一种基于SRAP标记的草果种质资源遗传多样性分析方法与流程
本发明属于分子生物学dna标记技术与应用领域,具体地说,涉及一种基于srap标记的草果种质资源遗传多样性分析方法。
背景技术:
草果(amomumtsaokocrevostetlemaire)是姜科豆蔻属植物中一种多年生草本植物,不仅是我国食品加工业和轻工业的重要原料,而且是一种传统的中药材,有燥湿、健胃、祛痰、温中、顺气、抗疟等功能,还是大众的调料食品,国际、国内市场需求量大。草果对生长环境条件要求较高,一般生长在海拔800~1500m的北热带和中、南亚热带中低山区,年降雨量在1200~1600mm,年平均气温在16~22℃,冬季多雾、湿度大、透光度在40%~50%的阴湿山坡沟谷森林环境下,主要分布在我国云南、广西和贵州三省局部地区,其中云南是草果主产区,产量约占全国的95%,主要分布在红河、文山、西双版纳、德宏、保山、思茅、临沧7个地(州)的31个县(市)。
分子标记是一种以dna序列差异性为标记的检测遗传多样性的方法,具有不受外界环境及基因表达与否的影响,不影响实验材料的性状、可标记数量大及分辨率高等特点,目前被广泛应用于生物遗传研究。srap(相关序列扩增多态性,sequence—relatedamplifiedpolymorphism)是一种新型的基于pcr的标记系统,由美国加州大学蔬菜系li与quiros博士(li,g.,quiros,c.f.,2001.sequence-relatedamplifiedpolymorphism(srap),anewmarkersystembasedonasimplepcrreaction:itsapplicationtomappingandgenetagginginbrassica.theor.appl.genet.103,455–461.)于2001年在芸薹属作物中开发出来。该标记通过独特的双引物设计对基因的orfs(开放式阅读框)的特定区域进行扩增,因不同个体以及物种的内含子、启动子与间隔区长度不同而产生多态性。该标记具有简便、高效、产率高、重复性好、易测序、便于克隆目标片段的特点。目前已成功地应用于作物遗传多样性分析、遗传图谱的构建、重要性状的标记以及相关基因的克隆等。但在草果中,应用srap分子标记技术进行种质资源鉴定、遗传多样性分析的方法尚未见报道。
技术实现要素:
有鉴于此,本发明针对目前对草果的鉴定仍采用传统的形态鉴定法、主要依据果实的形状、大小、色泽等进行划分,形态性状易受环境条件影响,实验误差较大,限制了人们对草果资源的鉴定和遗传多样性分析的问题,提供了一种相对科学合理有效的基于srap标记的草果种质资源遗传多样性分析方法,可以为后续的科学研究提供很好的技术指导和理论支持。
为了解决上述技术问题,本发明公开了一种用于草果遗传多样性分析的srap分子标记引物体系,包括12对引物,分别是:
me1-em11:其核苷酸序列如seqidno.1和seqidno.20所示;
me1-em12:其核苷酸序列如seqidno.1和seqidno.21所示;
me1-em15:其核苷酸序列如seqidno.1和seqidno.24所示;
me2-em11:其核苷酸序列如seqidno.2和seqidno.20所示;
me2-em12:其核苷酸序列如seqidno.2和seqidno.21所示;
me5-em5:其核苷酸序列如seqidno.5和seqidno.14所示;
me5-em6:其核苷酸序列如seqidno.5和seqidno.15所示;
me6-em2:其核苷酸序列如seqidno.6和seqidno.11所示;
me6-em5:其核苷酸序列如seqidno.6和seqidno.14所示;
me6-em14:其核苷酸序列如seqidno.6和seqidno.23所示;
me9-em6:其核苷酸序列如seqidno.9和seqidno.15所示;
me9-em11:其核苷酸序列如seqidno.9和seqidno.20所示。
本发明还公开了一种用于草果遗传多样性分析的srap-pcr反应体系,每25μl的反应体系中含有不含mg2+的10×pcrbuffer3.0μl、taq酶1u、mg2+1.5mmol/l、dntp0.25mmol/l、引物0.2μmol/l和模板dna30ng;将上述反应混合液按如下程序进行扩增:95℃预变性5min,95℃变性1min,35℃退火1min,72℃延伸1min,35个循环,最后72℃延伸10min,4℃保存。
