一种细胞中内源性表达RNA聚合酶的核酸构建物的制作方法
本发明涉及生物技术领域,较佳地,涉及一种细胞中内源性表达rna聚合酶的核酸构建物。
背景技术:
rna聚合酶是一种rna聚合酶(rnapolymerase):以一条dna链或rna为模板催化由核苷-5′-三磷酸合成rna的酶,是催化以dna为模板(template)、三磷酸核糖核苷为底物、通过磷酸二酯键而聚合的合成rna的酶,因为在细胞内与基因dna的遗传信息转录为rna有关,所以也称转录酶。
t7rna聚合酶(t7rnap)是来源于t7噬菌体的一种rna聚合酶,识别23nt的保守启动子序列(pt7)并提供较强的转录活性。t7rnap广泛应用于大肠杆菌(escherichiacoli)等原核生物中外源基因的表达,在真核生物中也有成功应用的报。在酵母系统中,将t7rnap在酿酒酵母(saccharomycescerevisiae)或者毕赤酵母(pichiapastoris)中表达时,均能实现靶基因的高效转录,虽然其转录产物并不能被有效翻译成蛋白质。目前,无论是大肠杆菌(de3质粒)还是酵母(2μ质粒)中,t7rnap均是以外源质粒的方式存在于细胞中的,尚无直接重组至其基因组中稳定高效率表达t7rnap的细胞系统。
蛋白质体外合成系统是指在细菌、真菌、植物细胞或动物细胞的裂解体系中,加入mrna或者dna模板、rna聚合酶及氨基酸和atp等组分,完成外源蛋白的快速高效翻译。目前,经常实验的商业化体外蛋白表达系统包括大肠杆菌系统(e.coliextract,ece)、兔网织红细胞(rabbitreticulocytelysate,rrl)、麦胚(wheatgermextract,wge)、昆虫(insectcellextract,ice)和人源系统。
克鲁维酵母(kluyveromyces)是一种子囊孢子酵母,其中的马克斯克鲁维酵母(kluyveromycesmarxianus)和乳酸克鲁维酵母(kluyveromyceslactis)是工业上广泛使用的酵母。例如乳酸克鲁维酵母是一种能够以乳酸作为其唯一的碳源和能源的酵母。与其他酵母相比,乳酸克鲁维酵母具有许多优点,如超强的分泌能力,良好的大规模发酵特性、食品安全的级别及同时具有蛋白翻译后修饰的能力等,其作为宿主系统表达药用蛋白也已显示出巨大的潜力。
t7rnap广泛应用于细胞体外表达系统,高效转录外源目的基因,最终实现蛋白的体外翻译。t7rnap的浓度,对蛋白的翻译效率也有显著影响。在现有的文献报导及商业试剂盒中,都是通过外源手动加入t7rnap蛋白质,包括大肠杆菌系统(e.coliextract,ece)、兔网织红细胞(rabbitreticulocytelysate,rrl)、麦胚(wheatgermextract,wge)、昆虫(insectcellextract,ice)和人源系统,对实验效率、成本、复杂性及稳定性都有不好的影响。
因此,本领域迫切需要开发一种能够增强蛋白质翻译效率的新的核酸构建物。
技术实现要素:
本发明的目的在于提供一种能够增强蛋白质翻译效率的新的核酸构建物。
本发明第一方面提供了一种核酸构建物,所述的构建物具有从5’至3’的式i结构:
z1-z2(i)
式中,
z1、z2分别为用于构成所述构建物的元件;
各“-”独立地为键或核苷酸连接序列;
z1为启动子元件,所述启动子元件选自下组:rnr2、adh1、gapdh、tef1、pgk1、sed1、或其组合;
