一种洛索洛芬钠开环杂质的制备方法与流程
本发明涉及一种洛索洛芬钠开环杂质的制备方法,属于原料药制备技术领域。
背景技术:
洛索洛芬钠(loxoprofensodium),其结构式如下:
化学名为2-[4-(2-氧代环戊烷-1-甲基)苯基]丙酸钠二水合物,属于二芳基丙酸类非甾体抗炎药,由日本三共株式会社首先研制,1986年在日本上市,现在日本为非甾体抗炎药中销量第一的品种。
各国药典关于洛索洛芬钠的质量标准,没有规定已知杂质,日本药典用薄层色谱(tlc)技术控制有关物质。提高临床用药质量,降低副作用,是药物工作者的努力方向。
随着分析技术的不断发展,洛索洛芬钠质量标准在不断升级,有关物质的检测逐渐被高效液相色谱技术(hplc)取代,完善洛索洛芬钠质量标准需要洛索洛芬钠工艺杂质对照品或降解杂质对照品。
式ⅴ所示化合物为洛索洛芬钠开环杂质,是洛索洛芬钠合成过程中工艺杂质,也是原研制剂(loxonin)中最大杂质,且其在洛索洛芬钠原料药和制剂稳定性实验中都会增长,因此对洛索洛芬钠原料药和制剂日常检验中需要合格的式ⅴ化合物作为对照品,式ⅴ化合物对照品国内没有销售,必须进口。经检索,没有该杂质制备的文献报道,且通过常规的酸碱、氧化、高温破坏都无法得到式ⅴ化合物,必须开辟新的路线定向合成此化合物。因此,提供一种洛索洛芬钠开环杂质的合成方法用于杂质标准品的制备具有重要的现实意义。
技术实现要素:
发明目的:本发明的目的是提供一种洛索洛芬钠开环杂质的制备方法。为洛索洛芬钠原料药生产企业和制剂生产企业提供合格的杂质对照品。
本发明的技术方案是:
本发明提供一种式ⅴ所示洛索洛芬钠开环杂质的制备方法,包括以下步骤:
第一步式ⅰ与六次亚甲基四胺在有机溶剂a中发生取代反应,经无机酸水解得到式ⅱ所示的化合物;
其中:式ⅰ中x为cl或br;
式ⅰ与六次亚甲基四胺投料物质量的比为1.0:1.0~3.0;
有机溶剂a选自二氯甲烷、氯仿、四氯化碳和冰乙酸中的一种;优选为氯仿;
无机酸为盐酸、硫酸和磷酸中一种;优选为盐酸。
第二步式ⅱ与式ⅲ在有机溶剂b中进行缩合反应得到式ⅳ所示的化合物;
其中:式ⅱ与式ⅲ投料物质量的比为1.0:1.0~2.0;
有机溶剂b选自二氯甲烷、氯仿、四氯化碳和甲苯中一种;优选为甲苯。
第三步式ⅳ所示的化合物在氧化剂和无机碱的条件下氧化开环得到式ⅴ所示的洛索洛芬钠杂质,合成路线如下所示。
其中:氧化剂选自双氧水、过硼酸钠和过硫酸氢钾复合盐中一种;优选为双氧水;
式ⅳ和氧化剂投料物质量的比为1.0:2.0~3.0;
无机碱选自碳酸钠、碳酸钾、氢氧化钠或氢氧化钾中的一种;优选为氢氧化钠;
反应温度为0~50℃,优选为20~30℃。
有益效果:
本发明技术方案提供了一种洛索洛芬钠开环杂质的制备方法,具有工艺简单,操作简便,收率良好,纯度高等优点。该杂质在洛索洛芬钠原料药和制剂中都存在,本发明能够为洛索洛芬钠的质量控制提供合格的对照品,对洛索洛芬钠的原料药和制剂的质量控制具有重要的意义。
(四)附图说明
图1实施例1中步骤(1)制得的2-(4-甲酰基苯基)丙酸核磁共振氢谱
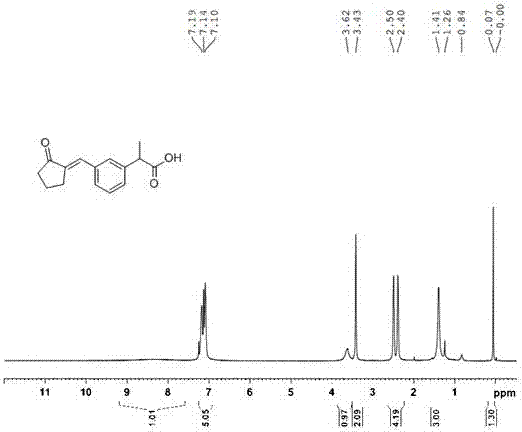
图2实施例1中步骤(2)制得2-(2-((2-氧代环戊基)甲基)苯基)丙酸核磁共振氢谱
图3实施例1中步骤(3)制得的6-(4-(1-羧乙基)苯基)-5-氧代己酸核磁共振氢谱图
(五)具体实施例:
实施例1