进一步地,引物选自上述me1-em11、me1-em12、me1-em15、me2-em11、me2-em12、me5-em5、me5-em6、me6-em2、me6-em5、me6-em14、me9-em6或me9-em11中任一对。
本发明还公开了一种利用srap分子标记分析草果遗传多样性的方法,其步骤包括:
(1)采用2*ctab法提取草果基因组dna以备用;
(2)采用srap-pcr反应体系对步骤(1)中提取的dna样品进行扩增;
(3)将pcr扩增产物在6%非变性聚丙烯酰胺凝胶上进行电泳分离,电泳结束后银染检测条带;
(4)草果遗传多样性分析:利用12对srap分子标记将不同来源的草果进行聚类及主坐标分析,并进行各遗传多样性参数的统计。
进一步地,步骤(1)中的dna的提取具体为:采用2*ctab法提取dna,并将提取的dna样品溶于te缓冲液后存储于-20℃备用;扩增前用双蒸水将dna样品稀释成20ng/μl的工作液,作为pcr扩增反应的模板。
进一步地,步骤(2)中的srap-pcr反应体系具体为:每25μl的反应体系中含有不含mg2+的10×pcrbuffer3.0μl、taq酶1u、mg2+1.5mmol/l、dntp0.25mmol/l、引物0.2μmol/l和模板dna30ng;将上述反应混合液按如下程序进行扩增:95℃预变性5min,95℃变性1min,35℃退火1min,72℃延伸1min,35个循环,最后72℃延伸10min,4℃保存。
进一步地,步骤(3)中的将pcr扩增产物在6%非变性聚丙烯酰胺凝胶上进行电泳分离,电泳结束后银染检测条带。具体为:电泳结束后,将胶体从玻璃板上取下,蒸馏水漂洗,转入含有0.2%硝酸银的染色液中,振荡10min,蒸馏水漂洗1次约1min,转入含2%氢氧化钠和0.4%甲醛的显色液中,轻轻振荡待条带显色完全,将胶体转入蒸馏水中。在凝胶成像系统下对胶体进行拍照保存。
进一步地,步骤(4)中的利用12对srap分子标记将不同来源的草果进行聚类及主坐标分析,并进行各遗传多样性参数的统计具体为:读取步骤(3)获得的谱带信息,将电泳图谱上100~2000bp范围内清晰且可重复出现的条带记为1,同一位置没有条带记为0,由此生成0和1原始矩阵;统计每对引物扩增出的总条带数和多态性条带数;用ntsys-pc(2.10e)软件中simqual程序计算相似系数矩阵,以clustering程序中shan进行upgma聚类;用treeplot模块生成聚类图,构建分子进化树;根据计算的相似系数矩阵进行decenter数据转换,进而进行主坐标分析;
根据01二元数据矩阵,统计srap扩增产物的条带总数和多态性条带数(npb),计算多态性条带所占的比率(ppb)和引物多态性信息含量(pic),pici=2fi(1-fi),公式中pici表示第i个位点的多态性信息含量,fi表示有带所占的频率,(1-fi)表示无带所占的频率;对每对引物来说,pic=∑pici/n,式中n表示每对引物的多态性条带数;对每个引物组合而言,标记指数(mi)可以按下述公式计算:mi=npb*pic;采用popgene32软件对所有试验材料进行遗传多样性参数计算:等位基因数、有效等位基因数、shannon's信息指数、基因多样性指数和多态位点比率。
与现有技术相比,本发明可以获得包括以下技术效果:
1)利用本发明所优化的pcr反应及反应程序进行srap分子标记试验,所得到的条带清晰、多态性好、重复性高,易于鉴别不同植株基因水平的差异,为草果基因水平研究奠定了良好的基础。
2)采用6%的非变性聚丙烯酰胺凝胶对srap-pcr扩增产物进行分离,分辨率较琼脂糖凝胶更高;克服了传统方法采用琼脂糖凝胶电泳对pcr产物进行分离,效果较差的问题。
3)利用优化的pcr反应体系,筛选出的12对srap引物在草果中扩增出的条带更具有多态性高、特异性强、背景清楚及稳定性强的优点;
4)本发明可加快草果资源的鉴定速度,缩短实验时间,结果稳定可靠,弥补了传统形态学鉴定方法的不足。
5)本发明成本低,在短时间内可完成大批试验材料鉴定。
6)本发明采用的srap分子标记是对基因组的重要组成部分orfs进行扩增,基因的多样性更能反映遗传资源的多样性;srap在基因组中分布均匀,它提供的信息比其他分子标记更为优良,用于草果遗传多样性评价可信度高。
