一种基于互穿网络结构的CO2响应型聚合物微球及其制备方法与流程
本发明属于智能高分子微球领域,涉及一种co2响应型聚合物微球及其制备方法。
背景技术:
智能高分子微球是一种能够感知外界环境变化,并通过自我响应调整而实现性能及尺寸变化的新材料。co2作为一种温室气体,不仅廉价易得,而且也是一种生物细胞代谢产物,具有很好的生物相容性和膜渗透性,将其作为刺激响应因子不仅操作简单,成本低廉,而且不会给体系带来新的污染,在药物控释、生物传感、能源环保、石油开采等领域都有具有潜在应用价值。目前co2气体刺激响应聚合物微球的制备主要包括两种途径:第一种是先制备微球,然后将co2响应单体在微球表面进行接枝共聚;第二种是通过自由基均聚或共聚方式直接制备co2响应型微球。第二种途径制备过程简便,更符合聚合物合成工艺的实际要求,有利于规模化生产,但存在因微球粘结抑制其响应性能的缺陷。因此,研究一种新的合成路线来制备具有优良分散性及co2响应性能,且co2响应性能可控的智能微球具有重要意义。
技术实现要素:
本发明的目的在于针对现有技术的不足,提供一种基于互穿网络结构的co2响应型聚合物微球及其制备方法,以提高co2响应型聚合物微球的分散性和co2响应性,且使co2响应性可控。
本发明所述基于互穿网络结构的co2响应型聚合物微球的制备方法,步骤如下:
(1)将10~15质量份化合物a与0.02~0.8质量份交联剂溶于去离子水中得到溶液ⅰ,将0.05~0.5质量份引发剂溶于去离子水中得到溶液ⅱ,在室温下分别向两种溶液中持续通入氮气以去除氧气;将0.2~1.0质量份乳化剂溶于有机溶剂中得到乳化剂溶液;在n2保护下将除氧后的溶液ⅰ和溶液ⅱ同步滴加到乳化剂溶液中,溶液ⅰ和溶液ⅱ滴加完后,在n2保护、40~80℃条件下搅拌反应1~7h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,得到骨架聚合物微球;
所述化合物a的结构式为:
上述结构式中,r1为h、-ch2ch3或-ch(ch3)2;
(2)将步骤(1)所得骨架聚合物微球0.5~2.0质量份与1.0~5.0质量份化合物b、0.01~0.05质量份交联剂、0.02~0.05质量份引发剂和去离子水混合后,于常温下溶胀0.5~5h得到溶胀混合物,将0.8~2.0质量份乳化剂溶于有机溶剂中得到乳化剂溶液,将溶胀混合物与乳化剂溶液混合,在n2保护、40~80℃条件下搅拌反应1~15h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得具有互穿网络结构的co2响应型聚合物微球;
所述化合物b的结构式为
结构式中,r2为-ch3或-ch2ch3。
上述方法中,所述乳化剂为span-60、span-65、span-80、span-85、tween-40、tween-60、tween-80、tween-85中的一种。
上述方法中,所述交联剂为甲撑双丙烯酰胺、双丙烯酸乙二醇酯、双丙烯酰胱胺中的一种。
上述方法中,所述引发剂为水溶性引发剂过硫酸铵(aps)、过硫酸钾(k2s2o4)、偶氮二氰基戊酸(acva)、偶氮二异丁基脒盐酸盐(aiba)、偶氮二咪唑啉基丙烷二盐酸盐(aibi)中的一种。
