的超敏反应(热痛觉过敏),如受注射爪第0天的热潜伏期减少所示(图6a)。通过皮下渗透微型泵以684μg/天和1824μg/天的量施用nv1d3034-oh完全逆转了热痛觉过敏(图4a和图4b)。684μg/天和1824μg/天的nv1d3368-oh完全逆转了cfa诱导的热痛觉过敏(图5a和图5b)。在cfa模型中nv1d2775-oh显示出较强的功效。在施用nv1d2775后,热潜伏期达到接近截止时的值(图6a和图6b,1824μg/天),表明在抗痛觉过敏效应的极点具有强痛觉丧失效应。此外,nv1d2775-oh逆转了cfa诱导的触觉异常痛敏(图6c和图6d,1824μg/天)。可在泵植入后4小时观察到nv1d2775-oh的抗痛觉过敏效应(图7a)。该效应在热和触觉测试中在8小时达到最大值(图7a和图7b),这两个测试均保持24小时。热潜伏期和触觉阈值在泵植入48h后恢复到对照水平(泵植入后大约24h被预测为空白)(图7a和图7b)。cfa诱导的热痛觉过敏易于被两个另外的肽逆转:nv1d3368-酰胺(nv1d3368-nh2)和nv1d3034-n-甲基酰胺(nv1d3034-nhme)。该实验的热mpe%汇总于表13中。表13.在热板测试中nv1d2775-oh还表现出强的剂量依赖性功效(图8)。以1824μg/天进行施用后,50℃和55℃下的潜伏期达到接近截止时的值。与pbs对照相比,228μg/天的nv1d2775-oh使得热潜伏期适度但显著地增加。在另一个炎性疼痛模型——角叉菜胶模型中评估nv1d2775-oh的功效。动物被植入nv1d2775-oh或pbs泵。在泵植入之前、以及植入之后第1天测量热退缩潜伏期。将λ-角叉菜胶注入后足,在角叉菜胶注入后2小时、3小时和4小时再次测量热潜伏期。1824μg/天的nv1d2775-oh产生了显著的痛觉丧失现象。(图9)。后足注入λ-角叉菜胶诱导了炎症(未示出),并且在hargreaves测试的4小时测试期内热退缩潜伏期缩短(图9,pbs组)。用1824μg/天的nv1d2775-oh预处理的动物完全避免了角叉菜胶诱导的痛觉过敏。实施例6.组合原毒素-ii变体的制备和表征生成针对原毒素-ii的氨基酸扫描库。在原毒素-ii的每个非半胱氨酸位置处(tyr1、gln3、lys4、trp5、met6、trp7、thr8、asp10、ser11、glu12、arg13、lys14、glu17、gly18、met19、val20、arg22、leu23、trp24、lys26、lys27、lys28、leu29和trp30),用天然残基置换以下残基:ala、asp、glu、phe、gly、his、ile、lys、leu、asn、pro、gln、arg、ser、thr、val和tyr。突变肽被表达为人血清白蛋白的重组融合物,并使用hrv3c位点特异性地酶切以生成如实施例1中所述的原毒素-ii变体。每个原毒素-ii变体从hsa切割后均具有来自切割位点的剩余n-末端gp。对于每种原毒素-ii变体,根据实施例3中所述的方案,使用fliprtetra或qpatch测量针对人nav1.7的ic50值。使用qpatch电生理学,对针对人nav1.7显示出ic50≤100nm的变体进行反筛选,以得到对另外hnav通道的选择性。选择性命中被鉴定并用于组合肽文库的设计,该组合肽文库使用重组表达和固相肽合成两者产生。使用与以上详述相同的策略筛选组合变体。基于结果,可以经突变以提高选择性的位置包括gln3、ser11、glu12、lys14、glu17、gly18、leu29和trp30(残基编号根据seqidno:1而定)。原毒素-ii的溶液结构通过nmr测定,并且在图10中表示为表面。图的左手侧显示了先前描述的(park等人,j.med.chem.2014,57:6623-6631),围绕m6的w5/w7/w24trp残基环。在分子的相对侧,使用诱变和nmr结构两者,在本研究中鉴定了由多个氨基酸位置组成的原毒素-ii的选择性面,该位置可突变以提高hnav1.7对其它钠通道亚型的选择性。位于选择性面上的残基包括残基ser11、glu12、lys14、glu17、gly18、leu29和trp30(残基编号根据seqidno:1而定)。早先未对选择性面和其内对nav1.7的选择性负责的多个位置的鉴定进行描述。在ser11处具有置换的原毒素ii变体的改善选择性是意想不到的,因为早先已经证明ser11的突变影响多个nav通道的活性,因此残基被认为在原毒素-ii对nav1.7的选择性中不起作用(park等人,j.med.chem.2014,57:6623-6631)。原毒素-ii变体合成生产中的关键步骤是形成二硫键配对的线性肽的氧化重折叠。