微滴式数字PCR探针法反应体系、检测方法和应用与流程
本发明涉及分子生物学领域,尤其是涉及一种微滴式数字pcr探针法反应体系、检测方法和应用。
背景技术:
数字pcr(digitalpcr,dpcr)方法发明于20世纪90年代末,并在此后获得迅速发展。与荧光定量pcr的整管pcr定量方式截然不同,数字pcr方法采用“有限稀释”分区和泊松分布算法等原理对初始模板进行绝对计数,从而达到临床需求的对肿瘤基因、病原基因等的绝对定量。将20-50μl含有待测模板、特异引物和探针的pcr体系均匀分散成少至768多至千万个分隔空间,并进行完整的pcr循环后,含有待测模板的分区会发出较强的荧光,而不含待测模板的分区则不会。通过泊松分布可以计算出加入体系中含有待测模板的绝对量,从而推算出病原体绝对数量、模板突变率、拷贝数变异、基因表达量等信息。当分区数量越多,待测模板分散越均匀,绝对定量也越精确。但随着分区的增加,数据量也呈几何级数增加,对设备和计算能力的要求越高。因此,在实际应用中,分区数与数据量之间通常会相互平衡。
数字pcr设备在21世纪初获得较大的进步,目前欧美国家推出了几款数字pcr仪,主要分为芯片式和微滴式。
2006年fluidigm公司推出了第一台商品化的基于芯片的商品化数字pcr系统。2008年德国inostics推出基于磁珠法乳浊液扩增和流式检测方式的beaming数字pcr检测服务。2013年下半年,life-technologies推出了quantstudio3d数字pcr系统,这也是目前数字pcr市场上最新型号的产品,其采用高密度的纳升流控芯片技术,样本均匀分配至20,000个单独的反应孔中。但这些方法无论在分散程度和数据群体的体量上都无法达到更加精细的要求,时间和耗材成本严重限制了数字pcr技术的发展。
近几年来,随着纳米制造技术和微流体技术的发展,数字pcr技术遇到了突破技术瓶颈的最佳契机。伴随二代测序技术发展起来的“油包水pcr”技术,可以一次生成数万乃至数百万个纳升甚至皮升级别的单个油包水微滴,可以作为数字pcr的样品分散载体。quantalife利用油包水微滴生成技术开发了微滴式数字pcr技术,并于2011年被bio-rad公司收购,其微滴式数字pcr仪产品更名为qx100型号仪继续在市场上销售。2013年bio-rad又推出了升级型号qx200。这也是最早出现的相对成熟的数字pcr平台。2012年,raindance公司推出raindrop型号数字pcr设备,这个设备将其原有的二代测序文库制备平台技术平移到数字pcr技术平台,在高压气体驱动下,将每个标准反应体系分割成包含100万至1000万个皮升级别微滴的反应乳液。
在基因组变异研究领域,群体基因组水平上的snp(singlenucleotidepolymorphism,单核苷酸多态性)检测面临的问题主要是突变序列往往与大量正常序列同时存在,竞争性反应严重影响突变序列的检测精度,数字pcr技术带来的极高的扩增特异性在稀有突变检测方面具有天然的优势,在癌症标志物检测、无创产前检查、线粒体突变检测等研究方向上具有极大优势。cnv(copynumbervariations,拷贝数变异)研究则需要极高的定量精度以区别不同拷贝数之间的微小差异,测序方法适用于高于30%的变异率检测,而qpcr的最高分辨在1.5倍左右,数字pcr通过直接计数目标基因与参照基因(拷贝数为1的基因,例如rnasep)的数目,计算比值,直接得到目标基因的拷贝数可以达到极高的拷贝数分辨精度。此外,数字pcr在病原微生物检测、microrna研究、基因表达研究、ngs测序文库的绝对定量、表观遗传学直接相关的dna甲基化定量检测、chip定量鉴定等领域的应用都令人极为期待,而在转基因成分鉴定、分子标准品精确定量、环境样本检测等具体的应用方向上,已经有大量实验数据和结果证明了数字pcr技术的优良性能。