上述方法中,所述乳化剂溶液的有机溶剂为环己烷、正己烷、煤油、液体石蜡油中的一种。
上述方法中,步骤(1)和步骤(2)中所述去离子水和有机溶剂的用量均以能使相应的加入其中的溶质溶解为限。
上述方法中,选用交联剂为甲撑双丙烯酰胺时,化学反应式为:
本发明还提供了上述方法制备的基于互穿网络结构的co2响应型聚合物微球。
与现有技术相比,本发明具有以下有益效果:
1、本发明所述co2响应型聚合物微球具有互穿网络结构,微球之间无粘结,分散性好。
2、本发明所述co2响应型聚合物微球在交替通入co2和n2条件下,能实现微球体系的ph值转变以及微球体积的可逆变化,表明其具有优良的co2响应性能。
3、本发明所述方法通过改变骨架聚合物微球交联度,以及改变溶胀过程中溶胀单体(化合物b)的浓度,可制备不同co2响应膨胀率的智能微球,实现响应膨胀性的可控刺激。
4、本发明所述方法合成co2响应型聚合物微球的反应条件温和,产率高,操作简单,有利于规模生产。
附图说明
图1为对实施例1~5制得的co2响应型聚合物微球的扫描电镜图。
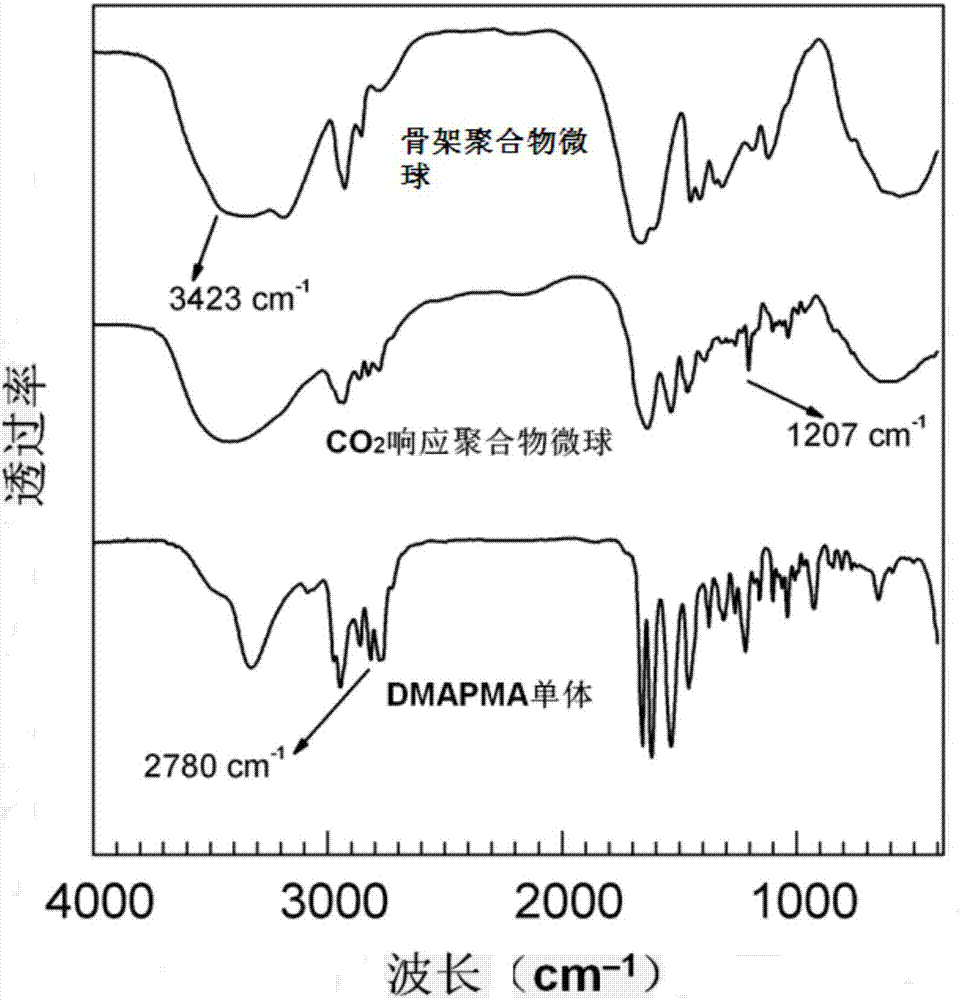
图2是实施例3制得的co2响应型聚合物微球的红外谱图。
图3为实施例3制得的co2响应型聚合物微球的响应前后微球体积及透明度变化的显微照片。
图4为实施例1~5制得的co2响应型聚合物微球在co2响应前后的尺寸及水分散体系酸碱度变化。
图5为实施例1~3中聚丙烯酰胺微球(骨架聚合物微球)的交联度对制得的互穿网络微球co2响应型膨胀率的影响。
图6为实施例3~5中化合物dmapma(溶胀单体)的浓度对制得的聚合物微球的co2响应膨胀率的影响。
具体实施方式
下面通过实施例对本发明所述基于互穿网络结构的co2响应型聚合物微球及其制备方法做进一步说明。
以下实施例中,去离子水和环己烷作为溶剂时的用量均以满足相应的溶质溶解即可。
实施例1
(1)将丙烯酰胺12.30g与交联剂甲撑双丙烯酰胺0.61g(交联度为4.9wt%)溶于去离子水中得到溶液ⅰ,将引发剂aibi0.1g溶于去离子水中得到溶液ⅱ,在室温下向两种溶液中通入氮气除氧0.5h,将乳化剂span-600.5g溶于环己烷中得到乳化剂溶液,在n2保护下将除氧后的溶液ⅰ和溶液ⅱ同步滴加到乳化剂溶液中,溶液ⅰ和溶液ⅱ滴加完后,在n2保护、60℃下搅拌反应6h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得骨架聚合物微球(聚丙烯酰胺微球);
(2)将1.05g步骤(1)所得骨架聚合物微球与2.8gdmapma(二甲氨基丙基甲基丙烯酰胺)、交联剂甲撑双丙烯酰胺0.03g、引发剂acva0.03g和27ml去离子水混合后(dmapma浓度为0.10g·ml-1),于室温下溶胀3h得到溶胀混合物,将乳化剂span-800.8g溶于环己烷中得到乳化剂溶液,将溶胀混合物与乳化剂溶液混合均匀,在n2保护、70℃条件下搅拌反应12h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得具有互穿网络结构的co2响应型聚合物微球,其扫描电镜观察结果如图1a所示,从图中可以看出,所获得的互穿网络微球为表面光滑的球体,并且分散性好。