7)利用该技术能很好的揭示草果居群间的遗传多样性,分辨草果种质资源间的亲缘关系,对草果资源的保护及利用具有重要意义。
当然,实施本发明的任一产品并不一定需要同时达到以上所述的所有技术效果。
附图说明
此处所说明的附图用来提供对本发明的进一步理解,构成本发明的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
图1是本发明1-24号样品me1em15引物组合扩增效果图;其中,m,700bpmarker,1-24号与表1的统一编号相对应;
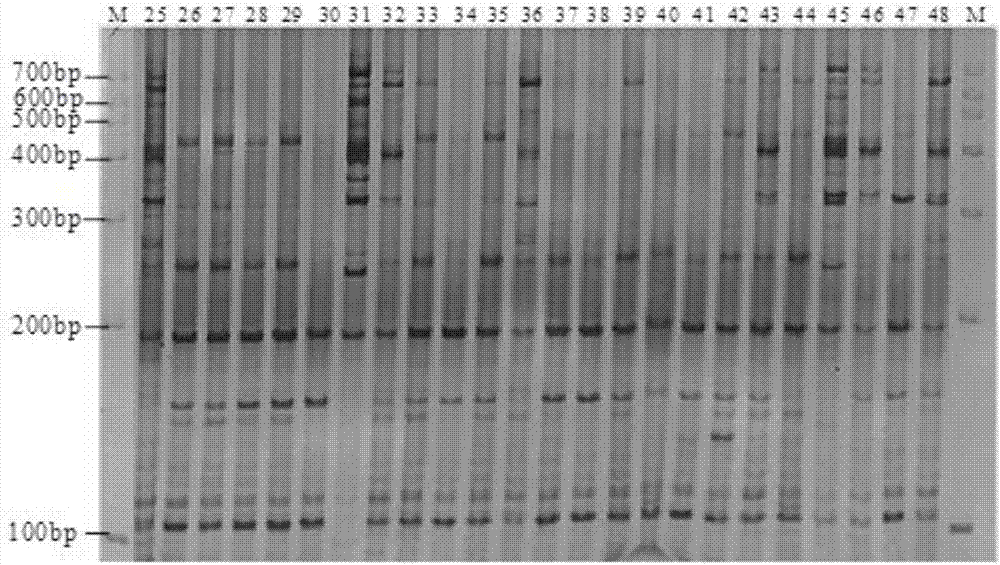
图2是本发明25-48号样品me1em15引物组合扩增效果图;其中,m,700bpmarker,25-48号与表1的统一编号相对应;
图3是本发明基于srap标记的96份草果样品聚类图;
图4是本发明基于srap标记的96份草果样品主坐标分析。
具体实施方式
以下将配合实施例来详细说明本发明的实施方式,藉此对本发明如何应用技术手段来解决技术问题并达成技术功效的实现过程能充分理解并据以实施。
实施例1基于srap标记的草果种质资源遗传多样性分析方法
(1)采用2*ctab法提取草果基因组dna以备用。
(2)草果srap-pcr反应体系优化,优化的反应体系其特征在于:25μl反应体系包括:10×pcrbuffer(不含mg2+)3.0μl、taq酶1u、mg2+1.5mmol/l、dntp0.25mmol/l、引物0.2μmol/l和模板dna30ng。
(3)pcr扩增反应程序的改良,改良后的程序:95℃预变性5min,95℃变性1min,35℃退火1min,72℃延伸1min,35个循环,最后72℃延伸10min,4℃保存。
(4)srap多态性引物筛选,从153对srap引物中筛选出12对带型稳定、清晰且重复性好的多态性引物不同居群的草果进行pcr扩增反应,扩增反应结束后加入2μl溴酚蓝和二甲苯青ff配制的双色指示剂,取2μl扩增产物在6%非变性聚丙烯酰胺凝胶电泳,电极缓冲液为0.5×tbe,染色后拍照。
(5)草果遗传多样性分析,利用12对srap分子标记可成功的将不同来源的草果进行聚类及主坐标分析,并进行各遗传多样性参数的统计。为草果种质资源鉴定及遗传多样性分析建立新的方法。
实施例2dna的提取
(1)实验材料
本试验所用的96份草果采自云南省8个草果产地,其中金平县13份、元阳县12份、绿春县11份、屏边县10份、普洱澜沧县14份、保山市11份、德宏梁河县13份、临沧云县12份,具体见表1和表2。
表1试验材料编号(1-48)及采样点
表2试验材料编号(49-96)及采样点
(2)dna的提取
在种质资源调查同时采集嫩叶用于全基因组dna提取。采用2*ctab法提取dna,并将提取的dna样品溶于te缓冲液后存储于-20℃备用。扩增前用双蒸水将dna样品稀释成20ng/μl的工作液,作为pcr扩增反应的模板。
实施例3pcr反应体系优化及引物筛选
采用正交设计方法,对mg2+、dntps和引物浓度、taqdna聚合酶及模板dna用量等5个因素进行筛选,得到适于草果的srap标记pcr反应体系。srap-pcr的正交设计如表3。经me1em15、me4em8、me7em1、me8em14对引物3次重复试验后,25μl反应体系中10×pcrbuffer(不含mg2+)3.0μl、taq酶1u、mg2+1.5mmol/l、dntp0.25mmol/l、引物0.2μmol/l和模板dna30ng综合扩增效果最好。
表3srap-pcr反应l16(45)正交试验设计表
上述反应体系的pcr扩增反应程序为改良的程序:95℃预变性5min,95℃变性1min,35℃退火1min,72℃延伸1min,35个循环,最后72℃延伸10min,4℃保存。
srap引物序列见表4。利用上述反应体系和反应程序共筛选出12对带型稳定、清晰且重复性好的多态性引物组合:me1em11、me1em12、me1em15、me2em11、me2em12、me5em5、me5em6、me6em2、me6em5、me6em14、me9em6、me9em11。图1和图2为me1em15对1-48号样品扩增效果图,可以得出本发明所建立的草果srap-pcr反应体系扩增出的条带具有多态性高、特异性强、背景清楚及稳定性强的优点。
表4srap引物序列
