基于多组学的宫颈癌特征获取方法和系统与流程
本发明涉及数据处理的技术领域,尤其是涉及一种基于多组学的宫颈癌特征获取方法和系统。
背景技术
宫颈癌患者之间存在的异质性使得患病人群中存在潜在的亚群,并且不同的亚群表现为不同的特征以及对治疗的不同反应。传统的宫颈癌患者的分类方法一般基于病人所处的临床阶段或对应癌组织具备的组织学特征,并未考虑病人间的异质性以及疾病在内在分子水平如dna、基因表达等展示的复杂性。其中,按照病人所处的临床阶段分类通常是采用国际妇产科联盟(internationalfederationofgynecologyandobstetrics,简称figo)系统,主要通过显微镜检查癌细胞的细胞核大小、染色质分布均匀等,以及癌细胞浸润的深度和宽度等参数来对病人肿瘤增生轻重程度来进行划分。
另外一种分类方法是根据患者对应癌症组织学特征来进行划分,主要划分为两类,一类是鳞状上皮细胞癌,另一类是腺癌。一般癌组织展现为鳞状上皮细胞癌类型的占主导,约占宫颈癌类型的80%。该分类方法通过宫颈刮片取样位置和显微镜检查的方式来进行判断,一般腺癌多发于子宫颈管部,显微镜下形态可分为腺样、乳头状及髓样,而鳞状细胞癌常发生于宫颈鳞状上皮和柱状上皮的交界区。
由于宫颈癌在早期阶段并无明显特征表现,并且鳞状上皮细胞癌和腺癌在临床表现上没有明显差别,现有分类方法均通过显微镜肉眼对癌细胞的细胞学特征进行判断,难免出现误诊,此外,对于腺癌而言,其往往生长在宫颈管里,取材方面可能会更容易被漏诊。
技术实现要素:
本发明的目的在于提供一种基于多组学的宫颈癌特征获取方法和系统,以缓解采用传统的方法对宫颈癌患者进行分类时准确度较差,以及特征提取不全面的技术问题。
根据本发明实施例的一个方面,提供了一种基于多组学的宫颈癌特征获取方法,该方法包括:获取待分析的甲基化数据,其中,所述甲基化数据为宫颈癌检测中的甲基化数据,包括待分析对象的目标甲基化位点的数值,所述待分析对象的数量为多个;根据各个所述待分析对象在目标甲基化位点的数值,对所述待分析对象进行亚群分类,得到多个亚群;基于所述甲基化数据对所述多个亚群进行第一特征提取,得到表观遗传特征,其中,所述表观遗传特征包括以下至少一种特征信息:差异甲基化位点、基因组分布特征和转录因子结合特征;基于每个所述待分析对象的基因表达数据对所述多个亚群进行第二特征提取,得到基因表达特征,其中,所述基因表达特征包括以下至少之一:差异表达基因和基因功能特征;其中,所述表观遗传特征中的转录因子结合特征和所述基因表达特征中的差异表达基因能够确定靶基因表达功能特征。
进一步地,获取待分析的甲基化数据包括:获取每个所述待分析对象进行宫颈癌检测过程中的甲基化图谱;从所述甲基化图谱中提取所述待分析对象在初始甲基化位点处的数值;对各个所述待分析对象在所述初始甲基化位点处的数值进行融合,得到第一融合矩阵,所述第一融合矩阵的行为各个所述初始甲基化位点,列为所述待分析对象;采用方差计算方法计算所述第一融合矩阵中每个所述初始甲基化位点在多个所述待分析对象之间的差异程度;基于所述差异程度对所述第一融合矩阵进行过滤,得到包含所述目标甲基化位点的所述甲基化数据,其中,所述目标甲基化位点为所述初始甲基化位点中差异程度最高的位点。
进一步地,根据各个所述待分析对象在目标甲基化位点的数值,对所述待分析对象进行亚群分类,得到多个亚群包括:对于所述甲基化数据中的每个目标甲基化位点,采用无监督聚类算法计算任意两个待分析对象之间的距离;将所述距离小于或者等于预设距离的待分析对象确定为同一类别的亚群,得到多个亚群。
进一步地,基于所述甲基化数据对所述多个亚群进行第一特征提取,得到表观遗传特征包括:通过多重校验方法对所述甲基化数据中的每个所述目标甲基化位点进行校正,得到校正后的概率p值;结合所述校正后的概率p值和任意两个亚群之间的甲基化水平差异值,按照第一阈值对所述目标甲基化位点进行过滤,得到差异甲基化位点;查看所述差异甲基化位点在对应的基因组区域中的分布特征,并将所述分布特征作为基因组分布特征,其中,所述基因组区域包括以下至少一个区域:3端非翻译区、5端非翻译区、启动子区域、外显子区域、内含子区域和基因间区;基于位于所述启动子区域的差异甲基化位点提取转录因子结合特征。
