氰基烯酮类三环二萜类似物及其制备方法和应用与流程
本发明属于医药及其制备和应用技术领域,具体涉及一种氰基烯酮类三环二萜类似物及其制备方法和应用。
背景技术:
结肠癌,也称结直肠癌、直肠癌或肠癌,是发生于结肠或直肠(部分为大肠)部位的消化道恶性肿瘤,它是由于细胞的异常生长进而能够入侵或扩散到身体的其他部位。在全球范围内,结肠癌是发病率和致死率第三的癌症,其致死率高达50%。bmi-1基因在结肠癌细胞中呈高表达状态,不仅对结肠癌肿瘤干细胞具有重要的调节作用,还可以促进结肠癌细胞的增殖,阻碍结肠癌细胞凋亡和衰老。此外,抑制bmi-1基因会降低结肠癌干细胞的自我更新能力,bmi-1已被证明可以作为有效治疗结肠癌的靶点。honda课题组合成了cddo-me,具有较好的抗肿瘤活性,并在很多动物模型中也表现出很好的疗效。现已被fda批准进入治疗实体瘤和淋巴恶性肿瘤i期临床研究;同时,研究发现cddo-me可以下调bmi-1的表达。
johne.dick课题组报道了化合物ptc-209,能够明显下调bmi-1的表达,对人结肠癌细胞hct-116有较好的抑制活性;同时,下调bmi-1的表达造成肿瘤生长的不可逆损伤。因此,靶向bmi-1可以作为治疗结肠癌的新方向。
技术实现要素:
本发明在寻找新型抗结肠癌药物的研究过程中,设计合成了一种具有氰基烯酮结构的三环二萜衍生物,其结构如式(i)所示:
其中,r选自羟基,氨基酸残基,酚基,取代的苯胺基,取代的苄胺基,取代的苯肼基。
其中,所述氨基酸残基为氨基酸中氨基缺少一个氢后形成的残基。
优选地,所述氨基酸残基为天然氨基酸残基,包括:甘氨酸残基、丙氨酸残基、缬氨酸残基、亮氨酸残基、异亮氨酸残基、苯丙氨酸残基、脯氨酸残基、色氨酸残基、丝氨酸残基、酪氨酸残基、半胱氨酸残基、蛋氨酸残基、天冬酰胺残基、谷氨酰胺残基、苏氨酸残基、天冬氨酸残基、谷氨酸残基、赖氨酸残基、精氨酸残基和组氨酸残基。
进一步优选地,所述氨基酸残基包括甘氨酸残基
其中,所述酚基包括苯酚基
其中,所述取代的苯胺基包括吸电子取代的苯胺基和给电子基在邻、间、对位取代的苯胺基,包括三氟甲氧基、乙酮、酰胺基、酯基、氰基、三氟甲基、氨基、卤素取代的苯胺基。
优选地,所述取代的苯胺基包括三氟甲氧基、乙酮、甲酰胺基、甲酯基、氰基、三氟甲基、氨基、卤素取代的苯胺基。
进一步优选地,所述取代的苯胺基包括
其中,所述取代的苄胺基包括三氟甲氧基、乙酮、酰胺基、酯基、氰基、三氟甲基、氨基、卤素取代的苯胺基。
优选地,所述取代的苄胺基包括三氟甲氧基、乙酮、甲酰胺基、甲酯基、氰基、三氟甲基、氨基、卤素取代的苄胺基。
进一步优选地,所述取代的苄胺基包括
其中,所述取代的苯肼基包括三氟甲氧基、乙酮、酰胺基、酯基、氰基、三氟甲基、氨基、卤素取代的苯胺基。
优选地,所述取代的苯肼基包括三氟甲氧基、乙酮、甲酰胺基、甲酯基、氰基、三氟甲基、氨基、卤素取代的苯肼基。
进一步优选地,所述取代的苯肼基包括
本发明还提出了一种氰基烯酮类三环二萜类似物的制备方法,以式(q1)所示的三环二萜衍生物为起始原料,经氧化脱氢反应、酯化反应、碘代反应、氰基取代反应、水解等五步反应,得到化合物q6;所述制备方法如路线(1)所示:
具体地,所述方法包含以下步骤:
(1)氧化脱氢反应
将化合物q1溶于有机溶剂中,加入氧化脱氢反应的试剂,进行氧化脱氢反应,得到化合物q2。
步骤(1)中,所述有机溶剂选自dmso、甲苯、四氢呋喃之任意的一种或多种;优选地,为dmso。
步骤(1)中,所述氧化脱氢反应的试剂为ibx、ddq;优选地,为ibx。
步骤(1)中,所述氧化脱氢反应的试剂的作用为氧化羟基及其α和β位。
步骤(1)中,所述化合物q1与氧化脱氢反应的试剂的摩尔比为1:(2~6);优选地,为1:4。
步骤(1)中,所述氧化脱氢反应的温度为20-85℃;优选地,为85℃。
步骤(1)中,所述氧化脱氢反应的时间为12-16h;优选地,为12h。
(2)酯化反应
将化合物q2溶于有机溶剂中,加入酯化反应所用的试剂,加热回流,得到化合物q3。
步骤(2)中,所述有机溶剂选自甲醇、四氢呋喃和甲醇混合溶剂之任意的一种或多种;优选地,为甲醇。
步骤(2)中,所述酯化反应所用的试剂为浓硫酸、对甲苯磺酸;优选地,为浓硫酸。
步骤(2)中,所述酯化反应所用的试剂的作用为促进酯化反应。
步骤(2)中,所述化合物q2与酯化反应所用的试剂的摩尔比为1:(0.5~2);优选地,为1:1。