z2为rnp蛋白的编码序列;
并且,所述z1、z2来源于酵母。
在另一优选例中,所述启动子元件选自下组:scrnr2、scadh1、scgapdh、sctef1、scpgk1、scsed1、klrnr2、kladh1、klgapdh、kltef1、klpgk1、klsed1、或其组合。
在另一优选例中,以所述启动子scrnr2为基准,所述启动子元件的强度为启动子scrnr2的±20%-±50%。
在另一优选例中,所述启动子scrnr2的强度以转录本的数量为参照。
在另一优选例中,所述启动强度是指,启动子调控其下游编码基因转录表达水平的能力,启动强度增强则下游编码基因的转录表达水平提高,启动强度减弱则下游编码基因的转录表达水平降低。
在另一优选例中,所述rnp蛋白选自下组:t7rnap蛋白、t3rnap、t4rnap、t5rnap、或其组合。
在另一优选例中,所述酵母选自下组:酿酒酵母、克鲁维酵母属酵母、或其组合。
在另一优选例中,所述克鲁维酵母属酵母选自下组:乳酸克鲁维酵母、马克斯克鲁维酵母、多布克鲁维酵母(kluyveromycesdobzhanskii)、或其组合。
在另一优选例中,所述核酸构建物的序列如seqidno.:1所示。
本发明第二方面提供了一种核酸构建物,所述的构建物具有从5’至3’的式ii结构:
z1-z2-z3(ii)
式中,
z1、z2、z3分别为用于构成所述构建物的元件;
各“-”独立地为键或核苷酸连接序列;
z1为启动子元件,所述启动子元件选自下组:scrnr2、scadh1、scgapdh、sctef1、scpgk1、scsed1、klrnr2、kladh1、klgapdh、kltef1、klpgk1、klsed1、或其组合;
z2为rnp蛋白的编码序列;
z3为外源蛋白的编码序列;
并且,所述z1、z2来源于酵母。
在另一优选例中,所述外源蛋白的编码序列来自原核生物、真核生物。
在另一优选例中,所述外源蛋白的编码序列来自动物、植物、病原体。
在另一优选例中,所述外源蛋白的编码序列来自哺乳动物,较佳地灵长动物,啮齿动物,包括人、小鼠、大鼠。
在另一优选例中,所述的外源蛋白的编码序列编码选自下组的外源蛋白:荧光素蛋白、或荧光素酶(如萤火虫荧光素酶)、绿色荧光蛋白、黄色荧光蛋白、氨酰trna合成酶、甘油醛-3-磷酸脱氢酶、过氧化氢酶、肌动蛋白、抗体的可变区域、萤光素酶突变体、α-淀粉酶、肠道菌素a、丙型肝炎病毒e2糖蛋白、胰岛素前体、干扰素αa、白细胞介素-1β、溶菌酶素、血清白蛋白、单链抗体段(scfv)、甲状腺素运载蛋白、酪氨酸酶、木聚糖酶、或其组合。
在另一优选例中,所述外源蛋白选自下组:荧光素蛋白、或荧光素酶(如萤火虫荧光素酶)、绿色荧光蛋白、黄色荧光蛋白、氨酰trna合成酶、甘油醛-3-磷酸脱氢酶、过氧化氢酶、肌动蛋白、抗体的可变区域、萤光素酶突变、α-淀粉酶、肠道菌素a、丙型肝炎病毒e2糖蛋白、胰岛素前体、干扰素αa、白细胞介素-1β、溶菌酶素、血清白蛋白、单链抗体段(scfv)、甲状腺素运载蛋白、酪氨酸酶、木聚糖酶、或其组合。
本发明第三方面提供了一种载体或载体组合,所述的载体或载体组合含有本发明第一方面或本发明第二方面所述的核酸构建物。
本发明第四方面提供了一种基因工程细胞,所述基因工程细胞的基因组的一个或多个位点整合有本发明第一方面或本发明第二方面所述的构建物,或者所述基因工程细胞中含有本发明第三方面所述的载体或载体组合。
在另一优选例中,所述基因工程细胞为酵母细胞。