进一步地,基于位于所述启动子区域的差异甲基化位点提取转录因子结合特征包括:提取位于所述启动子区域的差异甲基化位点,并将包含该差异甲基化位点的启动子区域作为待分析的启动子;计算所述启动子与所述启动子区域中其余基因之间的碱基距离;将多个所述碱基距离中的最短距离作为标准来获取所述启动子的靶基因;在数据库中获取转录因子的靶基因数据集;确定所述启动子的靶基因和所述靶基因数据集的重叠情况;基于所述重叠情况,通过统计学校验方法计算按照第二阈值确定所述转录因子在所述启动子上的富集情况,以及确定富集显著性,以获取转录因子结合特征。
进一步地,基于每个所述待分析对象的基因表达数据对所述多个亚群进行第二特征提取包括:按照基因名称对每个所述待分析对象的基因表达数据进行融合,得到第二融合矩阵,其中,所述第二融合矩阵的行为各个所述基因名称,列为所述待分析对象;通过多重校验方法对所述基因表达数据中的每个基因进行校正,得到校正后的概率p值;结合所述校正后的概率p值和任意两个亚群之间的表达水平差异值,按照第三阈值对所述基因表达数据进行过滤,得到所述差异表达基因;基于所述差异表达基因提取基因功能特征。
进一步地,基于所述差异表达基因提取基因功能特征包括:在数据库中获取基因功能模块,其中,所述基因功能模块的数量至少为一个;通过统计学算法计算所述差异表达基因在所述基因功能模块的富集情况;通过多重校验方法对所述基因功能模块进行校正,得到校正后的p值;结合所述校正后的p值和富集情况确定每个所述基因功能模块的富集显著性,以根据所述富集显著性确定差异表达基因功能模块,其中,所述差异表达基因功能模块包括用于表示所述多个亚群之间差异表达基因的基因功能特征。
进一步地,在基于每个所述待分析对象的基因表达数据对所述多个亚群进行第二特征提取,得到基因表达特征之后,所述方法还包括:获取所述差异表达基因功能模块和每个所述待分析对象的临床数据;采用生物学计算方法和cox比例风险回归模型计算出所述差异表达基因功能模块中与所述待分析对象预后相关联的基因集,并将所述预后相关联的基因集作为预后分子标志物特征。
进一步地,在基于每个所述待分析对象的基因表达数据对所述多个亚群进行第二特征提取,得到基因表达特征之后,所述方法还包括:在数据库中提取转录因子集;采用生物学计算方法和cox比例风险回归模型计算出所述转录因子集和所述待分析对象的预后相关联情况;结合所述转录因子集在所述多个亚群之间的基因表达情况,筛选出所述多个亚群之间的预后分子标志物。
根据本发明实施例的另一个方面,还提供了一种基于多组学的宫颈癌特征获取系统,该系统包括:第一获取单元,用于获取待分析的甲基化数据,其中,所述甲基化数据为宫颈癌检测中的甲基化数据,包括待分析对象的目标甲基化位点的数值,所述待分析对象的数量为多个;分类单元,用于根据各个所述待分析对象在目标甲基化位点的数值,对所述待分析对象进行亚群分类,得到多个亚群;第一提取单元,用于基于所述甲基化数据对所述多个亚群进行第一特征提取,得到表观遗传特征,其中,所述表观遗传特征包括以下至少一种特征信息:差异甲基化位点、基因组分布特征和转录因子结合特征;第二提取单元,用于基于每个所述待分析对象的基因表达数据对所述多个亚群进行第二特征提取,得到基因表达特征,其中,所述基因表达特征包括以下至少之一:差异表达基因和基因功能特征;其中,所述表观遗传特征中的转录因子结合特征和所述基因表达特征中的差异表达基因能够确定靶基因表达功能特征。
在本发明实施例中,首先获取待分析的宫颈癌检测中的甲基化数据的甲基化数据;然后,甲基化数据中包含的各个待分析对象在目标甲基化位点的数值,对待分析对象进行亚群分类,得到多个亚群;接下来,基于甲基化数据对每个亚群进行第一特征提取,以及进行第二特征提取;最后,得到每个亚群的表观遗传特征和基因表达特征。通过上述处理方式,能够更加准确的对待分析对象进行亚群分类,并且能够更加准确的对多个亚群进行特征提取,进而缓解了采用传统的方法对宫颈癌患者进行分类时准确度较差,以及特征提取不全面的技术问题。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1是根据本发明实施例一中的一种基于多组学的宫颈癌特征获取方法的流程图;
图2是图1中步骤s102的实现方法流程图;
图3是图1中步骤s104的实现方法的流程图;
图4是图1中步骤s106的实现方法的流程图;
图5是图1中步骤s108的实现方法的流程图;
图6是根据本发明实施例一中第一种可选地基于多组学的宫颈癌特征获取方法的流程图;