步骤(2)中,所述酯化反应的温度为25-70℃;优选地,为70℃。
步骤(2)中,所述酯化反应的时间为2-12h;优选地,为2h。
(3)碘代反应
将化合物q3溶于吡啶与thf中,加入碘代反应所用的试剂、dmap,加热至50℃反应5小时。
步骤(3)中,所述有机溶剂选自吡啶、thf、二氯甲烷、甲苯之任意的一种或多种;优选地,为吡啶与thf的混合溶剂。
步骤(3)中,所述催化剂为dmap。
步骤(3)中,所述碘代反应所用的试剂为i2。
步骤(3)中,所述碘代反应所用的试剂的作用为取代化合物q3的a环羰基α位的烯氢。
步骤(3)中,所述碘代反应所用的催化剂的作用为促进碘代反应。
步骤(3)中,所述化合物q3与碘代反应所用的试剂、催化剂的摩尔比为1:(2~4):(0.2~1);优选地,为1:2:0.2。
步骤(3)中,所述碘代反应的温度为25-50℃;优选地,为50℃。
步骤(3)中,所述碘代反应的时间为5-12h;优选地,为5h。
(4)氰基取代反应
将化合物q4溶于有机溶剂中,加入氰基取代反应所用的试剂,加热进行氰基取代反应,得到化合物q5。
步骤(4)中,所述有机溶剂选自dmf、甲苯之任意的一种或多种;优选地,为dmf。
步骤(4)中,所述氰基取代反应所用的试剂为氰化亚铜(cucn)、氰化钠;优选地,为cucn。
步骤(4)中,所述氰基取代反应所用的试剂的作用为取代a环上的碘。
步骤(4)中,所述化合物q4与氰基取代反应所用的试剂的摩尔比为1:(1~1.5);优选地,为1:1.2。
步骤(4)中,所述氰基取代反应的温度为100-140℃;优选地,为140℃。
步骤(4)中,所述氰基取代反应的时间为1-2h;优选地,为2h。
(5)水解反应
将化合物q5溶于有机溶剂中,加入水解反应所用的试剂,进行水解反应,得到化合物q6。
步骤(5)中,所述有机溶剂选自甲醇、四氢呋喃之任意的一种或多种;优选地,为甲醇。
步骤(5)中,所述水解反应所用的试剂为氢氧化钠、氢氧化钾、碳酸钾;优选地,为氢氧化钠。
步骤(5)中,所述水解反应所用的试剂的作用为促进水解。
步骤(5)中,所述化合物q5与水解反应所用的试剂的摩尔比为1:(1~3);优选地,为1:3。
步骤(5)中,所述水解反应的温度为25-70℃;优选地,为室温25℃。
步骤(5)中,所述水解反应的时间为2-12h;优选地,为12h。
在一个具体实施方式中,所述化合物q6的制备过程为:
本发明还提供了一种氰基烯酮类三环二萜类似物的制备方法,以化合物q6为起始原料,在dcc、dmap催化下,与苯酚缩合,得到如式(q7)所示的氰基烯酮类三环二萜类似物;所述制备方法如路线(2)所示:
其中,所述有机溶剂选自dcm、无水dcm、四氢呋喃之任意的一种或多种;优选地,为无水dcm。
其中,所述dcc、dmap的作用为促进酯化反应。
其中,所述化合物q6与dcc、dmap的摩尔比为1:(1~2):(1.5~4);优选地,为1:2:4。
其中,所述酯化反应的温度为25-40℃;优选地,为室温25℃。
其中,所述酯化反应的时间为2-6h;优选地,为6h。
在一个具体实施方式中,化合物q7的制备过程如下所示:
本发明还提供了一种氰基烯酮类三环二萜类似物的制备方法,以化合物q6为起始原料,在edci、hobt、dmap催化下,与甘氨酸甲酯盐酸盐进行缩合反应,得到化合物q8,化合物q8再与氢氧化钠进行水解反应,得到如式(q9)所示的氰基烯酮类三环二萜类似物;所述制备方法如路线(3)所示:
其中,所述缩合反应中,所述有机溶剂选自dcm、无水dcm、四氢呋喃之任意的一种或多种;优选地,为无水dcm。
其中,所述缩合反应中,所述edci、hobt、dmap的作用为促进酰胺化反应。
其中,所述缩合反应中,所述化合物q6、edci、hobt、dmap及甘氨酸甲酯盐酸盐的摩尔比为1:(1~2):(1~2):(2~4):(1~2);优选地,为1:2:2:4:2。
其中,所述缩合反应的温度为25-40℃;优选地,为室温25℃。
其中,所述缩合反应的时间为6-12h;优选地,为12h。
其中,所述水解反应中,所述有机溶剂选自甲醇、四氢呋喃之任意的一种或多种;优选地,为甲醇。
其中,所述水解反应中,所述氢氧化钠的作用为促进水解。
其中,所述水解反应中,所述化合物q8、氢氧化钠的摩尔比为1:(1~5);优选地,为1:3。
其中,所述水解反应的温度为25-60℃;优选地,为室温25℃。
其中,所述水解反应的时间为2-6h;优选地,为6h。