步骤(1):向500ml的三口瓶中,加入对氯甲基异苯丙酸(80g,0.40mol)、六次亚甲基四胺(112.1g,0.80mol)、氯仿240ml,搅拌升温至回流(63~65℃),搅拌反应6h,取样点板反应完全,降温至室温,加入36%浓盐酸60g,搅拌升温至回流反应2h,降温至室温,分层得有机层,有机层用饱和碳酸钠水溶液200ml洗涤,分层得有机层,减压浓缩至干得2-(4-甲酰基苯基)丙酸66.2g,淡黄色固体,收率92.3%hplc纯度为94.6%。1h-nmr(400mhz,cdcl3)d11.31(1h,s),9.99(1h,d),7.86(2h,d),7.50(2h,d),3.84(1h,q),1.56(3h,d)。
步骤(2):向500ml的三口瓶中,加入4-甲酰基一苯丙酸(25g,0.14mol)、1-(4-吗啡啉)环戊烯(43g,0.28mol)、甲苯150ml,搅拌升温至回流(115℃),搅拌反应6h,取样点板反应完全,减压浓缩至干得淡黄色油状物,加入100ml正己烷搅拌析晶1h,抽滤得2-(2-((2-氧代环戊基)甲基)苯基)丙酸30g,类白色固体,收率87.8%,hplc纯度为98.3%。1h-nmr(400mhz,dmso-d6)d8.35(1h,s),7.19(2h,d),7.14(2h,d),7.10(1h,s),3.62(1h,q),3.43(2h,t),2.50(2h,t),2.40(2h,m),1.41(3h,d)。
步骤(3):向100ml的三口瓶中加入2-(2-((2-氧代环戊基)甲基)苯基)丙酸(10g,0.04mol)、乙醇50ml,搅拌升温至溶清,降温至30℃以下,于30℃以下滴加30%氢氧化钠溶液15g,滴毕,降温至0~5℃,于0~5℃滴加30%双氧水10g,滴毕,于25~30℃反应4h,加入亚硫酸钠淬灭反应,用浓盐酸调ph-1~2,减压浓缩至干,加入50ml水,用2*50ml二氯甲烷萃取水相2次,分层得二氯甲烷层,减压浓缩至干,经柱层析分离得6-(4-(1-羧乙基)苯基)-5-氧代己酸8.2g,收率72%,纯度99.1%
实施例2
步骤(1):向500ml的三口瓶中,加入对溴甲基异苯丙酸(96.84g,0.40mol)、六次亚甲基四胺(112.1g,0.80mol)、氯仿240ml,搅拌升温至回流(63~65℃),搅拌反应6h,取样点板反应完全,降温至室温,加入36%浓盐酸60g,搅拌升温至回流反应2h,降温至室温,分层得有机层,有机层用饱和碳酸钠水溶液200ml洗涤,分层得有机层,减压浓缩至干得2-(4-甲酰基苯基)丙酸67.56g,淡黄色固体,收率94.2%hplc纯度为94.6%。
步骤(2):向500ml的三口瓶中,加入4-甲酰基一苯丙酸(25g,0.14mol)、1-(4-吗啡啉)环戊烯(43g,0.28mol)、甲苯150ml,搅拌升温至回流(115℃),搅拌反应6h,取样点板反应完全,减压浓缩至干得淡黄色油状物,加入100ml正己烷搅拌析晶1h,抽滤得2-(2-((2-氧代环戊基)甲基)苯基)丙酸30g,类白色固体,收率87.8%,hplc纯度为98.3%。
步骤(3):向100ml的三口瓶中加入2-(2-((2-氧代环戊基)甲基)苯基)丙酸(10g,0.04mol)、乙醇50ml,搅拌升温至溶清,降温至30℃以下,于30℃以下滴加30%氢氧化钠溶液15g,滴毕,降温至0~5℃,于0~5℃滴加30%双氧水10g,滴毕,于25~30℃反应4h,加入亚硫酸钠淬灭反应,用浓盐酸调ph-1~2,减压浓缩至干,加入50ml水,用2*50ml二氯甲烷萃取水相2次,分层得二氯甲烷层,减压浓缩至干,经柱层析分离得6-(4-(1-羧乙基)苯基)-5-氧代己酸8.2g,收率72%,纯度99.1%
