聚酯酰胺及其制备方法以及由该聚酯酰胺制得的纤维与流程
本发明涉及高分子材料技术领域,具体地说,本发明涉及一种聚酯酰胺及其制备方法以及由该聚酯酰胺制得的纤维。
背景技术:
聚酯纤维由于其良好的性能得到了广泛的应用,但由于其缺少亲水基团,在工业丝方面表现为与橡胶亲和力差,需进行多次浸胶处理,在民用丝方面,必须利用分散染料在高温、高压染色,由于分散染料溶解度低,同时需要加入偶氮类助催化剂,目前涤纶纤维越来越多的用于与羊毛等天然蛋白质纤维混纺,用于制备精纺面料。然而,羊毛、真丝等天然蛋白质纤维,采用的均是价格低廉、色谱齐全、色泽鲜艳的酸性染料染色。若用普通聚酯纤维与羊毛、真丝等混纺,无法进行同浴匹染。为了解决这一问题,国内外很多学者专家进行了深入研究。目前主要采用共聚和共混两种方法解决聚酯纤维的可染性。
专利cn101942708b公开了一种聚酯-聚酰胺共聚物,其在酸性染料下上染率大于80%,然而所加入聚酰胺为己内酰胺预聚体,即需提前进行己内酰胺预聚,然后再融化加入共聚,众所周知,己内酰胺聚合过程中会存在约10%单体,进行纺丝前需先水洗去除,同时聚酰胺以低聚物形式加入,降低了聚酰胺对聚酯性能的改善效果。同时专利中尼龙低聚体加入方式为分批加入,给工艺操作带来一定难度与复杂性。
专利cn103232596a公开了一种脂肪族聚酰胺改性共聚酯提高其可染性的方法,专利中需加入间苯二甲酸乙二醇酯-5-磺酸碱金属盐做添加剂,该添加剂价格昂贵,难于合成,同时所加入的聚酰胺为特性粘度0.5~1.0dl/g的低聚物,这就决定聚酰胺只能以嵌段形式嵌入聚酯分子链,同时裸露的端氨基易导致聚酯氨解。
专利特开昭63-256716公报,通过将分子量为200以上的聚乙二醇与磺酸基间苯二甲酸金属盐一起共聚到聚酯分子链中,从而使得聚酯纤维实现了在常压条件下的阳离子染料可染。这是因为柔性的聚乙二醇分子链段的引入,使得聚酯纤维分子链的结构更为疏松、无定型区域增大、玻璃化温度降低,从而使得阳离子染料能够在相对较低的温度下实现上染,即可在常压沸染的条件下染色。但是聚酯分子链中的聚醚链段的引入,会使得聚酯纤维的耐热性差,纺丝不稳定,影响可纺性和织物性能。
专利cn1291081c公开了一种使用聚酰胺与聚乙烯-甲基丙烯酸盐组合形成染色改性剂后加入聚酯切片中制备得到酸性可染聚酯纤维的方法。但由于聚酯与聚酰胺相容性不好,尽管有聚乙烯-甲基丙烯酸盐的改善,其纺丝仍较困难,很难实现工业化纺丝。而且,从微观结构分析,聚酰胺仅仅通过聚乙烯-甲基丙烯酸盐接在聚酯的侧链,造成引入的碱性基团不充分,酸性染料上染能力有限。
专利cn1249141c公开了一种将含有仲胺或仲胺盐的一种或多种单体的聚合物作为改性剂与聚酯组成酸性可染组合物后制备成酸性可染改性聚酯纤维的方法。但由于制备出的含仲胺或仲胺盐的一种或多种单体的聚合物工艺复杂,成本较高,要实现工业化难度较大。
专利cn101585915b通过采用二酰胺二醇、二元酸和脂肪族二元醇进行熔融缩聚,制备同时带有端羧基和端羟基结构的聚酯酰胺预聚物,再以二酰基双内酰胺和二恶唑啉扩链剂进行扩链得到聚酯酰胺。该专利的原料二酰胺二醇非化工常用原料,需预先制备,同时但该方法存在着扩链反应的通病,即分子量分布不易控制,这样会对纺丝造成一定影响。
之前,我们申请的专利cn104892934a虽然解决了上述问题,但在深入研究过程中发现部分聚酯酰胺会出现凝胶问题导致纤维条干不匀及纺丝组件更换周期明显缩短。
技术实现要素:
本发明的目的在于克服现有技术不足,提供一种特定结构的聚酯酰胺,通过调控聚合工艺,有效克服了聚酯酰胺中存在凝胶和纤维条干不匀以及组件更换周期短等问题。
一方面,本发明提供一种由m为不同数值的多个下述结构单元组成的聚酯酰胺:
式中m为≥1的整数,x为2~18的整数,y为2~18的整数,r1~r4各自独立地选自h或c1~c4烷基。
本发明的聚酯酰胺,其核磁定量碳谱中,与酰胺键相连接苯环上,和r1、r3相连碳的积分面积之和与和r2、r4相连碳的积分面积之和的比值为1.2:1~1:1.2,优选为1.1:1~1:1.1,更优选为1.05:1~1:1.05。
在本发明的聚酯酰胺的另一个实施方式中,所述聚酯酰胺的特性粘度为0.3~1.8dl/g,优选为0.5~1.0dl/g。
在本发明的聚酯酰胺的另一个实施方式中,x为2~4的整数,y为4~6的整数。
另一方面,本发明还提供一种制备上述聚酯酰胺的方法,包括:
1)在氮气或者惰性气体的保护下,将二元醇、对苯二甲酸和/或其衍生物、二元胺和/或其衍生物加入反应容器中,控制升温程序,使90%~100%对苯二甲酸和/或其衍生物发生酯化及酰化反应;
2)加热反应容器至230~320℃,进行进一步缩聚,期间抽真空至真空度达30kpa以下,待反应产物特性粘度为0.3~1.8dl/g后,停止反应,得到聚酯酰胺。
在本发明方法的另一个实施方式中,所述二元醇选自碳链长度为2~18个碳原子的脂肪族二元醇中的一种或多种,包括乙二醇、丙二醇、丁二醇、戊二醇、己二醇、庚二醇、辛二醇、壬二醇、癸二醇、十一碳二元醇、十二碳二元醇、十三碳二元醇、十四碳二元醇、十五碳二元醇、十六碳二元醇、十七碳二元醇、十八碳二元醇,优选乙二醇、丙二醇、丁二醇;所述对苯二甲酸和/或其衍生物选自对苯二甲酸、对苯二甲酸单/二酯、对苯二甲酰氯、及以上化合物其苯环上的氢部分或全部被含1~4个碳原子烷烃取代的化合物中的一种或多种,所述二元胺选自碳链长度为2~18个碳原子的脂肪族二元胺中的一种或多种,包括乙二胺、丙二胺、丁二胺、戊二胺、己二胺、庚二胺、辛二胺、壬二胺、癸二胺、十一碳二元胺、十二碳二元胺、十三碳二元胺、十四碳二元胺、十五碳二元胺、十六碳二元胺、十七碳二元胺、十八碳二元胺,优选丁二胺、戊二胺、己二胺;所述二元胺衍生物选自2~18个碳原子的脂肪族二元胺与对苯二甲酸和/或对苯二甲酸的苯环上的氢部分或全部被含1~4个碳原子烷烃取代的二元酸形成的尼龙盐中的一种或多种。
在本发明方法的另一个实施方式中,所述的二元胺和/或其衍生物、二元醇、对苯二甲酸和/或其衍生物的摩尔比为(0.002~0.5):(0.002~3):1,优选为(0.005~0.3):(0.3~2.8):1,更优选为(0.01~0.15):(0.7~2.5):1。
在本发明方法的另一个实施方式中,步骤1)将原料加入反应容器后,首先在室温至150℃将原料搅拌均匀,氮气置换后,逐步升温,保持夹套温度与料温温差小于80℃,冷凝塔柱顶温度低于170℃,料温低于270℃。
在本发明方法的另一个实施方式中,步骤1)中使95%~100%对苯二甲酸和/或其衍生物发生酯化及酰化反应。
在本发明方法的另一个实施方式中,步骤2)中反应容器加热至250~300℃。
在本发明方法的另一个实施方式中,步骤2)中真空度控制在1kpa以下。
在本发明方法的另一个实施方式中,步骤2)中聚酯酰胺特性粘度为0.5~1.0dl/g后,停止反应。
在本发明方法的另一个实施方式中,在步骤1)添加原料时,加入一种或多种助剂,和/或在步骤2)加热反应容器前,加入一种或多种助剂。
在本发明方法的另一个实施方式中,所述助剂包括酯交换催化剂、酯化催化剂、醚化防止剂、聚合催化剂、热稳定剂、光稳定剂、抗氧化剂、耐候剂、润滑剂、结晶成核剂、导电填料或防静电填料、阻燃剂、填充物。
再一方面,本发明还提供一种纤维,所述纤维的原料包括上述聚酯酰胺。
在本发明的纤维的一个实施方式中,所述纤维为聚酯酰胺初生纤维、聚酯酰胺纤维长丝、聚酯酰胺poy纤维、聚酯酰胺加弹丝、聚酯酰胺fdy或聚酯酰胺短纤维。
在本发明的纤维的另一个实施方式中,所述纤维的纤度为0.2~10dtex,优选为0.5~7.0dtex。
在本发明的纤维的另一个实施方式中,所述纤维的强度为1.0~8.0cn/dtex,优选为1.5~5.5cn/dtex。
在本发明的纤维的另一个实施方式中,所述纤维的断裂伸长率5~400%,优选15~130%。
在本发明的纤维的另一个实施方式中,所述纤维的条干不匀率0.1~3.0%,优选0.3~2.0%。
本发明的聚酯酰胺可有效降低聚酯纤维分子结构的紧密性,利于解决染料难以进入无定形区的问题,实现了聚酯酰胺中酰胺链段聚合度的可控性,即均以单结构单元嵌入到聚酯分子链上,克服了以往聚酯-聚酰胺共聚物中仅能将聚酰胺齐聚物嵌段到聚酯链上的弊端,可以以更少的酰胺键、更低的成本实现聚酯性能优化。解决了部分聚酯酰胺凝胶和纺丝过程中纤维不匀率偏高以及纺丝组件更换周期偏短等问题。另外由于控制了酰胺结构的分散性使得整个分子链结构均一,在纺丝过程中可有效提高可纺性,防止断丝、毛丝现象的发生。同时,本发明的聚酯酰胺与聚酯和聚酰胺混合物相比,克服了混合物的相容性差、难于纺丝的问题。
附图说明
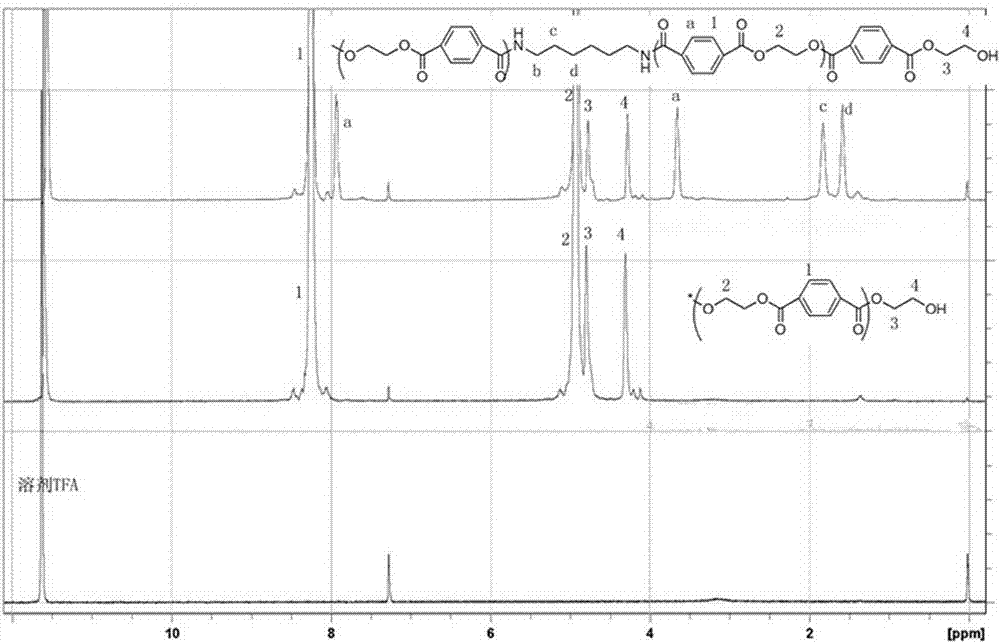
图1为实施例5的聚酯酰胺的h-nmr谱图(由上至下:peta、pet、空白)。
图2为实施例5的聚酯酰胺的c-nmr谱图(由上至下:peta、pet、空白)。
图3为比较例2(左)与实施例2(右)凝胶情况对比。