在本发明的一个具体实施方式中,化合物q9的制备方法包括:将化合物q6,edci(2eq),hobt(2eq),dmap(4eq)及甘氨酸甲酯盐酸盐(2eq)溶于无水dcm中,室温25℃下反应12小时,得到化合物q8;溶于甲醇,加入氢氧化钠(3eq),室温25℃下反应6小时,得到化合物q9。
本发明还提供了一种氰基烯酮类三环二萜类似物的制备方法,以q6为起始原料,在edci、hobt、dmap催化下,与n-boc-对苯二胺进行缩合反应,得到化合物q10,化合物q10与四丁基氟化铵进行脱保护反应,得到如式(q11)所示氰基烯酮类三环二萜类似物;所述制备方法如路线(4)所示:
其中,所述缩合反应中,所述有机溶剂选自dcm、无水dcm、thf之任意的一种或多种;优选地,为无水dcm。
其中,所述缩合反应中,所述edci、hobt、dmap的作用为促进酰胺化反应。
其中,所述缩合反应中,所述n-boc-对苯二胺的作用为与化合物q6的羧基进行酰胺化反应。
其中,所述缩合反应中,所述化合物q6、edci、hobt、dmap及n-boc-对苯二胺的摩尔比为1:(1~2):(1~2):(2~4):(1~2);优选地,为1:2:2:4:2。
其中,所述缩合反应的温度为25-40℃;优选地,为室温25℃。
其中,所述缩合反应的时间为6-12h;优选地,为12h。
其中,所述脱保护反应中,所述有机溶剂选自四氢呋喃、无水四氢呋喃、乙醚、二氯甲烷之任意的一种或多种;优选地,为无水四氢呋喃。
其中,所述脱保护反应中,所述四丁基氟化铵的作用为脱去boc保护基。
其中,所述脱保护反应中,所述化合物q10、四丁基氟化铵的摩尔比为1:(1~5);优选地,为1:5。
其中,所述脱保护反应的温度为0-25℃;优选地,为室温25℃。
其中,所述脱保护反应的时间为6-8h;优选地,为6h。
在一个具体的实施方式中,所述化合物q11的制备方法包括:将化合物q6,edci(2eq),hobt(2eq),dmap(4eq)及n-boc-对苯二胺(2eq)溶于无水dcm中,室温25℃下反应12小时,得到化合物q10,溶于无水四氢呋喃,加入四丁基氟化铵(5eq),室温25℃下反应6小时,得到化合物q11。
本发明还提供了一种氰基烯酮类三环二萜类似物的制备方法,以化合物q6为起始原料,在edci、hobt、dmap催化下,与r'-nh2或其相应的盐酸盐进行酰胺化反应,分别得到如式(ii)所示的氰基烯酮类三环二萜类似物;所述制备方法如路线(5)所示:
其中,r’-nh-基团选自取代的苯胺基,取代的苄胺基,取代的苯肼基。
其中,所述取代的苯胺基包括吸电子取代的苯胺基和给电子基在邻、间、对位取代的苯胺基;包括三氟甲氧基、乙酮、酰胺基、酯基、氰基、三氟甲基、氨基、卤素取代的苯胺基。
优选地,所述取代的苯胺基包括三氟甲氧基、乙酮、甲酰胺基、甲酯基、氰基、三氟甲基、氨基、卤素取代的苯胺基。
进一步优选地,所述取代的苯胺基包括
其中,所述取代的苄胺基包括三氟甲氧基、乙酮、酰胺基、酯基、氰基、三氟甲基、氨基、卤素取代的苯胺基。
优选地,所述取代的苄胺基包括三氟甲氧基、乙酮、甲酰胺基、甲酯基、氰基、三氟甲基、氨基、卤素取代的苄胺基。
进一步优选地,所述取代的苄胺基包括
其中,所述取代的苯肼基三氟甲氧基、乙酮、酰胺基、酯基、氰基、三氟甲基、氨基、卤素取代的苯胺基。
优选地,所述取代的苯肼基包括三氟甲氧基、乙酮、甲酰胺基、甲酯基、氰基、三氟甲基、氨基、卤素取代的苯肼基。
进一步优选地,所述取代的苯肼基包括
优选地,本发明所述r’-nh2选自:对氨基苯乙酮
其中,所述有机溶剂选自dcm、无水dcm、四氢呋喃、dmf之任意的一种或多种;优选地,为无水dcm。
其中,所述edci、hobt、dmap的作用为促进酰胺化反应。
其中,所述化合物q6、edci、hobt、dmap的摩尔比为1:(1~2):(1~2):(2~4);优选地,为1:2:2:4。
其中,所述酰胺化反应的温度为25-60℃;优选地,为室温25℃。
其中,所述酰胺化反应的时间为6-12h;优选地,为12h。
在一个具体实施方式中,所述氰基烯酮类三环二萜类似物的制备方法包括:将化合物q6,edci(2eq),hobt(2eq),dmap(4eq)及r’-nh2或其相应的盐酸盐(2eq)溶于无水dcm中,室温25℃下反应12小时,得到化合物q12、q13、q14、q15、q16、q17、q18、q19、q20、q21、q22、q23。