实施例3
步骤(1):向500ml的三口瓶中,加入对氯甲基异苯丙酸(80g,0.40mol)、六次亚甲基四胺(56.05g,0.40mol)、氯仿240ml,搅拌升温至回流(63~65℃),搅拌反应6h,取样点板反应完全,降温至室温,加入36%浓盐酸60g,搅拌升温至回流反应2h,降温至室温,分层得有机层,有机层用饱和碳酸钠水溶液200ml洗涤,分层得有机层,减压浓缩至干得2-(4-甲酰基苯基)丙酸61.4g,淡黄色固体,收率85.6%。hplc纯度为94.0%。
步骤(2):向500ml的三口瓶中,加入4-甲酰基一苯丙酸(25g,0.14mol)、1-(4-吗啡啉)环戊烯(43g,0.28mol)、二氯甲烷150ml,搅拌升温至回流(40℃),搅拌反应10h,取样点板反应完全,减压浓缩至干得淡黄色油状物,加入100ml正己烷搅拌析晶1h,抽滤得2-(2-((2-氧代环戊基)甲基)苯基)丙酸28.29g,类白色固体,收率82.8%,hplc纯度为97.6%。
步骤(3):向100ml的三口瓶中加入2-(2-((2-氧代环戊基)甲基)苯基)丙酸(10g,0.04mol)、乙醇50ml,搅拌升温至溶清,降温至30℃以下,于30℃以下滴加30%氢氧化钠溶液15g,滴毕,降温至0~5℃,于0~5℃分批投入过硼酸钠13.54g,投毕,于25~30℃反应4h,加入亚硫酸钠淬灭反应,用浓盐酸调ph=1~2,减压浓缩至干,加入50ml水,用2*50ml二氯甲烷萃取水相2次,分层得二氯甲烷层,减压浓缩至干,经柱层析分离得6-(4-(1-羧乙基)苯基)-5-氧代己酸7.72g,收率67.8%,纯度98.4%
实施例4
步骤(1):向500ml的三口瓶中,加入对氯甲基异苯丙酸(80g,0.40mol)、六次亚甲基四胺(168.2g,0.12mol)、二氯甲烷240ml,搅拌升温至回流(40℃),搅拌反应6h,取样点板反应完全,降温至室温,加入36%浓盐酸60g,搅拌升温至回流反应2h,降温至室温,分层得有机层,有机层用饱和碳酸钠水溶液200ml洗涤,分层得有机层,减压浓缩至干得2-(4-甲酰基苯基)丙酸65.48g,淡黄色固体,收率91.3%hplc纯度为94.6%。
步骤(2):向500ml的三口瓶中,加入4-甲酰基一苯丙酸(25g,0.14mol)、1-(4-吗啡啉)环戊烯(21.5g,0.14mol)、甲苯150ml,搅拌升温至回流(115℃),搅拌反应6h,取样点板反应完全,减压浓缩至干得淡黄色油状物,加入100ml正己烷搅拌析晶1h,抽滤得2-(2-((2-氧代环戊基)甲基)苯基)丙酸30g,类白色固体,收率87.8%,hplc纯度为98.3%。
步骤(3):向100ml的三口瓶中加入2-(2-((2-氧代环戊基)甲基)苯基)丙酸(10g,0.04mol)、乙醇50ml,搅拌升温至溶清,降温至30℃以下,于30℃以下滴加30%氢氧化钠溶液15g,滴毕,降温至0~5℃,于0~5℃滴加30%双氧水13.64g,滴毕,于45~50℃反应3h,加入亚硫酸钠淬灭反应,用浓盐酸调ph=1~2,减压浓缩至干,加入50ml水,用2*50ml二氯甲烷萃取水相2次,分层得二氯甲烷层,减压浓缩至干,经柱层析分离得6-(4-(1-羧乙基)苯基)-5-氧代己酸7.9g,收率69.35%,纯度99.0%