具体实施方式
下面根据具体实施例对本发明的技术方案做进一步说明。本发明的保护范围不限于以下实施例,列举这些实例仅出于示例性目的而不以任何方式限制本发明。
本发明提供一种由m为不同数值的多个下述结构单元组成的聚酯酰胺:
式中m为≥1的整数,x为2~18的整数,优选2~4的整数,更优选为2;y为2~18的整数,优选4~6的整数,更优选为5;r1~r4各自独立地选自h或c1~c4烷基。
在本发明的聚酯酰胺的另一个实施方式中,所述聚酯酰胺的特性粘度为0.3~1.8dl/g,优选为0.5~1.0dl/g。在本发明的聚酯酰胺的另一个实施方式中,x为2~4的整数,y为4~6的整数。
本发明的聚酯酰胺,其核磁定量碳谱中,与酰胺键相连的苯环上,和r1、r3相连碳的积分面积之和与和r2、r4相连碳的积分面积之和的比值为1.2:1~1:1.2,优选为1.1:1~1:1.1,更优选为1.05:1~1:1.05,其比例基本为1:1,所述偏差由核磁碳谱积分造成,表明聚酰胺单元均以单重复单元嵌入到聚酯片段中。
聚酯酰胺的特性粘度为0.3~1.8dl/g,优选为0.5~1.0dl/g。特性粘度过低会影响力学性能,实用价值降低,特性粘度过高则难于制备。
本发明还提供一种制备上述聚酯酰胺的方法,包括:
1)在氮气或者惰性气体的保护下,将二元醇、对苯二甲酸和/或其衍生物、二元胺和/或其衍生物加入反应容器中,控制升温程序,常压或低压下分馏出低沸点组分(水等小分子产物),使90%~100%对苯二甲酸和/或其衍生物发生酯化及酰化反应,其中上述二元胺和/或其衍生物也可以在酯化及酰化反应完成90%~100%后加入反应容器中;
2)加热反应容器至230~320℃,进行进一步缩聚,期间逐步抽真空至真空度达30kpa以下,待反应产物特性粘度为0.3~1.8dl/g后,停止反应,得到聚酯酰胺。
本发明的聚酯酰胺中可同时存在x、y和m为不同数值的多个结构单元。
步骤1)中所述二元醇选自碳链长度为2~18个碳原子的脂肪族二元醇中的一种或多种,包括乙二醇、丙二醇、丁二醇、戊二醇、己二醇、庚二醇、辛二醇、壬二醇、癸二醇、十一碳二元醇、十二碳二元醇、十三碳二元醇、十四碳二元醇、十五碳二元醇、十六碳二元醇、十七碳二元醇、十八碳二元醇,优选乙二醇、丙二醇、丁二醇。
步骤1)中可同时加入两种或两种以上的二元醇,以形成不同结构的聚酯酰胺。
步骤1)中所述对苯二甲酸和/或其衍生物包括对苯二甲酸,对苯二甲酸单/二酯,对苯二甲酰氯,及以上化合物其苯环上的氢部分或全部被含1~4个碳原子烷烃取代的化合物中的一种或多种。
步骤1)中所述二元胺选自碳链长度为2~18个碳原子的脂肪族二元胺中的一种或多种,包括乙二胺、丙二胺、丁二胺、戊二胺、己二胺、庚二胺、辛二胺、壬二胺、癸二胺、十一碳二元胺、十二碳二元胺、十三碳二元胺、十四碳二元胺、十五碳二元胺、十六碳二元胺、十七碳二元胺、十八碳二元胺,优选丁二胺、戊二胺、己二胺。所述的戊二胺可以是通过化学法制备,也可以采用生物法制备。二元胺的衍生物为上述的二元胺与对苯二甲酸和/或对苯二甲酸的苯环上的氢部分或全部被含1~4个碳原子烷烃取代的二元酸形成的尼龙盐;优选为戊二胺与对苯二甲酸形成的尼龙盐。
步骤1)中可同时加入两种或两种以上的二元胺和/或其衍生物,以形成不同结构的聚酯酰胺。
步骤1)中,所述的二元胺和/或其衍生物、二元醇、对苯二甲酸和/或其衍生物的摩尔比为(0.002~0.5):(0.002~3):1,优选为(0.005~0.3):(0.3~2.8):1,更优选为(0.01~0.15):(0.7~2.5):1。
步骤1)中所述升温程序具体为首先在室温至150℃将原料搅拌均匀,氮气置换后,逐步升温,保持夹套温度与料温温差小于80℃,冷凝塔柱顶温度低于170℃,料温低于270℃。
根据本发明,步骤1)存在酯化和酰化两种反应,而酰化的反应速率常数(约300~400)远高于酯化反应速率常数(约4),因此对苯二甲酸和/或其衍生物会首先与二元胺和/或其衍生物发生酰化反应,我们通过摩尔比和特定升温程序调节可以使聚酰胺的片段仅有一个重复单元,并在后续酯化过程中连接到聚酯中,巧妙地实现了酰胺链段聚合度的可控性,同时有效地控制副反应,避免了凝胶产生,降低了聚酯酰胺纤维的条干不匀率。
根据本发明,步骤1)中酯化终点设定为对苯二甲酸和/或其衍生物发生酯化及酰化反应的比例为90%~100%,优选为95%~100%,酯化及酰化比例高,利于后续缩聚反应的进行,易于控制副反应。酯化率及酰化率可以根据反应体系排出的水来测量。