本发明制备方法中,上述反应通过薄板层析法跟踪测定反应的进度,反应完毕后采用的后处理方法通常包括浓缩、萃取、柱层析分离等,最终产物以核磁共振谱来验证。
本发明还提供了式(i)所示的氰基烯酮类三环二萜类似物在制备bmi-1基因表达的抑制剂中的应用。
本发明还提供了式(i)所示的氰基烯酮类三环二萜类似物在制备抗肿瘤药物中的应用。
本发明还提供了式(i)所示的氰基烯酮类三环二萜类似物在制备抑制肿瘤的增殖、生长、侵袭、迁移中的应用。其中,所示肿瘤包括结肠癌、乳腺癌;优选地,为结肠癌;其中,所述氰基烯酮类三环二萜类似物用于hct116、ht29、hct8和ct26结肠癌细胞具有显著的有益效果。
本发明的有益效果在于,本发明通过对c环羧基上进行结构修饰改造,将羧基与不同的胺或酚缩合,或通过酰胺化反应、酯化反应脱保护等反应合成了一系列新型氰基烯酮类三环二萜类似物。本发明制备方法的反应条件温和、所用试剂便宜易得、合成路线较短、合成方法简便。制备得到的氰基烯酮类三环二萜类似物能显著下调bmi-1基因的表达,可以用于制备抗癌药物,对结肠癌细胞具有较好的抑制活性,具有良好的应用前景。
附图说明
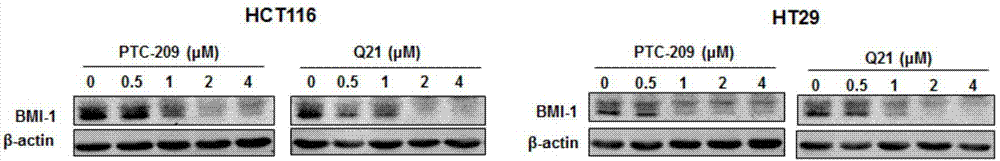
图1为实施例7中化合物q12、q19、q20、q21、q22、q23、q24、q25对bmi-1蛋白表达的影响的westernblot图。
图2为实施例7中化合物q21能够剂量依赖地下调bmi-1蛋白水平的westernblot图。
图3为实施例8中化合物q21能抑制hct116,ht29和hct8三种结肠癌细胞的克隆形成。
图4为实施例8中化合物q21能抑制ct26,hct116和hct8三种结肠癌细胞的迁移。
具体实施方式
结合以下具体实施例和附图,对本发明作进一步的详细说明。实施本发明的过程、条件、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。
下述实施例中化合物结构由核磁共振仪测定;试剂主要由上海国药化学试剂公司提供了;产品纯化主要通过柱色谱,硅胶(200‐300目)由青岛海洋化工厂生产。
实施例1化合物q6的制备
于250ml单口烧瓶中加入化合物q1(10g,31.4mmol)、ibx(35g,125.6mmol)、dmso(60ml),85℃反应12h。tlc检测原料反应完全,冷却至室温后加入水(100ml)、乙酸乙酯(50ml),室温搅拌30min,抽滤除去固体并用ea(30ml×2)淋洗,水相用ea(30ml×3)萃取,合并有机相,依次用水(50ml)、饱和nacl溶液(50ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=1:1)得到化合物q2(8.9g白色固体,产率90%)。1hnmr(400mhz,cdcl3)δ10.73(s,1h),7.94(s,1h),7.50(d,j=10.4hz,1h),7.04(s,1h),6.08(d,j=10.4hz,1h),4.11(s,3h),3.04(dd,j=17.2,6.4hz,1h),2.96–2.87(m,1h),2.14(dd,j=12.0,2.4hz,1h),2.00–1.86(m,2h),1.43(s,3h),1.22(s,3h),1.20(s,3h).
于250ml单口烧瓶中加入化合物q2(8.9g,28.3mmol),溶于meoh(50ml),逐滴加入浓硫酸(1.5ml,28.3mmol),加热至回流2h,tlc检测反应完全后,减压除去甲醇,加入水(30ml),用ea(30ml×3)萃取水相。合并有机相,分别用水(50ml)、饱和nacl溶液(50ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=3:1)得到化合物q3(8.8g白色固体,产率95%).1hnmr(400mhz,cdcl3)δ7.56(s,1h),7.54(d,j=10.0hz,1h),6.97(s,1h),6.06(d,j=10.0hz,1h),3.92(s,3h),3.87(s,3h),2.98(dd,j=16.8,6.4hz,1h),2.91–2.83(m,1h),2.14(dd,j=12.0,2.8hz,1h),1.97–1.81(m,2h),1.41(s,3h),1.22(s,3h),1.19(s,3h).