在另一优选例中,所述酵母细胞选自下组:酿酒酵母、克鲁维酵母属酵母、或其组合。
在另一优选例中,所述克鲁维酵母属酵母选自下组:乳酸克鲁维酵母、马克斯克鲁维酵母、多布克鲁维酵母(kluyveromycesdobzhanskii)、或其组合。
本发明第五方面提供了一种试剂盒,所述试剂盒中包含的试剂选自下组中的一种或多种:
(a)本发明第一方面或本发明第二方面所述的构建物;
(b)本发明第三方面所述的载体或载体组合;和
(c)本发明第四方面所述的基因工程细胞;
在另一优选例中,所述试剂盒还包括(d)酵母体外蛋白合成体系。
在另一优选例中,所述酵母体外蛋白合成体系为克鲁维酵母体外蛋白合成体系(优选乳酸克鲁维酵母体外蛋白合成体系)。
本发明第六方面提供了一种如本发明第一方面或本发明第二方面所述的构建物、本发明第三方面所述的载体或载体组合、本发明第四方面所述的基因工程细胞或本发明第五方面所述试剂盒的用途,用于进行高通量的体外蛋白合成。
本发明第七方面提供了一种体外高通量的外源蛋白合成方法,包括步骤:
(i)在酵母体外蛋白合成体系存在下,提供本发明第一方面或本发明第二方面所述的核酸构建物;
(ii)在适合的条件下,孵育步骤(i)的酵母体外蛋白合成体系一段时间t1,从而合成所述外源蛋白。
在另一优选例中,所述方法还包括:(iii)任选地从所述酵母体外蛋白合成体系中,分离或检测所述外源蛋白。
在另一优选例中,所述酵母体外蛋白合成体系为克鲁维酵母体外蛋白合成体系(优选乳酸克鲁维酵母体外蛋白合成体系)。
在另一优选例中,所述外源蛋白的编码序列来自原核生物、真核生物。
在另一优选例中,所述外源蛋白的编码序列来自动物、植物、病原体。
在另一优选例中,所述外源蛋白的编码序列来自哺乳动物,较佳地灵长动物,啮齿动物,包括人、小鼠、大鼠。
在另一优选例中,所述的外源蛋白的编码序列编码选自下组的外源蛋白:荧光素蛋白、或荧光素酶(如萤火虫荧光素酶)、绿色荧光蛋白、黄色荧光蛋白、氨酰trna合成酶、甘油醛-3-磷酸脱氢酶、过氧化氢酶、肌动蛋白、抗体的可变区域、萤光素酶突变体、α-淀粉酶、肠道菌素a、丙型肝炎病毒e2糖蛋白、胰岛素前体、干扰素αa、白细胞介素-1β、溶菌酶素、血清白蛋白、单链抗体段(scfv)、甲状腺素运载蛋白、酪氨酸酶、木聚糖酶、或其组合。
在另一优选例中,所述外源蛋白选自下组:荧光素蛋白、或荧光素酶(如萤火虫荧光素酶)、绿色荧光蛋白、黄色荧光蛋白、氨酰trna合成酶、甘油醛-3-磷酸脱氢酶、过氧化氢酶、肌动蛋白、抗体的可变区域、萤光素酶突变、α-淀粉酶、肠道菌素a、丙型肝炎病毒e2糖蛋白、胰岛素前体、干扰素αa、白细胞介素-1β、溶菌酶素、血清白蛋白、单链抗体段(scfv)、甲状腺素运载蛋白、酪氨酸酶、木聚糖酶、或其组合。
在另一优选例中,所述步骤(ii)中,反应温度为20-37℃,较佳地,22-35℃。
在另一优选例中,所述步骤(ii)中,反应时间为1-10h,较佳地,2-8h。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
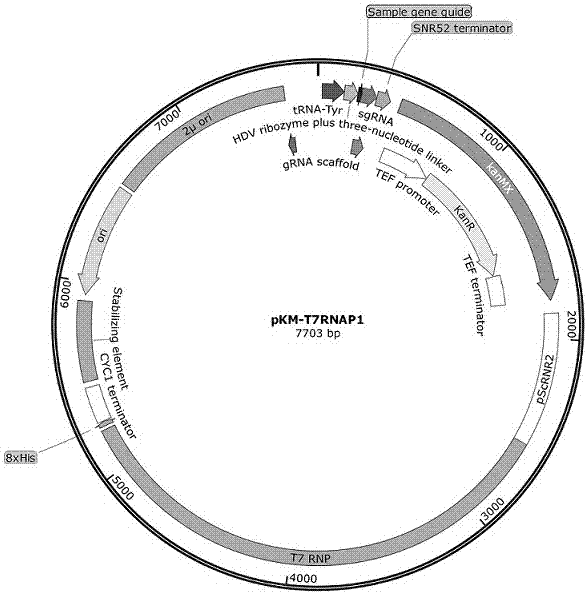
图1显示了pkm-t7rnap1质粒结构示意图;
图2显示了pkm-cas1.0-kltdh3-1质粒结构示意图;
图3显示了pkm-cas1.0-kltdh3-2质粒结构示意图;
图4显示了pkm-kltdh3-1-t7-dd质粒结构示意图;