本发明的聚酯酰胺制备方法中,步骤2)的缩聚温度优选为230~320℃,低温导致缩聚速度降低,高温导致副反应增加,因此更优选缩聚温度为250~300℃。步骤2)中的缩聚真空度优选控制在30kpa以下,为促进缩聚反应的进行得到更高特性粘度的聚酯酰胺,缩聚真空度更优选控制在1kpa以下,为使聚合度逐步上升其减压过程可以采用逐步降低的方式。为使聚酯酰胺具备实际生产意义,待聚酯酰胺特性粘度为0.3~1.8dl/g后,停止反应,更优选特性粘度为0.5~1.0dl/g。
本发明的聚酯酰胺制备方法中,步骤2)缩聚过程中由于二元胺和/或其衍生物的氨基已经完全酰化,会大幅降低其他专利中加入聚酰胺齐聚物端氨基导致聚酯分子链断裂的现象。
步骤2)所得聚酯酰胺为熔体状态,可以通入氮气加压,拉丝、造粒,优选氮气压力为0.05~0.8mpa,切片干燥后可以利用单螺杆纺丝机纺丝,干燥温度优选70~140℃,在中试连续聚合装置上也可以直接将该熔体利用熔体泵输送至纺丝箱制成纤维。
根据本发明,步骤1)和步骤2)中所述的一种或多种助剂包括制造聚酯酰胺化合物时的酯交换催化剂、酯化催化剂、醚化防止剂、或聚合中使用的聚合催化剂、热稳定剂、光稳定剂等各种稳定剂、聚合调节剂等。根据对聚酯酰胺最终性能的需要,该助剂也可选择抗氧化剂、耐候剂、防粘剂、润滑剂、结晶成核剂、增塑剂、抗静电剂、导电填料或防静电填料、阻燃剂、填充物及其他缩聚改良材料等。这些添加剂可以在不损害本发明的效果的范围内根据需要添加。其加入方式可以使用现有公知的方法。
作为酯交换催化剂及酯化催化剂,可例示锰、钴、锌、钛、钙等化合物。作为醚化防止剂,可例示胺化合物等。作为聚合催化剂,可例示含有锗、锑、钛、铝等的化合物。例如,作为含有锗的化合物,可列举出无定形二氧化锗、结晶性二氧化锗、氯化锗、四乙氧基锗、四正丁氧基锗、亚磷酸锗等,其用量优选以聚酯酰胺化合物中的锗原子浓度计设定为5~150ppm,更优选为10~100ppm,进一步优选为15~70ppm。作为含有锑的化合物,可列举出三氧化锑、乙酸锑、酒石酸锑、酒石酸锑钾、氯氧化锑、乙二醇锑、五氧化锑、三苯基锑等,其用量优选以聚酯酰胺化合物中的锑原子浓度计设定为10~400ppm,更优选为20~350ppm,进一步优选为30~300ppm。作为含有钛的化合物,可列举出钛酸四乙酯、钛酸四异丙酯、钛酸四正丙酯、钛酸四正丁酯等钛酸四烷基酯及它们的部分水解产物,草酸钛、草酸钛铵、草酸钛钠、草酸钛钾、草酸钛钙、草酸钛锶等草酸钛化合物,偏苯三酸钛、硫酸钛、氯化钛等,其用量优选以聚酯酰胺化合物中的钛原子浓度计设定为0.5~300ppm,更优选为1~200ppm,进一步优选为3~100ppm。作为含有铝的化合物,可列举出甲酸铝、乙酸铝、丙酸铝、草酸铝等羧酸盐,氧化物、氢氧化铝、氯化铝、氢氧化氯化铝、碳酸铝等无机酸盐,甲醇铝、乙醇铝等烷醇铝,乙酰丙酮铝、乙酰乙酸铝等铝络合物化合物,三甲基铝、三乙基铝等有机铝化合物及它们的部分水解产物等,其用量优选以聚酯酰胺化合物中的铝原子浓度计设定为1~400ppm,更优选为3~300ppm,进一步优选为5~200ppm。另外,本发明的聚酯酰胺化合物的制造中,可以使用碱金属化合物或碱土金属化合物。作为碱金属化合物或碱土金属化合物,可列举出碱金属或碱土金属的羧酸盐、醇盐等。其用量优选以聚酯酰胺化合物中的碱金属或碱土金属原子浓度计设定为0.1~200ppm,更优选为0.5~150ppm,进一步优选为1~100ppm。
另外,本发明的聚酯酰胺化合物的制造中,作为热稳定剂,可以使用1种以上磷酸、亚磷酸、次亚磷酸、膦酸、及它们的衍生物。例如可列举出磷酸、磷酸三甲酯、磷酸三乙酯、磷酸三丁酯、磷酸三苯酯、磷酸单甲酯、磷酸二甲酯、磷酸单丁酯、磷酸二丁酯、亚磷酸、次亚磷酸钠、亚磷酸三甲酯、亚磷酸三乙酯、亚磷酸三丁酯、甲基膦酸、甲基膦酸二甲酯、乙基膦酸二甲酯、苯基膦酸二乙酯、苯基膦酸二苯酯等。其用量优选以聚酯酰胺化合物中的磷原子浓度计设定为1~200ppm,更优选为2~150ppm,进一步优选为3~100ppm。
另外,本发明的聚酯酰胺化合物的制造中,为了调节重均分子量,可以添加月桂醇这样的高级醇。另外,为了改良物性,也可以添加甘油这样的多元醇。
所述抗氧剂包括铜系抗氧化剂、受阻酚系抗氧化剂、受阻胺系抗氧化剂、磷系抗氧化剂、硫系抗氧化剂等,其中,优选受阻酚系抗氧化剂、磷系抗氧化剂。
所述耐候剂,包括苯二酚系化合物、水杨酸酯系化合物、苯并三唑系化合物、二苯甲酮系化合物、受阻胺系化合物等。