与传统qpcr技术相比,数字pcr技术具有极高的灵敏度、特异性和精确性,但截止目前而言,数字pcr仍然是一种全新的检测方法,是一种全新的技术思路和手段。数字pcr技术相关的仪器、耗材、试剂和方法的开发与创新,将具有无有止境的空间。但目前数字pcr技术的应用成本相对较高,耗材使用不方便,导致数字pcr设备推广速度较慢。数字pcr试剂体系封闭,导致基于该体系的试剂开发和优化严重受到制约。尤其是目前商品化的数字pcr试剂均存在一些问题,如芯片式数字pcr的试剂不适用于微滴式数字pcr设备,只适用于科研实验室进行低通量数字pcr实验,不适合临床检测体系的开发,也不适合用于基因突变检测试剂盒的开发;又如微滴式数字pcr仪目前只有bio-rad和raindance两家,其中bio-rad的试剂采用封闭式系统(封闭的pcrmix体系及配套封闭的dropletoil体系,不允许用户更改配方以获得相应的kit),而raindance则采用开放式系统(兼容的qpcrmix和第三方采购的dropletoil),但qpcrmix并不针对数字pcr进行优化,会导致“rain”效应,即二维散点弥散,尤其是在raindance的微滴数达到千万级的情况下,rain效应将严重干扰数据的正常分析。
因此,开发一种新的开放式数字pcr试剂体系及其方法对于数字pcr设备和方法的推广极其重要。
技术实现要素:
基于此,本发明提供一种可以降低rain效应的微滴式数字pcr探针法反应体系、检测方法和应用。
一种微滴式数字pcr探针法反应体系,包括连续相试剂和分散相试剂;所述连续相试剂包括氟油与氟表面活性剂,所述表面活性剂为全氟聚醚和/或含全氟聚醚的嵌段氟化合物;所述分散相试剂为微滴式数字pcr反应试剂。
在其中一个实施例中,所述氟油选自c9h5f15o、c12f27n及c9f21n中的至少一种。
在其中一个实施例中,所述氟表面活性剂选自如下结构式表示的二嵌段共聚物化合物中的至少一种:
且所述氟表面活性剂的平均分子量为4000-9000,其中n选自20-60的整数,m选自9-40的整数。
在其中一个实施例中,所述氟表面活性剂选自三嵌段共聚物pfpe-peg-pfpe及pfpe-lpg-pfpe中的至少一种。
在其中一个实施例中,pfpe-peg-pfpe的结构式如下面所示:
其中,n选自20-80,m选自9-40,p选自20-80。
在其中一个实施例中,所述连续相试剂为包含氟油与氟表面活性剂的混合液,其中,所述氟油与所述氟表面活性剂的质量比为98:2至90:10。
在其中一个实施例中,所述分散相试剂中dntp的浓度为0.2-0.4mm,tris-hcl的浓度为10-100mm,(nh4)2so4的浓度为10-30mm,mgcl2的浓度为1.5-5mm,热启动taq酶的浓度为0.5-1u/μl,bsa的浓度为0.05-0.5wt%。
一种检测试剂盒,含有上述任一实施例所述的微滴式数字pcr探针法反应体系。
一种微滴式数字pcr探针法检测方法,使用上述任一实施例所述的微滴式数字pcr探针法反应体系,所述微滴式数字pcr探针法检测方法包括如下步骤:
向所述分散相试剂中加入引物对、探针和模板,所述引物对的两条引物的浓度分别为200-800nm,所述探针的浓度为200-800nm;
将所述连续相试剂及添加有引物对、探针和模板的分散相试剂分别通入微滴生成芯片的油相通道和水相通道中;
将所述微滴生成芯片中的分散相和连续相连续通过相应的通道,生成直径为20~40μm的微滴;