(3)电泳和银染体系
pcr扩增产物在6%非变性聚丙烯酰胺凝胶上进行电泳分离。电泳结束后银染检测条带,具体操作如下:电泳结束后,将胶体从玻璃板上取下,蒸馏水漂洗,转入含有0.2%(质量百分浓度)硝酸银的染色液中,振荡10min,蒸馏水漂洗1次约1min,转入含2%氢氧化钠和0.4%甲醛(10gnaoh定容至500ml,使用前加入2ml甲醛)的显色液中,轻轻振荡待条带显色完全,将胶体转入蒸馏水中。在凝胶成像系统下对胶体进行拍照保存。
实施例4srap标记在96份草果品种遗传多样性分析中的应用
(1)聚类分析和主坐标分析
将电泳图谱上100~2000bp范围内清晰且可重复出现的条带记为1,同一位置没有条带记为0,由此生成0和1原始矩阵。统计每对引物扩增出的总条带数和多态性条带数。用ntsys-pc(2.10e)软件中simqual程序计算相似系数矩阵,以clustering程序中shan进行upgma(unweightedpair-groupmethodwitharithmeticmeans)聚类;用treeplot模块生成聚类图,构建分子进化树。根据计算的相似系数矩阵进行decenter数据转换,进而进行主坐标分析。
基于srap标记,利用ntsyspc2.10e软件对8个居群的96份草果进行聚类分析和主坐标分析。聚类分析中除lc50、p1、p19、d16没能被成功聚类外,其余样本根据亲缘关系远近均被分到不同类群中,在阈值0.82处可将样本分为5类,来自金平的j108、j109、j118、j119和j128聚在一起,lc33、xy8和xy16聚在一类,绿春的l28、l34聚在一起,j8、j48、j55、j64、p48、lc2、j84、j91聚在一起,剩余的74份样本聚在一起,其中p17、yx10、lc31具有高度的一致性(图3)。
对96份草果样本的srap标记的原始矩阵进行主坐标分析,srap标记提取的前2个主坐标所能解释的遗传变异分别为11.11%和3.51%,二维排序图能够直观的表示96份样本之间的亲缘关系远近,主坐标分析结果与聚类结果基本一致(图4)。
(2)遗传多样性分析
根据01二元数据矩阵,统计srap扩增产物的条带总数和多态性条带数(npb),计算多态性条带所占的比率(ppb)和引物多态性信息含量(pic),pici=2fi(1-fi),公式中pici表示第i个位点的多态性信息含量,fi表示有带所占的频率,(1-fi)表示无带所占的频率;对每对引物来说,pic=∑pici/n,式中n表示每对引物的多态性条带数;对每个引物组合而言,标记指数(mi)可以按下述公式计算:mi=npb*pic。假定所研究的草果居群都处于hardy-weinberg平衡,釆用popgene32软件对所有试验材料进行遗传多样性参数计算:等位基因数、有效等位基因数、shannon's信息指数、基因多样性指数、多态位点比率等。12对srap引物共扩增534个条带,其中多态性条带527条,占总条带的98.69%,平均每对引物扩增44.5条带;不同引物所揭示的pic(多态性信息含量)在0.128~0.250之间,平均为0.161;mi(标记指数)最高的是引物me1em12,为8.794,表明该引物在12对引物中的扩增效率最高;有效等位位点数ne、nei基因多样度及shannon's信息指数i最高的引物为me5em5,分别为1.222、0.165和0.288,最低的为me6em14,分别为1.097、0.078和0.152(表5)。srap引物扩增多态性表明云南草果遗传多样性水平较低。居群间比较分析表明遗传多样性最高的是绿春和金平居群,最低的是保山居群。在物种水平上,多态性位点百分率p、观测等位基因数na、有效等位位点数ne、nei基因多样度h和shannon's信息指数i分别为98.57%、1.9857、1.1261、0.0983、0.1852(表6)。表明草果无论在居群水平还是物种水平遗传多样性都较低,亟需我们加大保护力度。
表512对srap引物扩增结果
注:tnp:扩增总条带数;npb:多态性条带数;ppb:多态性比率;na:观测等位基因数;ne:有效等位基因数;h:nei基因多样度;i:shannon's信息指数;pic:多态性信息含量;mi:标记指数。
表6草果居群srap遗传多样性分析
注:p:多态性位点百分率;na:观测等位基因数。