图5显示了pkm-kltdh3-2-t7-dd质粒结构示意图;
图6显示了pkm-cas1.0-klxrn1质粒结构示意图;
图7显示了pkm-klxrn1-t7-dd质粒结构示意图;
图8显示了转化pkm-t7rnap1质粒菌株体外翻译活性测定示意图;
图9显示了kltdh3-1-t7rnap融合蛋白菌株体外翻译活性测定示意图;
图10显示了kltdh3-2-t7rnap融合蛋白菌株体外翻译活性测定示意图;
图11显示了klxrn1δ-pscrnr2-t7rnap改造菌株体外翻译活性测定示意图。
具体实施方式
经过广泛而深入的研究,通过大量筛选和摸索,首次意外地发现了一种可大幅度增强蛋白质翻译效率的新型核酸构建物,本发明的核酸构建物由具有特定启动强度的启动子(如rnr2、adh1、gapdh、tef1、pgk1、sed1等)和rnap(如t7rnap、t3rnap、t4rnap、t5rnap等)蛋白的编码序列构成,在酵母体外蛋白质合成体系中应用本发明的核酸构建物,所合成的荧光素酶活性的相对光单位值(rlu)非常高,可达到与外源加入t7rnap相同效果,可高达6.6×107。在此基础上,本发明人完成了本发明。
酵母体外蛋白质合成体系
酵母(yeast)兼具培养简单、高效蛋白质折叠、和翻译后修饰的优势。其中酿酒酵母(saccharomycescerevisiae)和毕氏酵母(pichiapastoris)是表达复杂真核蛋白质和膜蛋白的模式生物,酵母也可作为制备体外翻译系统的原料。
克鲁维酵母(kluyveromyces)是一种子囊孢子酵母,其中的马克斯克鲁维酵母(kluyveromycesmarxianus)和乳酸克鲁维酵母(kluyveromyceslactis)是工业上广泛使用的酵母。与其他酵母相比,乳酸克鲁维酵母具有许多优点,如超强的分泌能力,更好的大规模发酵特性、食品安全的级别、以及同时具有蛋白翻译后修饰的能力等。
在本发明中,酵母体外蛋白质合成体系不受特别限制,一种优选的酵母体外蛋白质合成体系为克鲁维酵母表达系统(更佳地,乳酸克鲁维酵母表达系统)。
在本发明中,所述酵母体外蛋白质合成体系包括:
(a)酵母细胞提取物;
(b)聚乙二醇;
(c)任选的外源蔗糖;和
(d)任选的溶剂,所述溶剂为水或水性溶剂。
在一特别优选的实施方式中,本发明提供的体外蛋白合成体系包括:酵母细胞提取物,4-羟乙基哌嗪乙磺酸,醋酸钾,醋酸镁,腺嘌呤核苷三磷酸(atp),鸟嘌呤核苷三磷酸(gtp),胞嘧啶核苷三磷酸(ctp),胸腺嘧啶核苷三磷酸(ttp),氨基酸混合物,磷酸肌酸,二硫苏糖醇(dtt),磷酸肌酸激酶,rna酶抑制剂,荧光素,萤光素酶dna,rna聚合酶。
在本发明中,rna聚合酶没有特别限制,可以选自一种或多种rna聚合酶,典型的rna聚合酶为t7rna聚合酶。
在本发明中,所述酵母细胞提取物在体外蛋白合成体系中的比例不受特别限制,通常所述酵母细胞提取物在体外蛋白质合成蛋白合成体系中所占体系为20-70%,较佳地,30-60%,更佳地,40-50%。
在本发明中,所述的酵母细胞提取物不含完整的细胞,典型的酵母细胞提取物包括用于蛋白翻译的核糖体、转运rna、氨酰trna合成酶、蛋白质合成需要的起始因子和延伸因子以及终止释放因子。此外,酵母提取物中还含有一些源自酵母细胞的细胞质中的其他蛋白,尤其是可溶性蛋白。