将生成的微滴接入低吸附pcr管中,并在pcr仪上进行pcr反应;
pcr反应完成后进行荧光强度检测分析。
在其中一个实施例中,所述微滴式数字pcr探针法检测方法还包括在通入分散相试剂和连续相试剂之前,对所述微滴生成芯片进行亲氟化处理的步骤。
在其中一个实施例中,在pcr反应时,pcr程序要求全程升降温速率控制在2-4℃/s,具体的程序是:94℃预处理5-10分钟;94℃、10-20秒,60-65℃、60-90秒,共25-50个循环;4℃冷却1-5分钟。
通过在微滴式数字pcr探针法反应体系的连续相试剂中添加全氟聚醚和/或含全氟聚醚的嵌段氟化合物的氟表面活性剂,该氟表面活性剂可以作为微滴稳定剂,降低微滴的表面张力,使得在检测时生成的微滴具有均一直径(cv值小于5%)、热稳定性高等特性。
进一步,通过加入常规用量两倍左右的dntp和探针,在不改变热启动taq酶结构,并使用常规探针的情况下,可实现微滴式数字pcr在更少循环数下达到更高荧光强度的效果,从而可以从信噪比强度角度来减弱rain效应。
本发明的微滴式数字pcr探针法反应体系是一种开放式体系,适合根据不同需求对体系进行进一步优化从而获得专一性的各类数字pcr检测试剂盒。
附图说明
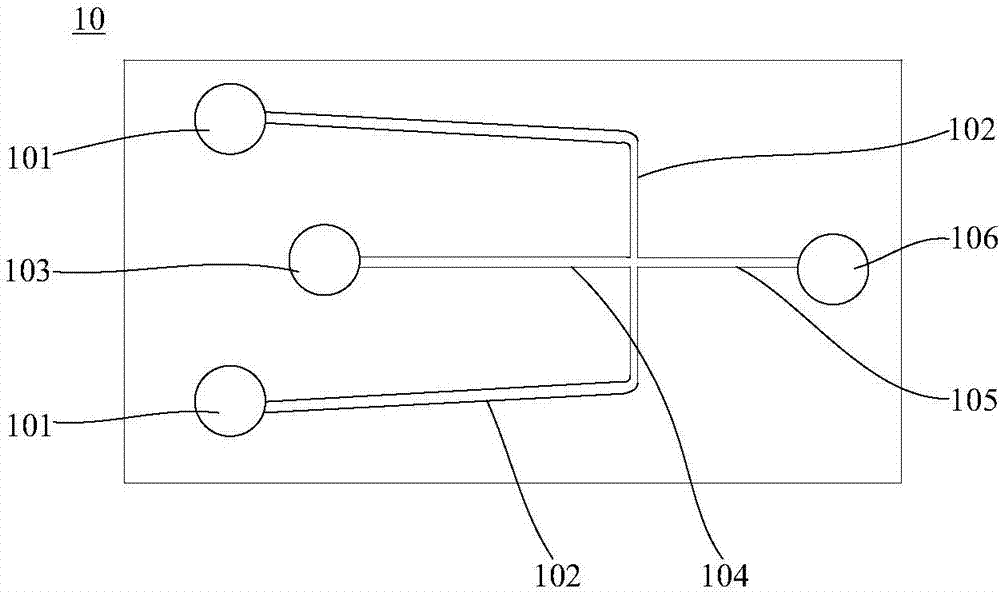
图1为一实施方式所使用的微滴生成芯片的结构示意图。
图2为实施例1中反应原料与产物红外光谱对比图,其中:实线表示产物pfpe-tris红外光谱,虚线表示原料pfpe-cooh红外光谱。
图3为实施例1中产物pfpe-tris氟谱图。
图4为实施例3中产物pfpe-tris氢谱图。
图5为实施例2中三段式氟表面活性剂产物氢谱图。
图6为实施例2中三段式氟表面活性剂产物氟谱图。
图7为实施例4中微滴生成后及时取样显微镜下观察图,图中图例长度为200μm。
图8为实施例4中微滴生成静置7天后取样显微镜下观察图,图中图例长度为200μm。
图9为实施例4中微滴生成后及时取样显微镜下观察图,图中图例长度为100μm。
图10为实施例4中微滴生成静置7天后取样显微镜下观察图,图中图例长度为100μm。
图11为实施例4中微流控芯片生成均一的油包水微滴示意图。
图12为实施例4中pcr扩增热循环前显微镜下观察图,图中图例长度为200μm。
图13为实施例4中pcr扩增热循环后显微镜下观察图,图中图例长度为200μm。
图14为图13虚线框放大视野图,图中图例长度为100μm。
图15为图14在汞灯环境下调整曝光率后微滴荧光示意图。
图16为微滴进行荧光信号采集波形图。
图17为图15经滤波处理后的波形图。
图18为实施例5中ddpcr(微滴式数字pcr)后fam、vic两通道检测信号波形局部示意图。
图19为实施例5中ddpcr后fam、vic两通道结果二维散点分析示意图。
图20为对比例1中同样dna浓度下不同浓度的dntp、引物与探针的荧光定量pcr结果示意图。
具体实施方式
为了便于理解本发明,下面将参照相关附图对本发明进行更全面的描述。附图中给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
本发明一实施方式的微滴式数字pcr探针法反应体系,包括连续相试剂和分散相试剂。连续相试剂包括氟油与氟表面活性剂。分散相试剂为微滴式数字pcr反应试剂。
在一个实施例中,氟油选自c9h5f15o、c12f27n及c9f21n中的至少一种。
表面活性剂为全氟聚醚和/或含全氟聚醚的嵌段氟化合物。在一个实施例中,氟表面活性剂选自如下结构式表示的二嵌段共聚物化合物中的至少一种:
且氟表面活性剂的平均分子量为4000-9000,优选为6000-9000,其中n选自20-60的整数,m选自9-40的整数。