于250ml单口烧瓶中加入化合物q3(8.8g,26.7mmol)、i2(13.6g,53.5mmol)、dmap(653mg,5.34mmol),加入吡啶(5ml)、thf(50ml),加热至50℃反应5h后,tlc检测反应完全。加入饱和naso3溶液(50ml),用ea(30ml×3)萃取水相。合并有机相,分别用水(50ml)、饱和nacl溶液(50ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=3:1)得到化合物q4(11.2g白色固体,产率92%)。1hnmr(400mhz,cdcl3)δ7.56(s,1h),7.54(d,j=10.0hz,1h),6.97(s,1h),6.06(d,j=10.0hz,1h),3.92(s,3h),3.87(s,3h),2.98(dd,j=16.8,6.4hz,1h),2.91–2.83(m,1h),2.14(dd,j=12.0,2.8hz,1h),1.97–1.81(m,2h),1.41(s,3h),1.22(s,3h),1.19(s,3h).
于250单口烧瓶中加入化合物q4(11.2g,24.4mmol)和cucn(2.6g,29.3mmol),加入dmf(50ml),加热至140℃搅拌2h后,tlc检测反应完全。加入饱和nh4cl(50ml)搅拌,用ea(50ml×3)萃取水相,合并有机相,分别用水(50ml×2)、饱和nacl溶液(50ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=3:1)得到化合物q5(7.7g白色固体,产率88%)。1hnmr(400mhz,cdcl3)δ8.29(s,1h),7.58(s,1h),6.89(s,1h),3.95(s,3h),3.89(s,3h),2.98(dd,j=16.8,6.0hz,1h),2.93–2.82(m,1h),2.19(d,j=12.0hz,1h),1.99–1.82(m,2h),1.48(s,3h),1.28(s,3h),1.24(s,3h).
于250单口烧瓶中加入化合物q5(7.6g,21.5mmol),溶于甲醇(60ml),加入naoh(1.72g,43.0mmol),室温搅拌10h后,tlc检测反应完全,减压除去甲醇,加入2mhcl至酸性,用ea(50ml×3)萃取水相,合并有机相,分别用水(50ml)、饱和nacl溶液(50ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=1:1)得到化合物q6(7.0g白色固体,产率95%)。1hnmr(400mhz,cdcl3)δ10.66(s,1h),8.31(s,1h),7.96(s,1h),7.01(s,1h),4.15(s,3h),3.05(dd,j=16.0,6.3hz,1h),2.96–2.87(m,1h),2.20(dd,j=16.0,4.0hz,1h),2.02–1.85(m,2h),1.50(s,3h),1.29(s,3h),1.25(s,3h).
实施例2化合物q7的制备
于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、dcc(121.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及苯酚(55.5mg,0.59mmol)置于单颈瓶,加入无水dcm(10ml),室温搅拌6h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析,得到化合物q7(109mg白色固体,89%)。1hnmr(400mhz,cdcl3)δ8.31(s,1h),7.80(s,1h),7.41(d,j=6.4hz,2h),7.21(d,j=6.4hz,2h),6.95(s,1h),3.99(s,3h),3.48(d,j=4.0hz,1h),3.10–2.90(m,2h),2.23(d,j=12.0hz,1h),1.99–1.92(m,2h),1.52(s,3h),1.30(s,3h),1.26(s,3h).
实施例3化合物q9的制备
于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及甘氨酸甲酯盐酸盐(70.1mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩得到粗产品q8,将q8溶于甲醇(10ml),加入氢氧化钠(34.8mg,0.87mmol),室温搅拌6h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=1:1)得到化合物q9(98mg白色固体,产率84%)。1hnmr(400mhz,cdcl3)δ8.54(t,j=4.8hz,1h),8.30(s,1h),7.97(s,1h),6.92(s,1h),4.29(dd,j=4.8,3.2hz,2h),4.06(s,3h),3.03(dd,j=16.0,4.0hz,1h),2.94–2.85(m,1h),2.19(dd,j=12.0,2.4hz,1h),2.00–1.84(m,2h),1.49(s,3h),1.28(s,3h),1.24(s,3h).
实施例4化合物q11的制备
于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及n-boc-对苯二胺(122.8mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩得到化合物q10粗产品。将化合物q10,溶于无水四氢呋喃,加入四丁基氟化铵(379mg,1.45mmol),室温25℃下反应6小时,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩硅胶柱层析得到化合物q11(101mg白色固体,80%)。1hnmr(400mhz,cdcl3)δ9.55(s,1h),8.31(s,1h),8.05(s,1h),7.45(d,j=8.0hz,2h),6.93(s,1h),6.70(d,j=8.0hz,2h),4.09(s,3h),3.63(s,2h),3.07(dd,j=16.0,4.0hz,1h),2.96–2.89(m,1h),2.22(d,j=12.0hz,1h),2.00–1.86(m,2h),1.50(s,3h),1.29(s,3h),1.25(s,3h).