实施例2
(1)将丙烯酰胺12.30g与交联剂甲撑双丙烯酰胺0.025g(交联度为0.2wt%),溶于去离子水中得到溶液ⅰ,将引发剂aibi0.1g溶于去离子水中得到溶液ⅱ,在室温下向两种溶液中通入氮气除氧0.5h,将乳化剂span-600.5g溶于环己烷中得到乳化剂溶液,在n2保护下将除氧后的溶液ⅰ和溶液ⅱ同步滴加到乳化剂溶液中,溶液ⅰ和溶液ⅱ滴加完后,在n2保护、60℃下搅拌反应6h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得骨架聚合物微球(聚丙烯酰胺微球);
(2)将1.05g步骤(1)所得骨架聚合物微球与2.8gdmapma、交联剂甲撑双丙烯酰胺0.03g、引发剂acva0.03g和27ml去离子水混合后(dmapma浓度为0.10g·ml-1),于室温下溶胀3h得到溶胀混合物,将乳化剂span-800.8g溶于环己烷中得到乳化剂溶液,将溶胀混合物与乳化剂溶液混合均匀,在n2保护、70℃条件下搅拌反应12h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得具有互穿网络结构的co2响应型聚合物微球,其扫描电镜观察结果如图1b所示,从图中可知所制备的互穿网络微球表面会附着少许膜状物质,但仍然具有优良的分散性,没有粘结。分析原因为:溶胀过程中太低的交联度导致吸收进过多的单体,以至于聚丙烯酰胺种子微球所提供的氢键不足以稳定住如此多单体,单体易向种子微球表面迁移。当升温至反应温度时,易发生单体在聚丙烯酰胺微球表面聚合,形成最终的微球表面附着薄膜的形貌。
实施例3
(1)将丙烯酰胺12.30g与交联剂甲撑双丙烯酰胺0.12g(交联度为0.97wt%),溶于去离子水中得到溶液ⅰ,将引发剂aibi0.1g溶于去离子水中得到溶液ⅱ,在室温下向两种溶液中通入氮气除氧0.5h,将乳化剂span-600.5g溶于环己烷中得到乳化剂溶液,在n2保护下将除氧后的溶液ⅰ和溶液ⅱ同步滴加到乳化剂溶液中,溶液ⅰ和溶液ⅱ滴加完后,在n2保护、60℃下搅拌反应6h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得骨架聚合物微球(聚丙烯酰胺微球),红外谱图见图2;
(2)将步骤(1)所得骨架聚合物微球1.05g与2.8gdmapma(红外谱图见图2)、交联剂甲撑双丙烯酰胺0.03g、引发剂acva0.03g和27ml去离子水混合后(dmapma浓度为0.10g·ml-1),于室温下溶胀3h得到溶胀混合物,将乳化剂span-801.0g溶于环己烷中得到乳化剂溶液,将溶胀混合物与乳化剂溶液混合均匀,在n2保护、70℃条件下搅拌反应12h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得具有互穿网络结构的co2响应型聚合物微球(红外谱图见图2),其扫描电镜观察结果如图1c所示,从图中可知,得到的互穿网络结构微球具有光滑的球形结构,并且具有优良的分散性。