pfpe-tris可以通过但不限于如下方法制备得到:以4-二甲氨基吡啶作为反应的催化剂,在惰性气体保护下,将全氟聚醚pfpe-cooh、三羟甲基氨基甲烷(tris)与4-二甲氨基吡啶溶于反应溶剂中,在60℃-120℃的范围下搅拌反应12hr-36hr,回收溶剂,即得。优选的,在反应后还包括纯化步骤。在纯化步骤中,将反应得到的产物再次加入氟油溶解,然后滴加二氯甲烷,使产物中的pfpe-tris析出,干燥后即得。其中,所述反应溶剂为甲氧基-九氟代丁烷等氟油溶剂,并进一步包括有选自三氟甲苯、二甲基甲酰胺、四氢呋喃和二甲基亚砜中的至少一种的极性有机溶剂。
pfpe-peg的制备原料是全氟聚醚pfpe-cooh与聚乙二醇(peg),制备过程与pfpe-tris的制备过程类似。
在另一个实施例中,氟表面活性剂选自pfpe-peg-pfpe及pfpe-lpg-pfpe中的至少一种。其中,pfpe-peg-pfpe的结构式如下面所示:
其中,n选自20-80,m选自9-40,p选自20-80。
该三段式氟表面活性剂的制备方法,所选用的原料含氟聚醚在80℃-100℃下为液态,聚氧乙烯(聚乙二醇二胺)也为液态,可直接混合,在无溶剂的条件下即可反应,且不需要生成酰氯,直接就合成了全氟聚醚聚氧乙烯的酰胺键,缩短了合成路线。
在一个实施例中,连续相试剂为包含氟油与氟表面活性剂的混合液,其中,氟油与氟表面活性剂的质量比为98:2-90:10,优选95:5。
在一个实施例中,分散相试剂中dntp的浓度为0.2-0.4mm,优选0.4mm,tris-hcl的浓度为10-100mm,(nh4)2so4的浓度为10-30mm,mgcl2的浓度为1.5-5mm,热启动taq酶的浓度为0.5-1u/μl,优选0.5u/μl,bsa的浓度为0.05-0.5wt%,优选0.1wt%。
本发明还提供了一种微滴式数字pcr探针法检测方法,其使用上述微滴式数字pcr探针法反应体系,包括如下步骤:
步骤一:向分散相试剂中加入引物对、探针和模板,引物对的两条引物的浓度分别为200-800nm,探针的浓度为200-800nm,优选400nm。
步骤二:将连续相试剂及添加有引物对、探针和模板的分散相试剂分别通入微滴生成芯片的油相通道和水相通道中。
优选的,在一个实施例中,在通入分散相试剂和连续相试剂之前,优选对微滴生成芯片进行亲氟化处理。
本实施方式所使用的微滴生成芯片的结构可以但不限于图1所示,在图1所示的微滴生成芯片10中,其包括连续相加样槽101、连续相微通道102、分散相加样槽103、分散相微通道104、微滴生成通道105及收集槽106等结构。其中,连续相微通道102有两条,两条连续相微通道102与一条分散相微通道104形成十字交叉结构,微滴生成通道105位于连续相微通道102与分散相微通道104的交叉部位的后方,且与连续相微通道102与分散相微通道104连通。分散相微通道中流通的水相液体在两侧的连续相微通道102中的油相液体的剪切力的作用下,形成微滴,通过控制对连续相加样槽101和分散相加样槽103施加的压力,可以控制相应液体的流速,得到不同尺寸的微滴样品,并最终汇聚在收集槽106中。
步骤三:将微滴生成芯片中的分散相和连续相连续通过相应的通道,生成直径为20~40μm的微滴。
步骤四:将生成的微滴接入低吸附pcr管中,并在pcr仪上进行pcr反应。
在一个实施例中,在pcr反应时,pcr程序要求全程升降温速率控制在2-4℃/s,具体的程序是:94℃预处理5-10分钟;94℃、10-20秒,60-65℃、60-90秒,共25-50个循环;4℃冷却1-5分钟。优选的,在另一个实施例中,在pcr反应时,pcr程序要求全程升降温速率控制在2℃/s,具体的程序是:94℃预处理10分钟;94℃、10秒,60℃、60秒,共25-45个循环;72℃延伸。