对再折叠之后纯化的天然原毒素-ii的rp-hplc追踪反映出在不同保留时间处的多个峰,这些峰为正确的质量但示出不同的活性水平,表示肽折叠错误。通过在各种原毒素-ii位置处进行置换可改善rp-hplc主峰的相对丰度,从而改善正确折叠的肽的相对丰度。trp7或trp30的突变改善了所得的原毒素-ii变体的折叠。trp7和trp30两者组合的突变进一步改善了所得的原毒素-ii变体的折叠,并且可以拯救难以重折叠的原毒素-ii变体的折叠。具有改善选择性(gln3、ser11、glu12、lys14、glu17、gly18和leu29)的一个或多个置换以及在trp7和trp30处的突变的组合突变体肽的生产得到了具有改善的选择性和改善的重折叠特性两者的肽。原毒素-ii属于抑制型半胱氨酸结多肽的家族3(klint等人,toxicon60:478–491,2012)。trp7在所有家族3成员中是保守的,并且在此位置处的置换以及在另一个家族3抑制型半胱氨酸结肽敬钊毒素-v中的trp5和met6处的置换导致效力丧失,表明在敬钊毒素-v中的位置5、6和7处的疏水性残基对于敬钊毒素-vnav1.7抑制型效力至关重要(国际专利公布2014/165277)。trp5/met6/trp7在原毒素-ii中也是保守的,因此意想不到的是,trp7处的极性置换可以在不损失原毒素-ii活性的情况下进行,具有显著改善的重折叠特性。trp30处的置换显示出同时改善变体肽的nav1.7选择性和重折叠特性,并且是意想不到的,因为单个有利的置换通常仅改善单个参数。表13示出所选生成的原毒素-ii变体的氨基酸序列。表13.使用如实施例3所述的fliprtetra或qpatch,对所选变体对于其对nav1.7的抑制进行表征。表14示出了所获得的ic50值。对于一些变体,记录qpatch结果的某些浓度下的抑制%(原毒素ii的%)。表14.针对各种人nav1.x通道测试所选变体的选择性。表15示出那些实验的结果。使用qpatch测量每个通道的ic50值。表15.如实施例1所述表达并纯化原毒素-ii变体,或通过标准固相合成法合成该变体。将重组或合成肽的收率与野生型原毒素的收率进行比较。表16示出所选原毒素-ii变体的收率显著大于原毒素-ii的收率,表明变体的折叠特性改善。固相合成的规模为0.1mmol。表16.实施例7.鞘内施用后原毒素-ii变体在体内疼痛模型中有效评估所选原毒素-ii变体在鞘内给药后减轻疼痛的功效。在研究中使用肽nv1d2775-oh、nv1d3034和63955918。使用测量急性热疼痛(甩尾和热板)和损伤诱发的疼痛(福尔马林退缩)的动物模型。甩尾测试:将动物放在甩尾装置(ugobasile)上。该装置具有焦点红外线加热区域(直径5毫米)。将动物的尾巴(离远侧端部1/3-1/2处)置于焦点加热区域。调节热源的温度,以在10秒内在用媒介物处理的动物中引发甩尾。使用15秒的截止时间来防止组织损伤,如文献中的标准那样。自动测量热刺激开始和任何回避响应之间的时间,并记录测试组。热板测试:将动物置于4个树脂玻璃壁(高15英寸)围绕的10"×10"金属板上,并保持在48-55℃的温度下。如果动物舔后足、跳跃或发声,则将其从板上移走并记录响应潜伏期。如果动物并未在20-90秒内(截止时间)显示出任何响应,则将其从板上移走以防止任何可能的组织损伤。福尔马林退缩:后足注射福尔马林诱导的疼痛行为(即退缩)使用自动化“退缩响应”测量装置ucsd测量。该装置使用安装在装置底部下方的运动传感器来检测胶合到动物的一个后足上的金属带的任何突然移动。在福尔马林注射前半小时至一小时,使用小滴氰基丙烯酸酯将小金属带附着到一个后足的足底表面,并将动物放置在测试室中以适应环境。金属带的附接并未显示出对动物有刺激。用金属带将福尔马林(2.5%,50μl)经皮下注射到足背。足底注射福尔马林后立即将动物放入定制的圆柱体(25×10×20cm,sandiegoinstrument)中。自动记录足退缩。在急性热疼痛模型中,原毒素-ii变体63955918产生效力和长效镇痛,如在单次鞘内给药后在甩尾测试(图11a和图11b)和热板测试(图11c、图11d)中升高的潜伏期所指示的那样。镇痛的显著性和持续时间是剂量依赖性的。后足福尔马林注射是常用的损伤诱导的疼痛的模型。注射诱导特征性的双相退缩行为,其指示测试动物的疼痛。如图11e所示,用鞘内注射原毒素-ii变体63955918预处理的动物在福尔马林测试中显示较少的退缩,表明损伤诱导的疼痛的抑制。类似地,在单次鞘内给药后,肽nv1d2775-oh和nv1d3034在甩尾、热板和福尔马林测试(图12a、图12b、图12c、图12d、图12e、图13a、图13b、图13c、图13d、图13e)中显示出显著的功效。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1