实施例4
(1)将丙烯酰胺12.30g与交联剂甲撑双丙烯酰胺0.12g(交联度为0.97wt%)溶于去离子水中得到溶液ⅰ,将引发剂aibi0.1g溶于去离子水中得到溶液ⅱ,在室温下向两种溶液中通入氮气除氧0.5h,将乳化剂span-600.5g溶于环己烷中得到乳化剂溶液,在n2保护下将除氧后的溶液ⅰ和溶液ⅱ同步滴加到乳化剂溶液中,溶液ⅰ和溶液ⅱ滴加完后,在n2保护、60℃下搅拌反应6h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得骨架聚合物微球(聚丙烯酰胺微球);
(2)所步骤(1)所得骨架聚合物微球1.05g与2.8gdmapma、交联剂甲撑双丙烯酰胺0.03g、引发剂acva0.03g和36ml去离子水混合后(dmapma浓度为0.078g·ml-1),于室温下溶胀3h得到溶胀混合物,将乳化剂span-80溶于环己烷中得到乳化剂溶液,将溶胀混合物与乳化剂溶液混合均匀,在n2保护、70℃条件下搅拌反应12h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得具有互穿网络结构的co2响应型聚合物微球,其扫描电镜观察结果如图1d所示,从图中可知,最终得到的互穿网络结构微球具有光滑的球形结构,并且具有很好的分散性。
实施例5
(1)将丙烯酰胺12.30g与交联剂甲撑双丙烯酰胺0.12g(交联度为0.97wt%),溶于去离子水中得到溶液ⅰ,将引发剂aibi0.1g溶于去离子水中得到溶液ⅱ,在室温下向两种溶液中通入氮气除氧0.5h,将乳化剂span600.5g溶于环己烷中得到乳化剂溶液,在n2保护下将除氧后的溶液ⅰ和溶液ⅱ同步滴加到乳化剂溶液中,溶液ⅰ和溶液ⅱ滴加完后,在n2保护、60℃下搅拌反应6h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得骨架聚合物微球(聚丙烯酰胺微球);
(2)将步骤(1)所得骨架聚合物微球1.05g与2.8gdmapma、交联剂甲撑双丙烯酰胺0.03g、引发剂acva和18ml去离子水混合后(dmapma水溶液浓度为0.16g·ml-1),于室温下溶胀3h得到溶胀混合物,将乳化剂span-800.8g溶于环己烷中得到乳化剂溶液,将溶胀混合物与乳化剂溶液混合均匀,在n2保护、70℃条件下搅拌反应12h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得具有互穿网络结构的co2响应型聚合物微球,其扫描电镜观察结果如图1e所示,从图中可知,微球表面会出现膜状物质,但微球之间仍然保持良好的分散性,没有粘结。这是由于当溶胀单体浓度过高时,聚丙烯酰胺种子微球所提供的氢键不足以稳定住如此多单体,导致单体向种子微球表面迁移。当升温至反应温度时,易发生单体在聚丙烯酰胺微球表面聚合,形成最终的微球表面附着薄膜的形貌。
实施例6