步骤五:pcr反应完成后进行荧光强度检测分析。
本发明的反应体系通过在连续相试剂中添加全氟聚醚和/或含全氟聚醚的嵌段氟化合物的氟表面活性剂,该氟表面活性剂可以作为微滴稳定剂,降低微滴的表面张力,使得在检测时生成的微滴具有均一直径(cv值小于5%)、热稳定性高等特性。
进一步,通过加入常规用量两倍左右的dntp和探针,在不改变热启动taq酶结构,并使用常规探针的情况下,可实现微滴式数字pcr在更少循环数下达到更高荧光强度的效果,从而可以从信噪比强度角度来减弱rain效应,提高检测结果的精度。
本发明的微滴式数字pcr探针法反应体系是一种开放式体系,适合根据不同需求对体系进行进一步优化从而获得专一性的各类数字pcr检测试剂盒。
以下为具体实施例部分。
实施例1
一种氟表面活性剂pfpe-tris,通过以下方法制备得到:
一、反应。
按照如下反应线路进行:
所述反应的具体条件为:在惰性气体保护下,将式ii的全氟聚醚(购自杜邦,型号为fsh157,该全氟聚醚的平均分子量为7000-7500)、式iii的三羟甲基氨基甲烷(tris)与4-二甲氨基吡啶溶(dmap)于氟油(购自3m,型号为hfe-7100)与三氟甲苯的混合反应溶剂中,所述hfe-7100与所述三氟甲苯的体积比为1.6,在80℃-100℃的范围下搅拌反应24hr,抽滤后旋干溶剂。
二、纯化。
将步骤一得到的产物再次加入hfe-7100溶解,滴加二氯甲烷清洗将最终产物析出并进行旋蒸,然后真空干燥12小时后得到白色透明油状物的最终产物,即得式i的氟表面活性剂(pfpe-tris)。
三、验证。
将上述pfpe-tris进行红外图谱及核磁图谱(氟谱、氢谱)检测核实。
从图谱中可以看出,如图2所示,在红外图谱中可以清晰观察到在1675cm-1位置上有明显的产物酰胺c=o特征吸收峰。如图3所示,氟谱中虚线图框内即化学位移在-83~-74范围的一系列共振峰可以核实是pfpe该成分;此外图4氢谱中化学位移3.5~5范围中的两个单峰的积分面积比值约为3:4,可以证实为tris结构,即验证所得产物即为式i的pfpe-tris。
实施例2
一种氟表面活性剂pfpe-peg-pfpe,通过以下方法制备得到:
(1)反应。
首先称取式ii的含氟聚醚(购自krytox,型号为157fsh,n=32-48)60g,式iii的聚乙二醇二胺(平均分子量m=600,m约为11-14)3g,4-二甲氨基吡啶(dmap)1.22g,直接混合,在80℃真空度-0.05~-0.1mpa的条件下下搅拌10h。
(2)纯化。
然后加入混合溶剂,该混合溶剂包括20mlhfe7100和20mlthf(四氢呋喃),再在室温下搅拌2.5h,将反应液过滤,去除不溶物,得产物溶液,搅拌下加入100ml无水乙醇,取下层析出的粘稠物,真空干燥,即得到分子式为c3f7o(c3f6o)nconh(c2h4o)mch2ch2nh(c3f6o)nc3f7o的式i的三段式氟表面活性剂,其中n=32-48,m=11-14。
(3)验证。
将上述三段式氟表面活性剂进行核磁图谱(氟谱、氢谱)检测核实。
图5为三段式氟表面活性剂产物的氢谱,从图中可以看出,δ3.74-3.75处的峰代表聚乙二醇亚甲基峰,即表示产物结构中含聚乙二醇分子;图6为三段式氟表面活性剂产物的氟谱,从图中可以看出,δ-74.30~-74.37,-82.04~-82.09,-88.89处的峰代表-cf,-cf2,-cf3峰(-76.2,-77.3,-82.29为cf3cooh及hfe7100的峰),即表示产物中含氟聚醚结构;即验证所得产物即为式i结构的三段式氟表面活性剂。