所述防粘剂或润滑剂,包括脂肪族醇、脂肪族酰胺、脂肪族双酰胺、双脲、聚乙烯蜡等。
所述结晶成核剂,包括滑石、二氧化硅、高岭土、粘土、氮化硼等无机质微粒或金属氧化物、高熔点尼龙等。
所述增塑剂,包括对羟基苯甲酸辛酯、n-丁基苯磺酰胺等。
所述抗静电剂,包括烷基硫酸盐型阴离子系抗静电剂、季铵盐型阳离子系抗静电剂、聚氧乙烯山梨糖醇酐单硬脂酸酯等非离子系抗静电剂、甜菜碱系两性抗静电剂等。
所述阻燃剂,包括三聚氰胺氰尿酸盐、氢氧化物(例如氢氧化镁、氢氧化铝等)、多磷酸铵、溴化聚苯乙烯、溴化聚苯醚、溴化聚碳酸酯、溴化环氧树脂或这些溴系阻燃剂与三氧化锑的组合等。
所述填充剂,包括玻璃纤维、碳纤维、炭黑、黑墨、硫酸钡、硫酸镁、碳酸钙、碳酸镁、氧化锑、二氧化钛、氧化铝、氧化锌、氧化铁、硫化锌、锌、铅、镍、铝、铜、铁、不锈钢、膨润土、蒙脱土、制造云母等颗粒状、针状、板状填充材料。
所述其他缩聚物,包括其他的聚酰胺、聚乙烯、聚丙烯、聚酯、聚碳酸酯、聚苯醚、聚苯硫醚、液晶聚合物、聚砜、聚醚砜、abs树脂、as树脂、聚苯乙烯等。
本发明还提供了一种纤维,所述纤维的原料包括上述的的聚酯酰胺。根据需要,例如制备海岛纤维,可以在制备纤维的过程中还可添加其它必要的原料。
进一步,所述的纤维为聚酯酰胺初生纤维、聚酯酰胺纤维长丝、聚酯酰胺poy纤维、聚酯酰胺加弹丝、聚酯酰胺fdy或聚酯酰胺短纤维。
本发明的纤维纤度优选为0.2~10dtex,根据应用领域的不同,本发明装置和工艺可以调节单丝纤度。由于单丝纤度过低易出现断头等异常现象,同时单丝纤度过高又会造成织物弯曲强度过高,手感硬。因此,本发明的纤维的纤度更优选范围为0.5~7.0dtex。同时,如果上述单丝纤度保持在发明要求范围内,则可以根据用途选择合适的细丝数,以满足应用要求。
本发明的纤维的强度优选为1.0~8.0cn/dtex。强度过低,在织造过程中易出现绒毛,成品易破损,强度过高,纺丝时可纺性差,易出现断丝现象,根据使用领域的不同,通过聚合和纺丝工艺调节,纤维更优选强度范围为1.5~5.5cn/dtex。
本发明的纤维的断裂伸长率5~400%,优选15~130%。
本发明的纤维的条干不匀率0.1~3.0%,优选0.3~2.0%。
本发明对制备上述聚酰胺改性聚酯纤维的方法没有特别的限定,可以采用现有任一适用的技术,本领域技术人员可以知道确定合适的工艺参数。
在一个实施例中,将本发明的聚酯酰胺切片或熔体利用熔体泵或单螺杆导入纺丝箱,在210~300℃进行纺丝,更优选240~290℃进行纺丝,纺丝速度为200~1500m/min,优选纺丝速度为400~1200m/min,更优选500~1100m/min进行纺丝,得到聚酯酰胺初生纤维。
将聚酯酰胺在210~300℃进行纺丝,纺丝速度1500~3500m/min,更优选纺速为1800~3200m/min,得到聚酯酰胺poy纤维。
将聚酯酰胺在210~300℃进行纺丝,从喷丝板下来的纤维直接进入第一热盘,热盘温度75~100℃,速度800~3000m/min,然后进入第二热盘,热盘温度120~180℃,速度3200~5200m/min,第一第二热盘之间进行纤维的牵伸,牵伸倍数1.1~5倍;从第二导盘出来的纤维束进入卷绕机进行卷装,卷绕速度3100~5200m/min,卷绕后得到聚酯酰胺fdy纤维。
将所述的聚酯酰胺初生纤维在40~90℃进行一级牵伸,牵伸倍数1.5~6倍,80~120℃进行二级牵伸,牵伸倍数1.1~1.6倍,120~160℃进行热定型后得到纤维长丝。
将所述的聚酯酰胺初生纤维在40~90℃进行一级牵伸,牵伸倍数1.5~4倍,80~120℃进行二级牵伸,牵伸倍数1.1~2倍,之后将纤维进行卷曲,卷曲数10~20个/25cm,然后在120~180℃进行热定型15分钟,将定型后的纤维在切断机上切短、打包后得到聚酯酰胺短纤维。
将所述的聚酯酰胺poy纤维在加弹机上以300~1200m/min的速度牵伸1.3~3倍,预热箱温度为120~220℃,d/y为1.4~2.6,定型箱温度140~200℃,卷绕速度800~2200m/min,得到聚酯酰胺加弹丝。
本发明的聚酯酰胺可有效降低聚酯纤维分子结构的紧密型,利于解决染料难以进入无定形区的问题,同时本发明以巧妙的方式实现了聚酯酰胺中酰胺链段聚合度的可控性甚至可以降低到1,克服了以往聚酯-聚酰胺共聚物中仅能将聚酰胺齐聚物嵌段到聚酯链上的弊端,这样可以以更少的酰胺键、更低的成本实现聚酯性能优化、染色性提高的作用。另外由于控制了酰胺结构的分散性使得整个分子链结构均一,这样在纺丝过程中可有效提高可纺性,防止断丝、毛丝现象的发生。在缩聚过程中由于二元胺和/或其衍生物的氨基已经完全酰化,会大幅降低其他专利中加入聚酰胺齐聚物端氨基导致聚酯分子链断裂的现象。