上述说明示出并描述了发明的若干优选实施例,但如前所述,应当理解发明并非局限于本文所披露的形式,不应看作是对其他实施例的排除,而可用于各种其他组合、修改和环境,并能够在本文所述发明构想范围内,通过上述教导或相关领域的技术或知识进行改动。而本领域人员所进行的改动和变化不脱离发明的精神和范围,则都应在发明所附权利要求的保护范围内。
序列表
<110>红河学院
<120>一种基于srap标记的草果种质资源遗传多样性分析方法
<130>2017
<160>26
<170>patentinversion3.3
<210>1
<211>17
<212>dna
<213>人工序列
<400>1
tgagtccaaaccggata17
<210>2
<211>17
<212>dna
<213>人工序列
<400>2
tgagtccaaaccggagc17
<210>3
<211>17
<212>dna
<213>人工序列
<400>3
tgagtccaaaccggaat17
<210>4
<211>17
<212>dna
<213>人工序列
<400>4
tgagtccaaaccggacc17
<210>5
<211>17
<212>dna
<213>人工序列
<400>5
tgagtccaaaccggaag17
<210>6
<211>17
<212>dna
<213>人工序列
<400>6
tgagtccaaaccggtag17
<210>7
<211>17
<212>dna
<213>人工序列
<400>7
tgagtccaaaccggttg17
<210>8
<211>17
<212>dna
<213>人工序列
<400>8
tgagtccaaaccggtgt17
<210>9
<211>17
<212>dna
<213>人工序列
<400>9
tgagtccaaaccggtca17
<210>10
<211>18
<212>dna
<213>人工序列
<400>10
gactgcgtacgaattaat18
<210>11
<211>18
<212>dna
<213>人工序列
<400>11
gactgcgtacgaatttgc18
<210>12
<211>18
<212>dna
<213>人工序列
<400>12
gactgcgtacgaattgac18
<210>13
<211>18
<212>dna
<213>人工序列
<400>13
gactgcgtacgaatttga18
<210>14
<211>18
<212>dna
<213>人工序列
<400>14
gactgcgtacgaattaac18
<210>15
<211>18
<212>dna
<213>人工序列
<400>15
gactgcgtacgaattgca18
<210>16
<211>18
<212>dna
<213>人工序列
<400>16
gactgcgtacgaattatg18
<210>17
<211>18
<212>dna
<213>人工序列
<400>17
gactgcgtacgaattagc18
<210>18
<211>18
<212>dna
<213>人工序列
<400>18
gactgcgtacgaattacg18
<210>19
<211>18
<212>dna
<213>人工序列
<400>19
gactgcgtacgaatttag18
<210>20
<211>18
<212>dna
<213>人工序列
<400>20
gactgcgtacgaatttcg18
<210>21
<211>18
<212>dna
<213>人工序列
<400>21
gactgcgtacgaattgtc18
<210>22
<211>18
<212>dna
<213>人工序列
<400>22
gactgcgtacgaattggt18
<210>23
<211>18
<212>dna
<213>人工序列
<400>23
gactgcgtacgaattcag18
<210>24
<211>18
<212>dna
<213>人工序列
<400>24
gactgcgtacgaattctg18
<210>25
<211>18
<212>dna
<213>人工序列
<400>25
gactgcgtacgaattcgg18
<210>26
<211>18
<212>dna
<213>人工序列
<400>26
gactgcgtacgaattcca18
- 还没有人留言评论。精彩留言会获得点赞!