在本发明中,所述的酵母细胞提取物所含蛋白含量为20-100mg/ml,较佳为50-100mg/ml。所述的测定蛋白含量方法为考马斯亮蓝测定方法。
在本发明中,所述的酵母细胞提取物的制备方法不受限制,一种优选的制备方法包括以下步骤:
(i)提供酵母细胞;
(ii)对酵母细胞进行洗涤处理,获得经洗涤的酵母细胞;
(iii)对经洗涤的酵母细胞进行破细胞处理,从而获得酵母粗提物;
(iv)对所述酵母粗提物进行固液分离,获得液体部分,即为酵母细胞提取物。
在本发明中,所述的固液分离方式不受特别限制,一种优选的方式为离心。
在一优选实施方式中,所述离心在液态下进行。
在本发明中,所述离心条件不受特别限制,一种优选的离心条件为5000-100000×g,较佳地,8000-30000×g。
在本发明中,所述离心时间不受特别限制,一种优选的离心时间为0.5min-2h,较佳地,20min-50min。
在本发明中,所述离心的温度不受特别限制,优选的,所述离心在1-10℃下进行,较佳地,在2-6℃下进行。
在本发明中,所述的洗涤处理方式不受特别限制,一种优选的洗涤处理方式为采用洗涤液在ph为7-8(较佳地,7.4)下进行处理,所述洗涤液没有特别限制,典型的所述洗涤液选自下组:4-羟乙基哌嗪乙磺酸钾、醋酸钾、醋酸镁、或其组合。
在本发明中,所述破细胞处理的方式不受特别限制,一种优选的所述的破细胞处理包括高压破碎、冻融(如液氮低温)破碎。
所述体外蛋白质合成体系中的核苷三磷酸混合物为腺嘌呤核苷三磷酸、鸟嘌呤核苷三磷酸、胞嘧啶核苷三磷酸和尿嘧啶核苷三磷酸。在本发明中,各种单核苷酸的浓度没有特别限制,通常每种单核苷酸的浓度为0.5-5mm,较佳地为1.0-2.0mm。
所述体外蛋白质合成体系中的氨基酸混合物可包括天然或非天然氨基酸,可包括d型或l型氨基酸。代表性的氨基酸包括(但并不限于)20种天然氨基酸:甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸、苯丙氨酸、脯氨酸、色氨酸、丝氨酸、酪氨酸、半胱氨酸、蛋氨酸、天冬酰胺、谷氨酰胺、苏氨酸、天冬氨酸、谷氨酸、赖氨酸、精氨酸和组氨酸。每种氨基酸的浓度通常为0.01-0.5mm,较佳地0.02-0.2mm,如0.05、0.06、0.07、0.08mm。
在优选例中,所述体外蛋白质合成体系还含有聚乙二醇或其类似物。聚乙二醇或其类似物的浓度没有特别限制,通常,聚乙二醇或其类似物的浓度(w/v)为0.1-8%,较佳地,0.5-4%,更佳地,1-2%,以所述蛋白合成体系的总重量计。代表性的peg例子包括(但并不限于):peg3000,peg8000,peg6000和peg3350。应理解,本发明的体系还可包括其他各种分子量的聚乙二醇(如peg200、400、1500、2000、4000、6000、8000、10000等)。
在优选例中,所述体外蛋白质合成体系还含有蔗糖。蔗糖的浓度没有特别限制,通常,蔗糖的浓度为0.03-40wt%,较佳地,0.08-10wt%,更佳地,0.1-5wt%,以所述蛋白合成体系的总重量计。
一种特别优选的体外蛋白质合成体系,除了酵母提取物之外,还含有以下组分:22mm,ph为7.4的4-羟乙基哌嗪乙磺酸,30-150mm醋酸钾,1.0-5.0mm醋酸镁,1.5-4mm核苷三磷酸混合物,0.08-0.24mm的氨基酸混合物,25mm磷酸肌酸,1.7mm二硫苏糖醇,0.27mg/ml磷酸肌酸激酶,1%-4%聚乙二醇,0.5%-2%蔗糖,8-20ng/µl萤火虫荧光素酶的dna,0.027-0.054mg/mlt7rna聚合酶。