(1)将丙烯酰胺10g与交联剂双丙烯酸乙二醇酯0.02g,溶于去离子水中得到溶液ⅰ,将引发剂aibi0.05g溶于去离子水中得到溶液ⅱ,在室温下向两种溶液中通入氮气除氧0.5h,将乳化剂span-600.2g溶于环己烷中得到乳化剂溶液,在n2保护下将除氧后的溶液ⅰ和溶液ⅱ同步滴加到乳化剂溶液中,溶液ⅰ和溶液ⅱ滴加完后,在n2保护、70℃下搅拌反应5h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得骨架聚合物微球(聚丙烯酰胺微球);
(2)将步骤(1)所得骨架聚合物微球0.5g与1.0gdmapma、交联剂双丙烯酸乙二醇酯0.01g、引发剂acva和去离子水混合后,于室温下溶胀3h得到溶胀混合物,将乳化剂span800.8g溶于环己烷中得到乳化剂溶液,将溶胀混合物与乳化剂溶液混合均匀,在n2保护、80℃条件下搅拌反应12h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得具有互穿网络结构的co2响应型聚合物微球。
实施例7
(1)将丙烯酰胺15g与交联剂甲撑双丙烯酰胺0.8g,溶于去离子水中得到溶液ⅰ,将引发剂aibi0.5g溶于去离子水中得到溶液ⅱ,在室温下向两种溶液中通入氮气除氧0.5h,将乳化剂span601.0g溶于环己烷中得到乳化剂溶液,在n2保护下将除氧后的溶液ⅰ和溶液ⅱ同步滴加到乳化剂溶液中,溶液ⅰ和溶液ⅱ滴加完后,在n2保护、60℃下搅拌反应6h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得骨架聚合物微球(聚丙烯酰胺微球);
(2)将步骤(1)所得骨架聚合物微球2.0g与5.0gdmapma、交联剂甲撑双丙烯酰胺0.05g、引发剂acva和去离子水混合后,于室温下溶胀3h得到溶胀混合物,将乳化剂span802.0g溶于环己烷中得到乳化剂溶液,将溶胀混合物与乳化剂溶液混合均匀,在n2保护、70℃条件下搅拌反应12h,反应结束后分离出反应所得微球,用乙醇和去离子水交替洗涤微球去除杂质后真空干燥,即得具有互穿网络结构的co2响应型聚合物微球。
实施例8实施例1~5制备的聚合物微球的co2响应性能测试
1、光学显微镜观察
光学显微镜观察所用仪器型号为体视显微镜xtl-340(上海光学仪器有限公司)。测试过程为:向实施例3制备的co2响应型聚合物微球的水分散体系里先后通入足量的co2及n2,然后用吸管吸出一滴滴在载玻片上,用显微镜观察,并拍摄微球图片。
测试结果如图3,从图3可知,通入co2后的微球显示出明显的尺寸增加以及透明度的增强,通入n2后回复至初始状态。结果分析为:由于微球中的叔胺组分被co2质子化生成季铵盐,导致微球亲水性增强,更多水渗透进微球。导致最终的微球尺寸增加以及响应的透明度增强。通入n2后,质子化的季铵盐又回复到叔胺状态,导致微球疏水性增强,使微球回复到初始的较小尺寸与较弱透明度。
2、微球水分散体系ph值测试
微球水分散体系的ph值的测试所用仪器为ph计(sartoriusbasicphmeterpb-10)。测试过程为:分别取一定量各实施例制备的co2响应型聚合物微球与400ml去离子水倒入500ml烧杯,依次通入足量co2和n2后,将校正后的ph计插入微球水分散体系中,稳定后读取ph值。