实施例5
步骤(1):向500ml的三口瓶中,加入对氯甲基异苯丙酸(80g,0.40mol)、六次亚甲基四胺(112.1g,0.80mol)、四氯化碳240ml,搅拌升温至回流(75~80℃),搅拌反应5h,取样点板反应完全,降温至室温,加入36%浓盐酸60g,搅拌升温至回流反应2h,降温至室温,分层得有机层,有机层用饱和碳酸钠水溶液200ml洗涤,分层得有机层,减压浓缩至干得2-(4-甲酰基苯基)丙酸65.62g,淡黄色固体,收率91.5%hplc纯度为94.2%。
步骤(2):向500ml的三口瓶中,加入4-甲酰基一苯丙酸(25g,0.14mol)、1-(4-吗啡啉)环戊烯(32.25g,0.21mol)、甲苯150ml,搅拌升温至回流(115℃),搅拌反应6h,取样点板反应完全,减压浓缩至干得淡黄色油状物,加入100ml正己烷搅拌析晶1h,抽滤得2-(2-((2-氧代环戊基)甲基)苯基)丙酸28.6g,类白色固体,收率83.7%,hplc纯度为98.0%。
步骤(3):向100ml的三口瓶中加入2-(2-((2-氧代环戊基)甲基)苯基)丙酸(10g,0.04mol)、乙醇50ml,搅拌升温至溶清,降温至30℃以下,于30℃以下滴加30%碳酸钠溶液39.67g,滴毕,降温至0~5℃,于0~5℃滴加30%双氧水10g,滴毕,于25~30℃反应4h,加入亚硫酸钠淬灭反应,用浓盐酸调ph=1~2,减压浓缩至干,加入50ml水,用2*50ml二氯甲烷萃取水相2次,分层得二氯甲烷层,减压浓缩至干,经柱层析分离得6-(4-(1-羧乙基)苯基)-5-氧代己酸8.0g,收率70.24%,纯度98.9%
实施例6
步骤(1):向500ml的三口瓶中,加入对氯甲基异苯丙酸(80g,0.40mol)、六次亚甲基四胺(112.1g,0.80mol)、冰乙酸240ml,搅拌升温至回流(63~65℃),搅拌反应6h,取样点板反应完全,降温至室温,加入36%浓盐酸60g,搅拌升温至回流反应2h,降温至室温,分层得有机层,有机层用饱和碳酸钠水溶液200ml洗涤,分层得有机层,减压浓缩至干得2-(4-甲酰基苯基)丙酸64.62g,淡黄色固体,收率90.1%hplc纯度为94.0%。
步骤(2):向500ml的三口瓶中,加入4-甲酰基一苯丙酸(25g,0.14mol)、1-(4-吗啡啉)环戊烯(43g,0.28mol)、氯仿150ml,搅拌升温至回流(62℃),搅拌反应10h,取样点板反应完全,减压浓缩至干得淡黄色油状物,加入100ml正己烷搅拌析晶1h,抽滤得2-(2-((2-氧代环戊基)甲基)苯基)丙酸29.66g,类白色固体,收率86.8%,hplc纯度为98.1%。
步骤(3):向100ml的三口瓶中加入2-(2-((2-氧代环戊基)甲基)苯基)丙酸(10g,0.04mol)、乙醇50ml,搅拌升温至溶清,降温至30℃以下,于30℃以下滴加30%氢氧化钠溶液15g,滴毕,降温至0~5℃,于0~5℃分批加入过硫酸氢钾复合盐29.72g,投毕,于0~5℃反应10h,加入亚硫酸钠淬灭反应,用浓盐酸调ph=1~2,减压浓缩至干,加入50ml水,用2*50ml二氯甲烷萃取水相2次,分层得二氯甲烷层,减压浓缩至干,经柱层析分离得6-(4-(1-羧乙基)苯基)-5-氧代己酸7.69g,收率67.5%,纯度98.6%
实施例7