实施例3
一种连续相试剂,将实施例1制备得到的氟表面活性剂pfpe-tris按质量百分浓度2%的量溶于hfe-7500氟油(购自3m,型号为hfe-7500)中,即得。
实施例4
利用实施例3的连续相试剂进行微滴生成实验。
一、静置稳定性测试。
对实施例3的微滴生成油进行了微滴静置稳定性的测试,使用生成后立即取样的微滴与放置7天后的微滴进行对比,如图7-10所示,在显微镜可以观察到两者几乎没有区别,微滴照样能呈现蜂巢式均一性排布,可以说明该油相所制微滴具备优秀的静置稳定性能。
二、微滴式数字pcr测试。
对实施例3的微滴生成油进行微滴式数字pcr测试。具体为:以hotstartmastermix配合引物、探针及dna模板作为水相,如图11所示,在微滴生成芯片中生成均一的油包水微滴,进行pcr扩增热循环。
pcr扩增热循环前显微镜下观察到微滴形态如图12所示,pcr扩增热循环后显微镜下观察到微滴形态如图13所示。在显微镜下观察到,微滴经30至50圈热循环后,其均一性状态依然保持良好,仍然呈现规律的蜂窝形貌,破损及融合微滴的存在率(cv值)远低于5%。
将图13中虚线框的局部放大,得到图14的局部视图。在汞灯环境下调整曝光率,清晰观察到,扩增后呈阳性的微滴荧光强度相对其他阴性微滴更加明亮,如图15所示,该图15为图14相同视野图。
再对微滴荧光强度进行定量分析,利用光机模块对每个通过的微滴进行荧光信号采集并进行滤波处理,如图16-17所示,在随机一段采集片段中可以清晰看到,几个阳性峰的峰值强度是众多阴性峰值的4倍左右,符合显微镜观察结果。
实施例5
actb/gapdh双通道检测
一、pcr反应体系:
(1)连续相试剂:3mnovec7500氟油+实施例2制备的pfpe-peg-pfpe,其中pfpe-peg-pfpe质量百分含量为5%。
(2)分散相试剂:ddpcrmix+探针+引物对:
其中:
actb通用引物对(5’-3’):
正向引物:gatcagcaagcaggagtatgacg、
反向引物:gatgctcgctccaac;
gapdh通用引物对(5’-3’):
正向引物:gctccctctttctttgcagcaat、
反向引物:taccatgagtccttccacgatac。
按配方配制pcr预混液两组,其中一组为ntc组,每份20μl,加入2μl无菌水,共3份;另一组为样品组,每份20μl,加入人类基因组dna2μl(10ng/μl),共3份。
二、检测方法
(1)反应前确认微滴生成芯片表面已接受亲氟化处理。
(2)将连续相通入微滴生成芯片油相通道中。
(3)将分散相通入微滴生成芯片水相通道中。
(4)将连续相和分散相以4:2-4:3压力比连续通过微滴生成芯片,生成直径20-40μm微滴。每20μl样品可生成直径40μm微滴30万个,可生成直径20μm微滴240万个。
(5)将生成的直径40μm的微滴接入低吸附pcr管中,调整体积至100μl。
(6)将装有微滴的pcr管在pcr仪上进行pcr反应操作。本实施例的pcr程序要求升降温速率控制在2℃/s,pcr循环数可以25~45之间,pcr程序是:94℃预处理10分钟;94℃、10秒,60℃、60秒,共40个循环;4℃冷却3分钟。
(7)pcr反应完成后的每管微滴在数字pcr检测仪中直接进行荧光强度检测,也可以将pcr管置于4℃保存不超过24h后进行荧光强度检测。检测通道为fam通道和vic/hex通道。所得数据在软件中进行后续分析。
三、反应结果
结果如图18-19所示,图18所示的检测结果波形图阳性微滴和阴性微滴荧光强度比例大于4:1;图19所示的二维散点图阴性微滴和阳性微滴可以明显区分。