测试结果如图4所示,初始水分散体系的ph值为8.8左右,通入co2后水分散体系的ph值降到4.9左右,再通入n2排出co2后水分散体系的ph值又回复到8.7左右。机理分析:微球水分散体系中通入co2后,co2与水反应形成弱酸碳酸,并且与叔胺结合生成碳酸铵,导致体系ph值下降;通入n2后,弱酸碳酸与碳酸铵逐渐分解掉,并且以co2气体的形式从分散体系逸出,因而体系ph值回复。
测试结果表明,本发明所述co2响应型聚合物微球的水分散体系随着co2的通入与排出,可导致微球水分散体系ph值的可逆变化。
3、微球粒径测试分析
微球尺寸的测试所用仪器为马尔文2000激光粒度仪(malvern,uk),装备有633nm的氦-氖激光以及466nm的固态蓝色激光。对每个实施例制备的co2响应型聚合物微球进行如下操作:取一定量co2响应型聚合物微球与400ml去离子水混合于烧杯中,先向烧杯中通入足量co2并将马尔文粒度仪搅拌桨调速至1000rpm,点击测试并进行数据读取,测试完成后再通入n2,按照同样方法测试微球尺寸。
根据公式体积膨胀率=(co2响应后微球的直径/初始微球的直径)3计算各个实施例制备的co2响应型聚合物微球的体积膨胀率。
测试数据及结果:
实施例1:微球co2响应性能如图4-实施例1所示。微球直径变化为277μm到335μm,即所得聚合物微球的co2响应体积膨胀率为1.8。
实施例2:微球co2响应性能如图4-实施例2所示。微球直径变化为286μm到648μm,即所得聚合物微球的co2响应体积膨胀率为11.6。
实施例3:微球co2响应性能如图4-实施例3所示。微球直径变化为312μm到433μm,所得聚合物微球的co2响应体积膨胀率为2.7。
实施例4:微球co2响应性能如图4-实施例4所示。微球直径变化为307μm到385μm,所得聚合物微球的co2响应体积膨胀率为2.0。
实施例5:微球co2响应性能如图4-实施例5所示。微球直径变化为296μm到485μm,所得聚合物微球的co2响应体积膨胀率为4.4。
测试结果如图4所示,微球尺寸分布为单峰分散,说明微球之间未出现粘结;相对于初始微球,不同配方的微球在通入co2后(生成季铵盐)显示出不同程度的膨胀倍率,通入n2(季铵盐回复成叔胺)后微球尺寸回复到初始状态。
结果分析:实施例2,3,1随着聚丙烯酰胺骨架聚合物微球的交联度增大(从0.2wt%、0.97wt%到4.9wt%),制备得到的co2响应型聚合物微球中叔胺含量降低,导致微球响应膨胀率逐渐降低(从11.6、2.7到1.8),如图5所示。实施例4,3,5随着溶胀单体浓度增大(从0.078、0.10到0.16g·ml–1),制备得到的co2响应型聚合物微球中叔胺含量增高,导致微球响应膨胀率逐渐升高(从2.0、2.7到4.4),如图6所示。说明本专利所述方法可通过改变骨架聚合物微球交联度,改变溶胀过程中溶胀单体(化合物b)的浓度,制备不同co2响应膨胀率的智能微球,实现响应膨胀性的可控刺激。
- 还没有人留言评论。精彩留言会获得点赞!