本发明的聚酯酰胺可有效降低聚酯纤维分子结构的紧密性,利于解决染料难以进入无定形区的问题,实现了聚酯酰胺中酰胺链段聚合度的可控性,即均以单结构单元嵌入到聚酯分子链上,克服了以往聚酯-聚酰胺共聚物中仅能将聚酰胺齐聚物嵌段到聚酯链上的弊端,可以以更少的酰胺键、更低的成本实现聚酯性能优化。解决了部分聚酯酰胺凝胶和纺丝过程中纤维不匀率偏高以及纺丝组件更换周期偏短等问题。另外由于控制了酰胺结构的分散性使得整个分子链结构均一,在纺丝过程中可有效提高可纺性,防止断丝、毛丝现象的发生。
本发明所用原料均为普通大众化工原料,无需深加工后使用,聚合通过熔融聚合一步完成,工艺简单、生产效率高,在目前聚酯装置上简单改造即可直接实现生产。该纤维制备工艺简单,在普通聚酯纺丝装置能顺利进行纺丝,所得纤维性能良好,纤维断裂强度>1.0cn/dtex,断裂伸长率>5%,各项指标均能满足后续织造的要求,且该纤维生产操作简单,成本较低,适宜进行工业化生产。同时,本发明的聚酯酰胺与聚酯和聚酰胺混合物相比,克服了混合物的相容性及难于纺丝的问题。
除非另作限定,本发明所用术语均为本领域技术人员通常理解的含义。
以下通过实施例对本发明作进一步地详细说明。
实施例
在实施例和比较例中的各特性,按照以下方法及行业内公知方法测定:
特性粘度η(dl/g),测试方法:参照astmd4603-2003;
熔点tm(℃),测试方法:参照gb/t19466.3-2004;
断裂强度(cn/dtex),测试方法:参照gb/t3916-1997;
断裂伸长率(%),测试方法:参照gb/t3916-1997;
条干不匀率cv(%),测试方法:参照gb/t14346-1993;
纺丝组件更换周期(天):组件压力升至200atm更换;
凝胶含量(%):以苯酚、四氯乙烷混合液(重量比1:1)为溶剂,在90℃下溶解聚酯酰胺样品,不溶部分占初始聚酯酰胺重量百分比。
实施例1制备聚酯酰胺共聚物
将81.5kg对苯二甲酸、0.9kg戊二胺、37.4kg乙二醇、39g乙二醇锑和24.3g磷酸三甲酯,加入到事先在100℃预热的300l酯化反应釜中,搅拌10min后,氮气置换后充氮气至2atm,逐步升温至250℃,保持夹套温度与料温温差小于80℃,冷凝塔柱顶温度低于170℃,利用分馏塔分馏出低沸点组分,期间先在0.3atm下保压,后逐步降至常压,待馏分达到理论量的92%时,结束酯化、酰化反应,反应进行265分钟。将反应液转移至缩聚釜中,逐步升温,先程序低抽真空至1kpa,最后维持高抽,最终体系温度276℃,真空度50pa(绝压),反应进行168min,向聚合釜内充入0.3mpa氮气,拉丝造粒。所得聚酯酰胺特性粘度0.77dl/g,熔点为252℃,凝胶含量0%。
实施例2制备聚酯酰胺共聚物
将80kg对苯二甲酸、38.4kg乙二醇、4kg对苯二甲酸戊二胺盐(尼龙5t盐)加入到事先在100℃预热的300l酯化反应釜中,搅拌10min后,氮气置换后充氮气至2atm,逐步升温至260℃,保持夹套温度与料温温差小于80℃,冷凝塔柱顶温度低于170℃,利用分馏塔分馏出低沸点组分,期间先在0.3atm下保压,后逐步降至常压,待馏分达到理论量的96%时,结束酯化、酰化反应,反应进行290分钟。向体系中加入39g乙二醇锑、24.3g磷酸三甲酯,搅拌均匀后再将反应液转移至缩聚釜中,逐步升温,先程序低抽真空至1kpa,最后维持高抽,最终体系温度282℃,真空度56pa(绝压),反应进行175min,向聚合釜内充入0.3mpa氮气,拉丝造粒。所得聚酯酰胺特性粘度0.78dl/g,熔点为248.5℃,凝胶含量0%。
实施例3制备聚酯酰胺共聚物
将93.5kg对苯二甲酸二甲酯与37.4kg乙二醇和8kg对苯二甲酸戊二胺盐(尼龙5t盐)、钛酸四丁酯19g加入到事先在100℃预热的300l酯化反应釜中,搅拌10min后,氮气置换后充氮气至2atm,逐步升温至250℃,保持夹套温度与料温温差小于80℃,冷凝塔柱顶温度低于170℃,利用分馏塔分馏出低沸点组分,期间先在0.3atm下保压,后逐步降至常压,待馏分达到理论量的92%时,结束酯交换、酰化反应,反应进行265分钟。向体系中加入24g钛酸四丁酯缩聚催化剂,搅拌均匀后再将反应液转移至缩聚釜中,逐步升温,先程序低抽真空至1kpa,最后维持高抽,最终体系温度272℃,真空度25pa(绝压),反应进行188min,向聚合釜内充入0.3mpa氮气,拉丝造粒。所得聚酯酰胺特性粘度0.74dl/g,熔点为243℃,凝胶含量0%。