图7是根据本发明实施例一中第二种可选地基于多组学的宫颈癌特征获取方法的流程图;
图8是根据本发明实施例一中的一种可选地基于多组学的宫颈癌特征获取方法的示意图;
图9是根据本发明实施例二中的一种基于多组学的宫颈癌特征获取系统的示意图;
图10是根据本发明实施例二中第一种可选地基于多组学的宫颈癌特征获取系统的示意图;
图11是根据本发明实施例二中第二种可选地基于多组学的宫颈癌特征获取系统的示意图。
具体实施方式
下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
在本发明的描述中,需要说明的是,术语“中心”、“上”、“下”、“左”、“右”、“竖直”、“水平”、“内”、“外”等指示的方位或位置关系为基于附图所示的方位或位置关系,仅是为了便于描述本发明和简化描述,而不是指示或暗示所指的装置或元件必须具有特定的方位、以特定的方位构造和操作,因此不能理解为对本发明的限制。此外,术语“第一”、“第二”、“第三”仅用于描述目的,而不能理解为指示或暗示相对重要性。
在本发明的描述中,需要说明的是,除非另有明确的规定和限定,术语“安装”、“相连”、“连接”应做广义理解,例如,可以是固定连接,也可以是可拆卸连接,或一体地连接;可以是机械连接,也可以是电连接;可以是直接相连,也可以通过中间媒介间接相连,可以是两个元件内部的连通。对于本领域的普通技术人员而言,可以具体情况理解上述术语在本发明中的具体含义。
实施例一:
根据本发明实施例,提供了一种基于多组学的宫颈癌特征获取方法的实施例。
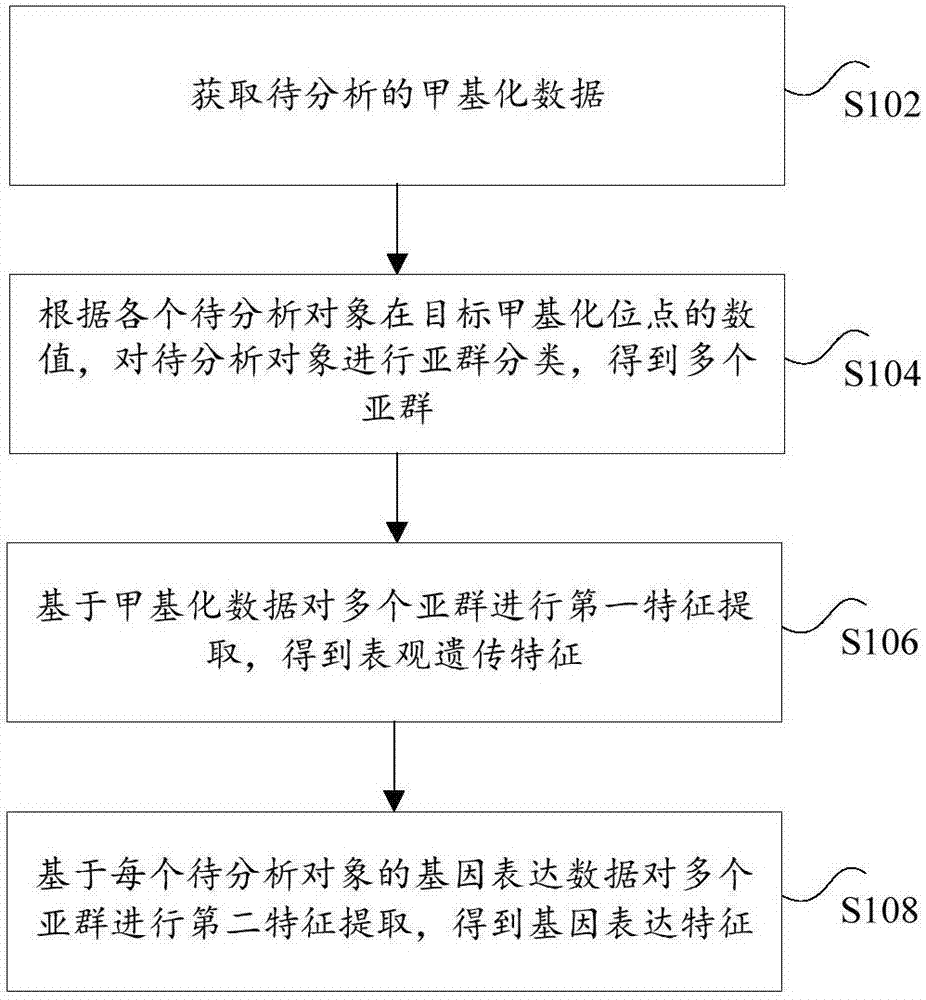
图1是根据本发明实施例的一种基于多组学的宫颈癌特征获取方法的流程图,如图1所示,该方法包括如下步骤:
步骤s102,获取待分析的甲基化数据,其中,甲基化数据为宫颈癌检测中的甲基化数据,包括待分析对象的目标甲基化位点的数值,待分析对象的数量为多个;
需要说明的是,在本发明实施例中,甲基化数据除了可以是宫颈癌检测中的甲基化数据之外,还可以是其他肿瘤组学研究过程中的甲基化数据。其中,在下述实施方式中,甲基化数据均以宫颈癌检测中的甲基化数据为例进行说明。
在本发明实施例中,待分析的甲基化数据为一个矩阵形式的数据,在该甲基化数据中包括每个待分析对象在每个目标甲基化位点的数值,其中,目标甲基化位点为在众多的甲基化位点中提取的有用的甲基化位点,具体提取过程将在下述实施例进行具体的介绍。
步骤s104,根据各个待分析对象在目标甲基化位点的数值,对待分析对象进行亚群分类,得到多个亚群;
在本发明实施例中,充分结合生物学水平因素,能够更加精准的对待分析对象进行亚群分类。在分类得到的多个亚群中,除了包括采用传统的方法得到的亚群分类之外,还包括采用传统的方法不能发现的亚群分类。
步骤s106,基于甲基化数据对多个亚群进行第一特征提取,得到表观遗传特征,其中,表观遗传特征包括以下至少一种特征信息:差异甲基化位点、基因组分布特征和转录因子结合特征;
步骤s108,基于每个待分析对象的基因表达数据对多个亚群进行第二特征提取,得到基因表达特征,其中,基因表达特征包括以下至少之一:差异表达基因和基因功能特征;其中,表观遗传特征中的转录因子结合特征和基因表达特征中的差异表达基因能够确定靶基因表达功能特征。
需要说明的是,在本发明实施例中,并不限制步骤s106和步骤s108的执行顺序,也就是说,可以先执行第一特征提取的步骤,然后执行第二特征提取的步骤;还可以先执行第二特征提取的步骤,再执行第一特征提取的步骤。
需要说明的是,上述步骤s102至步骤s108的执行主体可以为数据处理器或者数据分析仪,但不限于此。
在本发明实施例中,首先获取待分析的宫颈癌检测中的甲基化数据的甲基化数据;然后,甲基化数据中包含的各个待分析对象在目标甲基化位点的数值,对待分析对象进行亚群分类,得到多个亚群;接下来,基于甲基化数据对每个亚群进行第一特征提取,以及进行第二特征提取;最后,得到每个亚群的表观遗传特征和基因表达特征。通过上述处理方式,能够更加准确的对待分析对象进行亚群分类,并且能够更加准确的对多个亚群进行特征提取,进而缓解了采用传统的方法对宫颈癌患者进行分类时准确度较差,以及特征提取不全面的技术问题。
在本发明实施例的一个可选实施方式中,如图2所示,上述步骤s102获取待分析的甲基化数据包括如下步骤:
步骤s201,获取每个待分析对象进行宫颈癌检测过程中的甲基化图谱;
步骤s202,从甲基化图谱中提取待分析对象在初始甲基化位点处的数值;
步骤s203,对各个待分析对象在初始甲基化位点处的数值进行融合,得到第一融合矩阵,第一融合矩阵的行为各个初始甲基化位点,列为待分析对象;
步骤s204,采用方差计算方法计算第一融合矩阵中每个初始甲基化位点在多个待分析对象之间的差异程度;
步骤s205,基于差异程度对第一融合矩阵进行过滤,得到包含目标甲基化位点的甲基化数据,其中,目标甲基化位点为初始甲基化位点中差异程度最高的位点。
在本发明实施例中,首先向数据处理器中输入多个待分析对象在宫颈癌的检测过程中的甲基化图谱,其中,甲基化图谱中包含初始甲基化位点处的甲基化数值。但是,在实际操作过程中,每个待分析对象的甲基化数据通常以单独文件的形式存在,因此,在进行亚群分类和特征提取之前,需要对多个待分析对象的甲基化数据进行融合。
具体地,在融合之前,可以在每个待分析对象的甲基化图谱中提取初始甲基化位点处的数值;然后,根据初始甲基化位点对多个待分析对象在初始甲基化位点处的数值进行融合,融合得到一个矩阵形式的数据,即上述第一融合矩阵,在该第一融合矩阵中包括每个待分析对象在每个初始甲基化位点处的数值。在得到第一融合矩阵之后,可以对第一融合矩阵进行标准化处理,例如,进行log变化处理,得到标准化处理之后的第一融合矩阵。
需要说明的是,由于初始甲基化位点中包含的数量非常庞大,且在进行亚群分类和特征提取的过程中,初始甲基化位点中差异程度不大的位点多为一些无用的甲基化位点,因此,需要将初始甲基化位点中无用的位点进行过滤,得到有用的甲基化位点,以提高数据的处理效率和处理精度。
在本发明实施例中,首先采用统计学计算方法(例如,方差计算方法)计算每个初始甲基化位点在任意两个待分析对象之间的差异程度;然后,基于差异程度对标准化处理之后的第一融合矩阵按照一定的阈值进行过滤,得到包含目标甲基化位点的甲基化数据。
上述步骤s201至步骤s205为数据的清理和预处理过程,该处理过程能够保证甲基化数据的整齐性,一致性和有效性。由于多个待分析对象的甲基化图谱在融合为矩阵后,样本量以及对应的初始甲基化位点数量非常庞大,该庞大的数据使得数据处理效率大为降低,采取统计学方法从中过滤出甲基化差异度最高的位点的速度将大为提高。
在本发明实施例中,在获取待分析的甲基化数据之后,就可以根据各个待分析对象在目标甲基化位点的数值,对待分析对象进行亚群分类。
在本发明实施例的另一个可选实施方式中,如图3所示的步骤s104的实现方法流程图,即根据各个待分析对象在目标甲基化位点的数值,对待分析对象进行亚群分类,得到多个亚群包括如下步骤:
步骤s301,对于甲基化数据中的每个目标甲基化位点,采用无监督聚类算法计算任意两个待分析对象之间的距离;
步骤s302,将距离小于或者等于预设距离的待分析对象确定为同一类别的亚群,得到多个亚群。
在本发明实施例中,对于处理好的甲基化数据,通过无监督聚类方法和皮尔森相关系数,计算任意两个待分析对象之间的距离,并采用随机抽样(例如,随机抽样1000次),最终选取稳定性最高的分类。其中,稳定性最高的是指在多次随机抽样中任意两个待分析对象之间的距离都最大化多次满足小于或者等于预设距离。例如,待分析对象a和待分析对象b之间的距离为a,其中,a小于预设距离l,待分析对象a和待分析对象c之间的距离为b,其中,b大于预设距离l,那么此时,可以将待分析对象a和待分析对象b分为一类,但是,待分析对象c并不属于这类亚群。
在确定好每个待分析对象的亚群分类之后,就可以对每个待分析对象标记所属的亚群类别。在确定每个待分析对象的亚群类别之后,可以直接结合待分析对象的临床资料数据检查在传统分类基础上是否发现了新的亚群,还可采取统计方法(例如,t检验方法)对每个亚群的甲基化水平进行比较,查看亚群间差异的显著性。
在本发明实施例中,充分结合生物学水平因素,能够更加精准的对待分析对象进行亚群分类。在分类得到的多个亚群中,除了包括采用传统的方法得到的亚群分类之外,还包括采用传统的方法不能发现的亚群分类。且通过无监督聚类方法,距离计算方法以及随机抽样方法的使用进一步地保证了最终亚群分类结果的稳定性与准确性。
在对多个待分析对象进行亚群分类之后,就可以对每个亚群进行第一体征提取和第二特征提取,具体地,将在下述实施方式中具体介绍第一特征提取的过程和第二特征提取的过程。