实施例5化合物q12~q23的制备
化合物q12的制备:于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及对氨基苯乙酮(79.7mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=2:1),得到化合物q12(98mg白色固体,73%)。1hnmr(400mhz,cdcl3)δ9.99(s,1h),8.32(s,1h),8.06(s,1h),7.98(d,j=8.0hz,2h),7.77(d,j=8.0hz,2h),6.97(s,1h),4.15(s,3h),3.07(dd,j=16.0,4.0hz,1h),2.99–2.90(m,1h),2.60(s,3h),2.20(dd,j=12.0,2.0hz,1h),2.02–1.88(m,2h),1.52(s,3h),1.30(s,3h),1.26(s,3h).
化合物q13的制备:于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及对氨基苯甲酰胺(80.3mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(dcm:meoh=200:1),得到化合物q13(124mg白色固体,92%)。1hnmr(400mhz,cdcl3)δ9.96(s,1h),8.31(s,1h),8.05(d,j=8.0hz,3h),7.75(d,j=8.0hz,2h),6.96(s,1h),4.14(s,2h),3.91(s,3h),3.07(dd,j=16.0,4.0hz,1h),3.01–2.90(m,1h),2.22(dd,j=12.0,2.0hz,1h),2.02–1.86(m,2h),1.51(s,3h),1.30(s,3h),1.26(s,3h).
化合物q14的制备:于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及对氨基苯甲酸甲酯(89.2mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=2:1),得到化合物q14(125mg白色固体,90%)。1hnmr(400mhz,cdcl3)δ9.96(s,1h),8.31(s,1h),8.06(d,j=8.0hz,3h),7.76(d,j=8.0hz,2h),6.96(s,1h),4.14(s,3h),3.91(s,3h),3.10–3.04(m,1h),2.98–2.89m,1h),2.23–2.20(m,1h),2.02–1.90(m,2h),1.51(s,3h),1.30(s,3h),1.26(s,3h).
化合物q15的制备:于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及对氨基苯腈(69.7mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=2:1),得到化合物q15(117mg白色固体,90%)。1hnmr(400mhz,cdcl3)δ9.99(s,1h),8.31(s,1h),8.04(s,1h),7.80(d,j=8.0hz,2h),7.65(d,j=8.0hz,2h),6.98(s,1h),4.15(s,3h),3.07(dd,j=16.0,4.0hz,1h),2.98–2.90(m,1h),2.21(d,j=12.0hz,1h),2.04–1.88(m,2h),1.51(s,3h),1.30(s,3h),1.26(s,3h).
化合物q16的制备:于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及对氯苯胺(75.3mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=2:1),得到化合物q16(122mg白色固体,92%)。1hnmr(400mhz,cdcl3)δ9.77(s,1h),8.31(s,1h),8.05(s,1h),7.62(d,j=8.0hz,2h),7.32(d,j=8.0hz,2h),6.95(s,1h),4.12(s,3h),3.06(dd,j=16.0,4.0hz,1h),2.98–2.89(m,1h),2.21(dd,j=12.0,2.0hz,1h),2.02–1.87(m,2h),1.51(s,3h),1.30(s,3h),1.25(s,3h).
化合物q17的制备:于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及对溴苯胺(101.5mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=2:1),得到化合物q17(134mg白色固体,92%)。1hnmr(400mhz,cdcl3)δ9.77(s,1h),8.30(s,1h),8.05(s,1h),7.59(d,j=8.0hz,2h),7.48(d,j=8.0hz,2h),6.95(s,1h),4.12(s,3h),3.07(dd,j=16.9,5.7hz,1h),3.01–2.87(m,1h),2.21(dd,j=12.0,2.0hz,1h),2.02–1.88(m,2h),1.51(s,3h),1.30(s,3h),1.26(s,3h).
化合物q18的制备:于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及对三氟甲基苯胺(95mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=4:1),得到化合物q18(126.5mg白色固体,89%)。1hnmr(400mhz,cdcl3)δ9.93(s,1h),8.31(s,1h),8.06(s,1h),7.79(d,j=8.0hz,2h),7.63(d,j=8.0hz,2h),6.97(s,1h),4.15(s,3h),3.07(dd,j=16,5.8hz,1h),2.99–2.90(m,1h),2.27–2.18(m,1h),2.04–1.87(m,2h),1.52(s,3h),1.30(s,3h),1.26(s,3h).
化合物q19的制备:于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及对三氟甲氧基苯胺(104.5mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=4:1),得到化合物q19(133mg白色固体,92%)。1hnmr(400mhz,cdcl3)δ9.81(s,1h),8.32(s,1h),8.06(s,1h),7.71(d,j=8.0hz,2h),7.23(d,j=8.0hz,2h),6.96(s,1h),4.13(s,3h),3.10–3.04(m,1h),2.99–2.90(m,1h),2.24–2.20(m,1h),2.02–1.88(m,2h),1.52(s,3h),1.30(s,3h),1.26(s,3h).