步骤(1):向500ml的三口瓶中,加入对氯甲基异苯丙酸(80g,0.40mol)、六次亚甲基四胺(112.1g,0.80mol)、氯仿240ml,搅拌升温至回流(63~65℃),搅拌反应6h,取样点板反应完全,降温至室温,加入磷酸58g,搅拌升温至回流反应2h,降温至室温,分层得有机层,有机层用饱和碳酸钠水溶液200ml洗涤,分层得有机层,减压浓缩至干得2-(4-甲酰基苯基)丙酸64.98g,淡黄色固体,收率90.6%hplc纯度为94.4%。
步骤(2):向500ml的三口瓶中,加入4-甲酰基一苯丙酸(25g,0.14mol)、1-(4-吗啡啉)环戊烯(43g,0.28mol)、四氯化碳150ml,搅拌升温至回流(77℃),搅拌反应8h,取样点板反应完全,减压浓缩至干得淡黄色油状物,加入100ml正己烷搅拌析晶1h,抽滤得2-(2-((2-氧代环戊基)甲基)苯基)丙酸29.1g,类白色固体,收率85.16%,hplc纯度为98.0%。
步骤(3):向100ml的三口瓶中加入2-(2-((2-氧代环戊基)甲基)苯基)丙酸(10g,0.04mol)、乙醇50ml,搅拌升温至溶清,降温至30℃以下,于30℃以下滴加30%碳酸钾溶液51.83g,滴毕,降温至0~5℃,于0~5℃滴加30%双氧水10g,滴毕,于25~30℃反应4h,加入亚硫酸钠淬灭反应,用浓盐酸调ph=1~2,减压浓缩至干,加入50ml水,用2*50ml二氯甲烷萃取水相2次,分层得二氯甲烷层,减压浓缩至干,经柱层析分离得6-(4-(1-羧乙基)苯基)-5-氧代己酸7.82g,收率68.66%,纯度98.8%
实施例8
步骤(1):向500ml的三口瓶中,加入对氯甲基异苯丙酸(80g,0.40mol)、六次亚甲基四胺(112.1g,0.80mol)、氯仿240ml,搅拌升温至回流(63~65℃),搅拌反应6h,取样点板反应完全,降温至室温,加入浓硫酸58g,搅拌升温至回流反应2h,降温至室温,分层得有机层,有机层用饱和碳酸钠水溶液200ml洗涤,分层得有机层,减压浓缩至干得2-(4-甲酰基苯基)丙酸65.98g,淡黄色固体,收率92.0%hplc纯度为94.2%。
步骤(2):向500ml的三口瓶中,加入4-甲酰基一苯丙酸(25g,0.14mol)、1-(4-吗啡啉)环戊烯(43g,0.28mol)、甲苯150ml,搅拌升温至回流(115℃),搅拌反应6h,取样点板反应完全,减压浓缩至干得淡黄色油状物,加入100ml正己烷搅拌析晶1h,抽滤得2-(2-((2-氧代环戊基)甲基)苯基)丙酸30g,类白色固体,收率87.8%,hplc纯度为98.3%。1h-nmr(400mhz,dmso-d6)d8.35(1h,s),7.19(2h,d),7.14(2h,d),7.10(1h,s),3.62(1h,q),3.43(2h,t),2.50(2h,t),2.40(2h,m),1.41(3h,d)。
步骤(3):向100ml的三口瓶中加入2-(2-((2-氧代环戊基)甲基)苯基)丙酸(10g,0.04mol)、乙醇50ml,搅拌升温至溶清,降温至30℃以下,于30℃以下滴加30%氢氧化钾溶液21g,滴毕,降温至0~5℃,于0~5℃滴加30%双氧水10g,滴毕,于25~30℃反应4h,加入亚硫酸钠淬灭反应,用浓盐酸调ph-1~2,减压浓缩至干,加入50ml水,用2*50ml二氯甲烷萃取水相2次,分层得二氯甲烷层,减压浓缩至干,经柱层析分离得6-(4-(1-羧乙基)苯基)-5-氧代己酸7.9g,收率69.3%,纯度99.1%。
- 还没有人留言评论。精彩留言会获得点赞!