在本发明实施例的另一个可选实施方式中,如图4所示的步骤s106的实现方法流程图,即基于甲基化数据对多个亚群进行第一特征提取,得到表观遗传特征包括如下步骤:
步骤s401,通过多重校验方法对甲基化数据中的每个目标甲基化位点进行校正,得到校正后的概率p值;
步骤s402,结合校正后的p值和任意两个亚群之间的甲基化水平差异值,按照第一阈值对目标甲基化位点进行过滤,得到差异甲基化位点;
步骤s403,查看差异甲基化位点在对应的基因组区域中的分布特征,并将分布特征作为基因组分布特征,其中,基因组区域包括以下至少一个区域:3端非翻译区、5端非翻译区、启动子区域、外显子区域、内含子区域和基因间区;
步骤s404,基于位于启动子区域的差异甲基化位点提取转录因子结合特征。
在本发明实施例中,对于表观遗传特征提取,在步骤s201至步骤s205中提到了可采取统计方法计算待分析对象之间甲基化水平差异的显著性,为了准确知晓哪些甲基化位点发生了显著差异性,需要计算出差异甲基化位点。具体地,首先采用统计计算平台r中的常规生物信息计算软件包(例如,sam软件包),并通过多重检验方法(例如,falsediscoveryrate,简称fdr检验算法)对每个目标甲基化位点进行校正,得到校正后的概率p值。接下来,就可以结合亚群两两之间的甲基化水平差异值(例如,foldchange)以及校正后的概率p值,并按照一定的阈值(即,第一阈值)进行差异甲基化位点的过滤,其中,该第一阈值可以根据实际需要进行调整。
在确定差异甲基化位点之后,可以查看步骤s402中确定出的差异甲基化位点在对应的基因组区域中的分布特征,进而,确定基因组分布特征。具体地,可以通过目标甲基化位点的碱基位置查看在对应的基因组中分布特征,其中,一般选择的基因组区域包括:3’utr(untranslatedregions,3端非翻译区)、5’utr(untranslatedregions,5端非翻译区)、启动子区域、外显子区域、内含子区域以及基因间区,一般情况下,是看目标甲基化位点的位置是否与特定基因组区域的位置存在一个碱基的重叠。
可选地,基于位于启动子区域的差异甲基化位点提取转录因子结合特征具体过程可以为如下过程:
首先,提取位于启动子区域的差异甲基化位点,并将包含差异甲基化位点的启动子区域作为待分析的启动子;然后,计算启动子与启动子区域中其余基因之间的碱基距离,并将多个碱基距离中的最短距离作为标准来获取启动子的靶基因;接下来,在数据库中获取转录因子的靶基因数据集,其中,该数据库可以为分子特征数据库(molecularsignaturesdatabase,简称msigdb);并确定启动子的靶基因和靶基因数据集的重叠情况;基于重叠情况,通过统计学校验方法(例如,fisher精确检验方法)按照第二阈值确定转录因子在启动子上的富集情况,以及确定富集显著性,以获取转录因子结合特征,其中,该转录因子的结合特征可以理解为亚群之间转录因子在发生了差异甲基化水平的启动子上的结合特征。
通过上述描述可知,在本发明实施例中,通过第一特征提取,提取出多个亚群的表观遗传特征,其中,提取出的表观遗传特征包括:差异甲基化位点、基因组分布特征和转录因子结合特征。通过该提取方法能够从多层次对亚群特征进行提取,从而发现传统的特征提取方法所不能提取到的特征。
在本发明实施例的另一个可选实施方式中,如图5所示的步骤s108的实现方法流程图,即基于每个待分析对象的基因表达数据对多个亚群进行第二特征提取包括如下步骤:
步骤s501,按照基因名称对每个待分析对象的基因表达数据进行融合,得到第二融合矩阵,其中,第二融合矩阵的行为各个基因名称,列为待分析对象;
步骤s502,通过多重校验方法对基因表达数据中的每个基因进行校正,得到校正后的概率p值;
步骤s503,结合校正后的概率p值和任意两个亚群之间的表达水平差异值,按照第三阈值对基因表达数据进行过滤,得到差异表达基因;
步骤s504,基于差异表达基因提取基因功能特征。
在进行基因表达数据的提取之前,需要获取每个待分析对象的基因表达数据,其中,基因表达数据中包含至少一个基因。同样地,每个待分析对象的基因表达数据为单独的数据。因此,需要根据基因表达数据中的基因的名称对多个待分析对象的基因表达数据进行融合,以得到矩阵形式的数据,即,第二融合矩阵。