化合物q20的制备:于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及间三氟甲氧基苯胺(104.5mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=2:1),得到化合物q20(120mg白色固体,82%)。1hnmr(400mhz,cdcl3)δ9.85(s,1h),8.31(s,1h),8.06(s,1h),7.76(s,1h),7.48(d,j=8.0hz,1h),7.36(t,j=8.0hz,1h),6.99(d,j=8.0hz,1h),6.96(s,1h),4.14(s,3h),3.07(dd,j=16.0,4.0hz,1h),2.98–2.89(m,1h),2.22(dd,j=12.0,2.0hz,1h),2.02–1.85m,2h),1.51(s,3h),1.30(s,3h),1.26(s,3h).
化合物q21的制备:于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及邻三氟甲氧基苯胺(104.5mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=2:1),得到化合物q21(125mg白色固体,85%)。1hnmr(400mhz,cdcl3)δ10.41(s,1h),8.71(dd,j=8.0,1.2hz,1h),8.33(s,1h),8.09(s,1h),7.35–7.29(m,2h),7.14–7.10(m,1h),6.98(s,1h),4.13(s,3h),3.07(dd,j=16.0,4.0hz,1h),2.99–2.90(m,1h),2.22(dd,j=12.0,2.4hz,1h),2.02–1.86(m,2h),1.52(s,3h),1.30(s,3h),1.26(s,3h).
化合物q22的制备:于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及对三氟甲氧基苄胺(104mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=2:1),得到化合物q22(134mg白色固体,89%)。1hnmr(400mhz,cdcl3)δ8.33(s,1h),8.21(t,j=4.0hz,1h),8.01(s,1h),7.37(d,j=8.0hz,2h),7.18(d,j=8.0hz,2h),6.92(s,1h),4.72–4.61(m,2h),4.00(s,3h),3.06–3.01(m,1h),2.95–2.86(m,1h),2.19(dd,j=12.0,4.0hz,1h),1.99–1.82.(m,2h),1.49(s,3h),1.29(s,3h),1.24(s,3h).
化合物q23的制备:于100ml单口烧瓶中加入化合物q6(100mg,0.29mmol)、edci(113.1mg,0.59mmol),hobt(79.7mg,0.59mmol),dmap(144.2mg,1.18mmol)及对三氟甲氧基苯肼盐酸盐(137.2mg,0.59mmol),加入无水dcm(10ml),室温搅拌12h后,tlc检测原料反应完全,加20ml水,用dcm(20ml×3)萃取水相,合并有机相,分别用水(30ml)、饱和nacl溶液(30ml)洗涤,无水naso4干燥,减压浓缩后硅胶柱层析(pe:ea=2:1),得到化合物q23(124mg浅黄色固体,82%)。1hnmr(400mhz,cdcl3)δ9.40(d,j=4.0hz,1h),8.31(s,1h),7.97(s,1h),7.10(d,j=8.0hz,2h),6.96(s,1h),6.91–6.89(m,2h),6.46(d,j=4.0hz,1h),4.11(s,3h),3.06–3.01(m,1h),2.95–2.86(m,1h),2.20–2.18m,1h),2.01–1.85(m,2h),1.51(s,3h),1.30(s,3h),1.25(s,3h).
实施例6氰基烯酮类三环二萜类似物抗结肠癌细胞增殖实验
方法步骤:
1.细胞的培养
本发明中所用结肠癌细胞hct116,ht29,hct8和人成纤维细胞haf购自atcc细胞库。细胞培养于37℃恒温培养箱(湿度95%,co2浓度5%)中。hct116,ht29和hct8培养在含10%胎牛血清(gemini)的rpmi-1640(gibco)中,haf培养在含10%胎牛血清(gemini)和1%谷氨酰胺的dmem(gibco)中。
2.srb法对结肠癌细胞进行抗增殖活性测试。
磺酰罗丹明b(srb)是一种粉红色阴离子染料,易溶于水,在酸性条件下可特异性地与细胞内组成蛋白质的碱性氨基酸结合,其在515nm波长处产生吸收峰。在一定读值范围内,吸光值与细胞量成线性正相关,故可用作细胞数的定量检测。采用srb法在结肠癌细胞株hct-116和人成纤维细胞株haf中检测化合物对相应细胞增殖的抑制率。计算半数抑制浓度(ic50),并与阳性对照(ptc-209)进行比较。