实施例4制备聚酯酰胺共聚物
将80kg对苯二甲酸、37.4kg乙二醇和16kg对苯二甲酸戊二胺盐(尼龙5t盐)、39g乙二醇锑和32g亚磷酸磷酸三苯酯,加入到事先在100℃预热的300l酯化反应釜中,搅拌10min后,氮气置换后充氮气至2atm,逐步升温至250℃,保持夹套温度与料温温差小于80℃,冷凝塔柱顶温度低于170℃,利用分馏塔分馏出低沸点组分,期间先在0.3atm下保压,后逐步降至常压,待馏分达到理论量的96%时,结束酯化、酰化反应,反应进行248分钟。将反应液转移至缩聚釜中,逐步升温,先程序低抽真空至1kpa,最后维持高抽,最终体系温度268℃,真空度32pa(绝压),反应进行175min,向聚合釜内充入0.3mpa氮气,拉丝造粒。所得聚酯酰胺特性粘度0.74dl/g,熔点为239℃,凝胶含量0%。
实施例5制备聚酯酰胺共聚物
将80kg对苯二甲酸、37.4kg乙二醇和4kg对苯二甲酸己二胺盐(尼龙6t盐)、39g乙二醇锑和24.3g磷酸三甲酯,加入到事先在100℃预热的300l酯化反应釜中,搅拌10min后,氮气置换后充氮气至2atm,逐步升温至255℃,保持夹套温度与料温温差小于80℃,冷凝塔柱顶温度低于170℃,利用分馏塔分馏出低沸点组分,期间先在0.3atm下保压,后逐步降至常压,待馏分达到理论量的97%时,结束酯化、酰化反应,反应进行276分钟。将反应液转移至缩聚釜中,逐步升温,先程序低抽真空至1kpa,最后维持高抽,最终体系温度275℃,真空度36pa(绝压),反应进行184min,向聚合釜内充入0.3mpa氮气,拉丝造粒。所得聚酯酰胺特性粘度0.76dl/g,熔点为249℃,凝胶含量0%。
实施例6制备聚酯酰胺共聚物
将80kg对苯二甲酸、54.3kg丁二醇和4kg对苯二甲酸戊二胺盐(尼龙5t盐)、39g乙二醇锑和24.3g磷酸三甲酯,加入到事先在100℃预热的300l酯化反应釜中,搅拌10min后,氮气置换后充氮气至2atm,逐步升温至252℃,保持夹套温度与料温温差小于80℃,冷凝塔柱顶温度低于170℃,利用分馏塔分馏出低沸点组分,期间先在0.3atm下保压,后逐步降至常压,待馏分达到理论量的94%时,结束酯化、酰化反应,反应进行276分钟。将反应液转移至缩聚釜中,逐步升温,先程序低抽真空至1kpa,最后维持高抽,最终体系温度270℃,真空度36pa(绝压),反应进行180min,向聚合釜内充入0.3mpa氮气,拉丝造粒。所得聚酯酰胺特性粘度0.70dl/g,熔点为217℃,凝胶含量0%。
实施例7制备聚酯酰胺poy
实施例2得到的聚酯酰胺切片在120℃真空干燥10h后,在285℃下纺丝,然后经过侧吹风冷却,丝束上油后通过纺丝甬道在两个导丝辊的带动下进入卷绕辊,由卷绕辊对聚酰胺纤维丝束进行卷绕,卷绕时的环境温度为25℃,环境湿度为70%,卷绕辊的线速度为3000m/min,最终形成聚酯酰胺纤维卷装,该纤维断裂强度2.1cn/dtex,断裂伸长率130%。条干不匀率1.3%,组件更换周期12天,无毛丝。
实施例8制备聚酯酰胺长丝
实施例1得到的聚酯酰胺切片在120℃真空干燥10h后,在282℃下纺丝,卷绕速度600m/min,得到聚酯酰胺初生纤维,将该初生纤维75℃进行一级牵伸,牵伸倍数3倍,115℃进行二级牵伸,牵伸倍数1.4倍,150℃进行热定型后得到纤维长丝。该纤维断裂强度3.74cn/dtex,断裂伸长率31%。条干不匀率1.2%,组件更换周期15天,无毛丝。
实施例9制备聚酯酰胺加弹丝
实施例3得到的聚酯酰胺切片在120℃真空干燥12h后,在280℃下纺丝,卷绕速度2800m/min,得到聚酯酰胺共聚物poy纤维,将该纤维在加弹机上以469m/min的速度喂入,牵伸1.6倍,预热箱温度为180℃,d/y为1.6,卷绕速度750m/min,得到聚酯酰胺加弹丝。该纤维断裂强度2.81cn/dtex,断裂伸长率30%。条干不匀率1.3%,无毛丝。
实施例10制备聚酯酰胺fdy纤维