在得到第二融合矩阵之后,通过采用统计计算平台r中的常规生物信息计算软件包(例如,edger软件包),并通过多重检验方法(例如,fdr校验算法)对第二融合矩阵中的每个基因进行校正,得到校正后的概率p值。结合亚群两两之间的表达水平差异值(例如,log2foldchange)和校正后的概率p值,按照一定的阈值(第三阈值)对第二融合矩阵中的基因表达数据进行过滤,得到差异表达基因。在得到差异表达基因之后,就可以基于该差异表达基因提取功能基因特征。
可选地,基于差异表达基因提取基因功能特征的过程描述如下:
首先,在数据库中获取基因功能模块,其中,基因功能模块的数量至少为一个;例如,从msigdb数据库提取基因功能模块(例如,go,kegg通路等);然后,通过统计学算法(例如,fisher精确检验方法)计算差异表达基因在基因功能模块的富集情况;接下来,通过多重校验方法(例如,fdr校验方法)对基因功能模块进行校正,得到校正后的概率p值;结合校正后的p值和富集情况确定每个基因功能模块的富集显著性,以根据富集显著性确定差异表达基因功能模块,其中,差异表达基因功能模块包括用于表示亚群之间差异表达基因的基因功能特征。
在本发明实施例中,上述步骤s404在获取到转录因子结合特征之后,可以查看转录因子结合特征的靶基因集在步骤s503中得到的差异表达基因中的重叠情况,并基于差异表达基因提取基因功能特征的过程计算重叠靶基因的功能特征。
基于图1所示的方法,在本发明实施例的另一个可选实施方式中,如图6所示,在基于甲基化位点和每个待分析对象的基因表达数据对多个亚群进行第二特征提取,得到基因表达特征之后,该方法还包括如下步骤:
步骤s601,获取差异表达基因功能模块和每个待分析对象的临床数据;
步骤s602,采用生物学计算方法和cox比例风险回归模型计算出差异表达基因功能模块中与待分析对象预后相关联的基因集,并将预后相关联的基因集作为预后分子标志物特征。
步骤s601和步骤s602的计算过程既可作为本发明实施例中亚群间分子标志物特征的提取过程,也可作为是上述第二特征提取之后的延伸。
在本发明实施例中,首先获取上阶段中获取到的亚群间差异表达基因功能模块;然后,获取待分析对象的临床资料数据,采用统计计算平台r中的常规生物信息计算方法(例如,kaplan-meier方法和log-rank检验等方法),以及cox比例风险回归模型(即coxproportional-hazards模型),计算出差异表达基因功能模块中和待分析对象预后相关联的基因集,由于这些基因在亚群间表现为差异表达,因而,可以作为亚群间预后分子标志物特征。
其中,cox比例风险回归模型简称cox回归模型。该模型由英国统计学家d.r.cox于1972年提出,主要用于肿瘤和其它慢性病的预后分析,也可用于队列研究的病因探索。
基于图1所示的方法,在本发明实施例的另一个可选实施方式中,如图7所示,在基于甲基化位点和每个待分析对象的基因表达数据对多个亚群进行第二特征提取,得到基因表达特征之后,该方法还包括:
步骤s701,在数据库中提取转录因子集;
步骤s702,采用生物学计算方法和cox比例风险回归模型计算出转录因子集和待分析对象的预后相关联情况;
步骤s703,结合转录因子集在多个亚群之间的基因表达情况,筛选出亚群之间的预后分子标志物。
在本发明实施例中,由于根据差异表达基因功能模块可能无法筛选出预后基因,因此,本发明实施例中,做了一定的延伸,即,可以先从msigdb数据库中抽提特定基因集(例如,转录因子集),然后,采用生物学计算方法和cox比例风险回归模型计算转录因子集和待分析对象预后相关联情况;结合转录因子集在亚群之间的差异表达情况,同样可以筛选出亚群间的预后分子标志物。
由于亚群之间差异表达基因功能模块中所包括的基因往往数量很大,逐一进行病人预后关联计算将使得计算显得繁琐,盲目性比较高,结合亚群间差异表达基因以及对亚群间差异基因功能模块的判断,将使得基因集范围缩小,从而提高了效率,并使得预后分子标志物的筛查具备生物学意义。