各细胞以5×103个/孔密度接种到96孔板,经过24小时培养后,实验组给予不同浓度的化合物处理72小时,对照组加入培养基培养。药物作用72小时后,取出培养板,每孔加入25μl50%tca溶液固定细胞,4℃放置1小时以上。取出固定后的培养板,用水洗涤5遍,自然晾干或吹风机吹干后,每孔加入50μl0.4%srb溶液,染色10分钟,弃去染色液,1%冰醋酸洗涤5遍并自然晾干或吹风机吹干。用100μltris-base碱液(10mm)溶解与细胞蛋白结合的染料,采用酶标仪515nm处测定光吸收值。实验重复三次,ic50用graphpadprism软件计算得到。
各测试氰基烯酮类三环二萜类似物对结肠癌细胞hct-116的半数抑制浓度(ic50)见表1。表2为氰基烯酮类三环二萜类似物对结肠癌细胞(hct-116、ht-29、hct-8)、人成纤维细胞haf的抑制增殖活性及选择性指数si。
表1氰基烯酮类三环二萜类似物对结肠癌细胞hct-116增殖的半数抑制浓度
表1为氰基烯酮类三环二萜类似物对结肠癌细胞hct-116增殖的抑制活性,由表1可知,化合物q18、q19、q20、q21对hct-116的抑制效果远好于母体化合物q6,且好于阳性对照ptc-209。
表2氰基烯酮类三环二萜类似物对结肠癌细胞、人成纤维细胞增殖的半数抑制浓度及选择性指数si
选择性指数si=haf/平均值
由表2可知,相比母体结构发生改变的化合物q24,q25,化合物q19、q20、q21、q22、q23抑制结肠癌细胞(hct-116、ht-29、hct-8)增殖的活性均好于化合物q24、q25。化合物q21对结肠癌细胞株表现出较好的选择性,选择性指数(si)为8.36,阳性参照物ptc-209选择性指数(si)仅为1.79。
其中q24、q25结构为:
实施例7化合物对bmi-1蛋白表达的影响
方法步骤:
免疫印迹(westernblot)实验:
结肠癌细胞hct116经2μm化合物处理8小时后,裂解细胞提取蛋白,蛋白经煮沸变性后用聚丙烯酰胺凝胶page电泳分离蛋白质样品,然后转移到硝酸纤维素薄膜上,用5%bsa封闭1小时,之后首先用bmi-1抗体及β-actin的抗体(一抗)4℃孵育过夜,再用带有荧光标记的二抗孵育1小时,最后用扫膜仪odyssey检测蛋白的表达水平。
结果如图1所示,利用免疫印迹实验证明,相比化合物q24、q25,化合物q12、q19、q20、q21、q22、q23能够显著抑制bmi-1蛋白的表达,其中化合物q12和q21对bmi-1蛋白的抑制效果比阳性化合物ptc209和cddo-me更显著,说明本发明化合物更有效地下调了bmi-1蛋白的表达。
选取化合物q21作进一步研究,设置浓度梯度进行免疫印迹实验,结果如图2所示,化合物q21在1μm时能明显抑制bmi-1蛋白的表达,并且能够剂量依赖性地下调bmi-1蛋白水平。
实施例8化合物抑制结肠癌细胞的增殖和迁移
方法步骤:
(1)克隆形成实验
克隆形成实验是检测癌细胞体外增殖能力的有效方法。对处于对数生长期的结肠癌细胞进行消化处理,以每孔2×103个细胞均匀接种于六孔板中。待细胞贴壁后加入含有化合物的完全培养基。培养一周后,用吸泵吸掉原来的培养基,磷酸盐缓冲液清洗3次,4%多聚甲醛溶液固定细胞20min,2‰结晶紫染液将细胞染色5min后,用自来水洗掉未结合的结晶紫染液,室温自然干燥。显微镜下拍照,计算细胞克隆形成数量。
实验结果如图3所示,左侧为化合物q21对结肠癌细胞株hct116,ht29和hct8的抑制生长效果图,右侧为相应的统计图。化合物q21在0.2μm下即能显著抑制hct116,ht29和hct8三种结肠癌细胞的克隆形成;在0.4μm时完全抑制三株结肠癌细胞的生长,说明化合物q21具有很好的抗癌效果。
(2)transwell小室迁移实验
消化并计数处于对数生长期的结肠癌细胞,细胞重悬在无血清并溶有不同浓度(0.5μm,1μm)化合物q21的基础培养基(如型号为rpmi-1640的基础培养基)中,细胞以8×104个/孔(200μl)接种至transwell小室的上室中,对照组加入等量的dmso。下室中则加入600μl含有对应浓度化合物的完全培养基。置于细胞培养箱中培养12h至24h。取出transwell小室用多聚甲醛固定小室细胞,20min后,2‰结晶紫染液将细胞染色处理5min,清洗小室,将未结合的结晶紫洗掉,用棉签轻轻擦拭transwell小室的上侧,将未迁移至下侧的细胞擦掉。自然干燥,显微镜下拍照,统计多个视野的细胞数目。细胞迁移率(%)=加药物细胞迁移数/对照组细胞迁移数×100%。
肿瘤转移是肿瘤发展的重要环节。实验结果如图4所示,左侧为化合物q21对结肠癌细胞ct26,hct116和hct8的抑制迁移效果图,右侧为相应的统计图。与空白对照组相比,本发明化合物q21对三株结肠癌细胞迁移的半数抑制浓度大约在1μm左右,并且以剂量依赖性的方式抑制结肠癌细胞ct26,hct116和hct8的迁移。
本发明的保护内容不局限于以上实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。
- 还没有人留言评论。精彩留言会获得点赞!