实施例5得到的聚酯酰胺切片在120℃真空干燥12h后,在288℃下纺丝,第一热盘温度70℃,速度2500m/min,然后进入第二热盘,热盘温度130℃,速度4200m/min,第一第二热盘之间进行纤维的牵伸倍数1.68倍;从第二导盘出来的纤维束进入卷绕机进行卷装,卷绕速度4180m/min,卷绕后得到聚酯酰胺fdy纤维。该纤维断裂强度4.1cn/dtex,断裂伸长率32%。条干不匀率1.3%,组件更换周期13天,无毛丝。
实施例11制备聚酯酰胺短纤维
实施例4得到的聚酯酰胺切片在120℃真空干燥10h后,将经预处理过的聚酯酰胺共聚物在278℃下纺丝,卷绕速度800m/min,得到聚酯酰胺初生纤维集束后在55℃进行一级牵伸,牵伸倍数3.2倍,120℃进行二级牵伸,牵伸倍数1.3倍,之后将纤维进行卷曲,然后在160℃进行松弛热定型,将定型后的纤维在切断机上切短、打包后得到纤度为1.5dtex,长度为38mm的棉型聚酯酰胺共聚物短纤维。该纤维断裂强度2.84cn/dtex,断裂伸长率39%,卷曲数13个/25mm。条干不匀率1.4%,组件更换周期12天。
实施例12制备聚酯酰胺初生纤维
实施例6得到的聚酯酰胺切片在120℃真空干燥10h后,加入单螺杆挤出机中,温度278℃进行熔融挤出制备得到聚酯酰胺熔体,所得熔体经计量泵计量后进入纺丝箱体,在纺丝箱体的纺丝组件中经过熔体分配、匀化后从喷丝板孔中喷出形成熔体细流;所述熔体细流经过冷却、上油后在纺丝速度为900m/min进行卷绕,得到聚酯酰胺共聚物初生纤维。该初生纤维的断裂强度1.8cn/dtex,断裂伸长率130%。条干不匀率1.2%,组件更换周期16天,无毛丝。
比较例1制备聚酯酰胺共聚物
将81.5kg对苯二甲酸、37.4kg乙二醇、39g乙二醇锑和24.3g磷酸三甲酯,加入到事先在100℃预热的300l酯化反应釜中,搅拌10min后,氮气置换后充氮气至2atm,程序升温至250℃,利用分馏塔分馏出低沸点组分,期间先在0.3atm下保压,后逐步降至常压,待馏分达到理论量的94%时,结束酯化、酰化反应,反应进行265分钟。向体系中加入0.9kg戊二胺,搅拌均匀后再将反应液转移至缩聚釜中,逐步升温,先程序低抽真空至1kpa,最后维持高抽,最终体系温度278℃,真空度30pa(绝压),反应进行172min,向聚合釜内充入0.3mpa氮气,拉丝造粒。所得聚酯酰胺特性粘度0.78dl/g,熔点为251℃,凝胶含量1.2%。
比较例2制备聚酯酰胺共聚物
将80kg对苯二甲酸、38.4kg乙二醇加入到事先在100℃预热的300l酯化反应釜中,搅拌10min后,氮气置换后充氮气至2atm,程序升温至260℃,利用分馏塔分馏出低沸点组分,期间先在0.3atm下保压,后逐步降至常压,待馏分达到理论量的98%时,结束酯化反应,反应进行305分钟。向体系中加入39g乙二醇锑、24.3g磷酸三甲酯、4kg对苯二甲酸戊二胺盐(尼龙5t盐),搅拌均匀后再将反应液转移至缩聚釜中,逐步升温,先程序低抽真空至1kpa,最后维持高抽,最终体系温度280℃,真空度52pa(绝压),反应进行182min,向聚合釜内充入0.3mpa氮气,拉丝造粒。所得聚酯酰胺特性粘度0.80dl/g,熔点为248℃,凝胶含量3.2%。
比较例3制备聚酯酰胺长丝
比较例1得到的聚酯酰胺切片在120℃真空干燥10h后,在282℃下纺丝,卷绕速度600m/min,得到聚酯酰胺初生纤维,将该初生纤维75℃进行一级牵伸,牵伸倍数3倍,115℃进行二级牵伸,牵伸倍数1.4倍,150℃进行热定型后得到纤维长丝。该纤维断裂强度3.58cn/dtex,断裂伸长率30%。条干不匀率2.6%,组件更换周期0.4天,有毛丝。
比较例4制备聚酯酰胺poy
比较例2得到的聚酯酰胺切片在120℃真空干燥10h后,在285℃下纺丝,然后经过侧吹风冷却,丝束上油后通过纺丝甬道在两个导丝辊的带动下进入卷绕辊,由卷绕辊对聚酰胺纤维丝束进行卷绕,卷绕时的环境温度为25℃,环境湿度为70%,卷绕辊的线速度为3000m/min,最终形成聚酯酰胺纤维卷装,该纤维断裂强度2.0cn/dtex,断裂伸长率136%。条干不匀率2.8%,组件更换周期0.5天,有毛丝。
以上实施例只是对技术方案的解释,不构成对本发明技术方案的限制。本领域的技术人员根据现有聚酯酰胺制备方法的知识,可以通过调整制备过程中的原料配比,制备过程的温度、压力等,实现不同粘数的聚酯酰胺。
本领域技术人员应当注意的是,本发明所描述的实施方式仅仅是示范性的,可在本发明的范围内作出各种其他替换、改变和改进。因而,本发明不限于上述实施方式,而仅由权利要求限定。
- 还没有人留言评论。精彩留言会获得点赞!