综上各实施例提供的基于多组学的宫颈癌特征获取方法,为了直观理解上述过程,以图8所示的基于多组学的宫颈癌特征获取方法的示意图为例进行说明,该方法主要包括:甲基化数据输入,对甲基化数据进行分类计算、亚群构建、特征发现以及预后基因筛查,其中,特征发现包括:(1)特征模块一:表观遗传特征提取;(2)特征模块二:基因表达特征提取。如上所述,表观遗传特征提取包括:差异甲基化位点、基因组分布特征和转录因子结合特征;基因表达特征提取主要基于基因表达数据进行的分析,包括:差异表达基因和基因功能特征。其中,转录因子结合特征和差异表达基因用于确定靶基因集,进而得出靶基因表达功能特征;基因功能特征结合输入的临床资料数据和msigdb数据库中的数据进行预后基因筛查,得到预后分子标志物。本发明实施例的方法从全基因组甲基化图谱出发,进行分类计算得到稳定的亚群分类,在此基础上一方面进行表观遗传特征的提取,另一方面整合基因表达图谱,进行基因表达与功能特征的计算,最后结合病人临床资料,进行预后基因筛查,识别潜在有临床应用价值的预后分子标志物。整套技术方案均基于组学数据,并结合临床信息,采用生物信息学计算方法及统计方法进行操作,具体实现过程如上所述,这里不再赘述。
本发明实施例通过对甲基化位点的过滤,以及统计方法的应用将使得从执行效率上保证待分析对象分类的顺利进行,从分类的准确性能最大化反应病人间的差异性。在本发明实施例中,对亚群特征的提取步骤包括了表观遗传特征和基因表达特征的提取计算,特征提取的覆盖面要更为系统、全面,此外,这些特征的发现在本发明实施例中一次性进行计算,避免了单独计算带来的多余步骤,从而也降低了冗余度。
发明人对本发明实施例中提供的基于多组学的宫颈癌特征获取方法进行了验证,在验证过程中,采取公开的307个宫颈癌病人的数据,按照上述步骤s102至步骤s108中所描述的过程进行操作,结果如下:
第一,采用本发明实施例提供的基于多组学的宫颈癌特征获取方法成功将待分析对象划分为三类亚群,每类亚群的待分析对象数目依次为66,146,以及95。采取统计方法t检验对每个亚群的甲基化水平进行比较,在设置p值为0.01水平时,发现三类亚群间甲基化水平差异呈现显著性,从侧面验证了本发明实施例提供的基于多组学的宫颈癌特征获取方法获取的亚群分类能反应病人间整体甲基化水平的差异性。
第二,亚群两两之间进行差异甲基化修饰比较后,成功提取出亚群两两之间差异的转录因子结合特征,总共包括26个转录因子,其中,包括21个已知的转录因子,5个未知的转录因子,表明本发明实施例提供的基于多组学的宫颈癌特征获取方法可以实现新知识的获取。
第三,成功获取了亚群两两之间差异表达的基因,差异表达基因数目依次为:亚群一与亚群二之间存在593个基因显著高表达,974个基因显著低表达;亚群一与亚群三之间存在753个基因显著高表达,661个基因显著低表达;亚群二与亚群三之间存在224个基因显著高表达,7个基因显著低表达。差异表达的基因存在kegg信号通路上拥有的显著性富集特征。
第四,在亚群间差异表达的基因中,并且参与显著性富集的信号通路,成功筛选出24个基因,其高表达和病人良好的预后相关联,表明本发明实施例提供的基于多组学的宫颈癌特征获取方法可实现预后分子标志物的筛选。
实施例二:
本发明实施例还提供了一种基于多组学的宫颈癌特征获取系统,该基于多组学的宫颈癌特征获取系统主要用于执行本发明实施例上述内容所提供的基于多组学的宫颈癌特征获取方法,以下对本发明实施例提供的基于多组学的宫颈癌特征获取系统做具体介绍。
图9是根据本发明实施例的一种基于多组学的宫颈癌特征获取系统的示意图,如图9所示,该基于多组学的宫颈癌特征获取系统主要包括:第一获取单元91,分类单元92,第一提取单元93和第二提取单元94,其中:
第一获取单元91,用于获取待分析的甲基化数据,其中,甲基化数据为宫颈癌检测中的甲基化数据,包括待分析对象的目标甲基化位点的数值,待分析对象的数量为多个;
分类单元92,用于根据各个待分析对象在目标甲基化位点的数值,对待分析对象进行亚群分类,得到多个亚群;
第一提取单元93,用于基于甲基化数据对多个亚群进行第一特征提取,得到表观遗传特征,其中,表观遗传特征包括以下至少一种特征信息:差异甲基化位点、基因组分布特征和转录因子结合特征;
第二提取单元94,用于基于每个待分析对象的基因表达数据对多个亚群进行第二特征提取,得到基因表达特征,其中,基因表达特征包括以下至少之一:差异表达基因和基因功能特征;
其中,表观遗传特征中的转录因子结合特征和基因表达特征中的差异表达基因能够确定靶基因表达功能特征。
需要说明的是,上述第一获取单元91,分类单元92,第一提取单元93和第二提取单元94的执行主体可以为数据处理器或者数据分析仪,但不限于此。