对比例1
引物、探针和dntp加倍实验可减少数字pcr程序循环次数以减少对微滴的破坏,并提高数字pcr终点荧光强度。
一、pcr反应体系:
(1)连续相试剂:3mnovec7500氟油+实施例2制备的pfpe-peg-pfpe,其中pfpe-peg-pfpe质量百分含量为5%。
(2)分散相试剂:ddpcrmix+探针+引物对:
试剂组1
试剂组2
其中:
gapdh引物对(5’-3’):
正向引物:gctccctctttctttgcagcaat
反向引物:taccatgagtccttccacgatac
按配方配制pcr反应体系两组,其中一组为试剂组1,引物、探针和dntp为常规用量,每份20μl,共3份;另一组为试剂组2,每份20μl,共3份。
二、检测方法
(1)将装有上述反应体系的pcr管在荧光定量pcr仪上进行操作。本实施例的pcr程序要求升降温速率控制在4℃/s,pcr程序是:94℃预处理10分钟;94℃、10秒,60℃、60秒,共40个循环,每个循环结束时进行实时荧光强度扫描,荧光检测通道设置为vic/hex通道开启;4℃冷却3分钟。
(7)所得数据在软件中进行后续分析。
三、反应结果
结果如图20所示,引物、探针和dntp浓度为常规浓度时,荧光强度在25轮达到阈值(500rfu),至35轮以后达到峰值(6000rfu);引物、探针和dntp加倍后,荧光强度在第8轮达到阈值(500rfu),至第19轮达到6000rfu,至40轮达到9000rfu。由此,本例应用于数字pcr,引物、探针和dntp加倍,可显著减少数字pcr程序循环次数,以减少对微滴的破坏;或在同样循环次数条件下,显著提高数字pcr终点荧光强度,以提高信号分辨率。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
-
 0访客 来自[江苏省常州市电信] 2020年03月20日 14:31PCR(聚合酶链式反应)技术的基本原理类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性→退火→延伸三个基本反应步骤,荧光定量检测根据所使用的标记物不同可分为荧光探针(BIOGHC Elitefast SYBR Kit)和荧光染料(BIOGHSC Super Probe Kit),PCR的应用:核酸定量分析。 对传染性疾病进行定量定性分析,病原微生物或病毒含量的检测 , 比如近期流行的甲型H1N1流感, 转基因动植物基因拷贝数的检测,RNAi 基因失活率的检测等; 基因表达差异分析。 比较经过不同处理样本之间特定基因的表达差异 ( 如药物处理、物理处理、化学处理等 ) ,特定基因在不同时相的表达差异以及cDNA芯片或差显结果的确证。
0访客 来自[江苏省常州市电信] 2020年03月20日 14:31PCR(聚合酶链式反应)技术的基本原理类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性→退火→延伸三个基本反应步骤,荧光定量检测根据所使用的标记物不同可分为荧光探针(BIOGHC Elitefast SYBR Kit)和荧光染料(BIOGHSC Super Probe Kit),PCR的应用:核酸定量分析。 对传染性疾病进行定量定性分析,病原微生物或病毒含量的检测 , 比如近期流行的甲型H1N1流感, 转基因动植物基因拷贝数的检测,RNAi 基因失活率的检测等; 基因表达差异分析。 比较经过不同处理样本之间特定基因的表达差异 ( 如药物处理、物理处理、化学处理等 ) ,特定基因在不同时相的表达差异以及cDNA芯片或差显结果的确证。