在本发明实施例中,首先获取待分析的宫颈癌检测中的甲基化数据的甲基化数据;然后,甲基化数据中包含的各个待分析对象在目标甲基化位点的数值,对待分析对象进行亚群分类,得到多个亚群;接下来,基于甲基化数据对每个亚群进行第一特征提取,以及基于基因表达数据进行第二特征提取;最后,得到每个亚群的表观遗传特征和基因表达特征。通过上述处理方式,能够更加准确的对待分析对象进行亚群分类,并且能够更加准确的对多个亚群进行特征提取,进而缓解了采用传统的方法对宫颈癌患者进行分类时准确度较差,以及特征提取不全面的技术问题。
可选地,第一获取单元用于:获取每个待分析对象进行宫颈癌检测过程中的甲基化图谱;从甲基化图谱中提取待分析对象在初始甲基化位点处的数值;对各个待分析对象在初始甲基化位点处的数值进行融合,得到第一融合矩阵,第一融合矩阵的行为各个初始甲基化位点,列为待分析对象;采用方差计算方法计算第一融合矩阵中每个初始甲基化位点在多个待分析对象之间的差异程度;以及,基于差异程度对第一融合矩阵进行过滤,得到包含目标甲基化位点的甲基化数据,其中,目标甲基化位点为初始甲基化位点中差异程度最高的位点。
可选地,分类单元用于:对于甲基化数据中的每个目标甲基化位点,采用无监督聚类算法计算任意两个待分析对象之间的距离;以及,将距离小于或者等于预设距离的待分析对象确定为同一类别的亚群,得到多个亚群。
可选地,第一提取单元用于:通过多重校验方法对甲基化数据中的每个目标甲基化位点进行校正,得到校正后的概率p值;结合校正后的概率p值和任意两个亚群之间的甲基化水平差异值,按照第一阈值对目标甲基化位点进行过滤,得到差异甲基化位点;查看差异甲基化位点在对应的基因组区域中的分布特征,并将分布特征作为基因组分布特征,其中,基因组区域包括以下至少一个区域:3端非翻译区、5端非翻译区、启动子区域、外显子区域、内含子区域和基因间区;以及,基于位于启动子区域的差异甲基化位点提取转录因子结合特征。
可选地,第一提取单元还用于:提取位于启动子区域的差异甲基化位点,并将包含所述差异甲基化位点的启动子区域作为待分析的启动子;计算启动子与启动子区域中其余基因之间的碱基距离;将多个碱基距离中的最短距离作为标准来获取启动子的靶基因;在数据库中获取转录因子的靶基因数据集;确定启动子的靶基因和靶基因数据集的重叠情况;以及,基于重叠情况,通过统计学校验方法计算按照第二阈值确定转录因子在启动子上的富集情况,以及确定富集显著性,以获取转录因子结合特征。
可选地,第二提取单元用于:按照基因名称对每个待分析对象的基因表达数据进行融合,得到第二融合矩阵,其中,第二融合矩阵的行为各个基因名称,列为待分析对象;通过多重校验方法对基因表达数据中的每个基因进行校正,得到校正后的概率p值;结合校正后的概率p值和任意两个亚群之间的表达水平差异值,按照第三阈值对基因表达数据进行过滤,得到差异表达基因;以及,基于差异表达基因提取基因功能特征。
可选地,第二提取单元还用于:在数据库中获取基因功能模块,其中,基因功能模块的数量至少为一个;通过统计学算法计算差异表达基因在基因功能模块的富集情况;通过多重校验方法对基因功能模块进行校正,得到校正后的p值;以及,结合校正后的p值和富集情况确定每个基因功能模块的富集显著性,以根据富集显著性确定差异表达基因功能模块,其中,差异表达基因功能模块包括用于表示多个亚群之间差异表达基因的基因功能特征。
可选地,如图10所示,该系统还包括:第二获取单元101,用于在基于每个待分析对象的基因表达数据对多个亚群进行第二特征提取,得到基因表达特征之后,获取差异表达基因功能模块和每个待分析对象的临床数据;第一计算单元102,用于采用生物学计算方法和cox比例风险回归模型计算出差异表达基因功能模块中与待分析对象预后相关联的基因集,并将预后相关联的基因集作为预后分子标志物特征。
可选地,如图11所示,该系统还包括:提取单元111,用于在基于每个待分析对象的基因表达数据对多个亚群进行第二特征提取112,得到基因表达特征之后,在数据库中提取转录因子集;第二计算单元113,用于采用生物学计算方法和cox比例风险回归模型计算出转录因子集和待分析对象的预后相关联情况;筛选单元114,用于结合转录因子集在多个亚群之间的基因表达情况,筛选出亚群之间的预后分子标志物。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!