外源蛋白的编码序列(外源dna)
如本文所用,术语“外源蛋白的编码序列”与“外源dna”可互换使用,均指外源的用于指导蛋白质合成的dna分子。通常,所述的dna分子为线性的或环状的。所述的dna分子含有编码外源蛋白的序列。
在本发明中,所述的编码外源蛋白的序列的例子包括(但并不限于):基因组序列、cdna序列。所述的编码外源蛋白的序列还含有启动子序列、5'非翻译序列、3'非翻译序列。
在本发明中,所述外源dna的选择没有特别限制,通常,外源dna选自下组:编码荧光素蛋白、或荧光素酶(如萤火虫荧光素酶)、绿色荧光蛋白、黄色荧光蛋白、氨酰trna合成酶、甘油醛-3-磷酸脱氢酶、过氧化氢酶、肌动蛋白、抗体的可变区域的外源dna、萤光素酶突变体的dna、或其组合。
外源dna还可以选自下组:编码α-淀粉酶、肠道菌素a、丙型肝炎病毒e2糖蛋白、胰岛素前体、干扰素αa、白细胞介素-1β、溶菌酶素、血清白蛋白、单链抗体段(scfv)、甲状腺素运载蛋白、酪氨酸酶、木聚糖酶的外源dna、或其组合。
在一优选实施方式中,所述外源dna编码选自下组的蛋白:绿色荧光蛋白(enhancedgfp,egfp)、黄色荧光蛋白(yfp)、大肠杆菌b-半乳糖苷酶(β-galactosidase,lacz)、人赖氨酸-trna合成酶(lysine-trnasynthetase)、人亮氨酸-trna合成酶(leucine-trnasynthetase)、拟南芥甘油醛3-磷酸脱氢酶(glyceraldehyde-3-phosphatedehydrogenase)、鼠过氧化氢酶(catalase)、或其组合。
核酸构建物
本发明提供了一种核酸构建物,所述核酸构建物含有结构如式i所示的核酸序列:
z1-z2(i)
式中,
z1、z2分别为用于构成所述构建物的元件;
各“-”独立地为键或核苷酸连接序列;
z1为启动子元件,所述启动子元件选自下组:rnr2、adh1、gapdh、tef1、pgk1、sed1、或其组合;
z2为rnp蛋白的编码序列;
并且,所述z1、z2来源于酵母。
本发明还提供了一种核酸构建物,所述的构建物具有从5’至3’的式ii结构:
z1-z2-z3(ii)
式中,
z1、z2、z3分别为用于构成所述构建物的元件;
各“-”独立地为键或核苷酸连接序列;
z1为启动子元件,所述启动子元件选自下组:rnr2、adh1、gapdh、tef1、pgk1、sed1、或其组合;
z2为rnp蛋白的编码序列;
z3为外源蛋白的编码序列;
并且,所述z1、z2来源于酵母。
在本发明中,所述外源蛋白的编码序列的选择没有特别限制,通常,外源蛋白的编码序列选自下组:编码荧光素蛋白、或荧光素酶(如萤火虫荧光素酶)、绿色荧光蛋白、黄色荧光蛋白、氨酰trna合成酶、甘油醛-3-磷酸脱氢酶、过氧化氢酶、肌动蛋白、抗体的可变区域的外源dna、萤光素酶突变体的dna、或其组合。
外源蛋白的编码序列还可以编码选自下组的蛋白:α-淀粉酶、肠道菌素a、丙型肝炎病毒e2糖蛋白、胰岛素前体、干扰素αa、白细胞介素-1β、溶菌酶素、血清白蛋白、单链抗体段(scfv)、甲状腺素运载蛋白、酪氨酸酶、木聚糖酶、或其组合。
此外,本发明的所述核酸构建物可以是线性的,也可以是环状的。本发明的所述核酸构建物可以是单链的,也可以是双链的。本发明的所述核酸构建物可以是dna,也可以是rna,或dna/rna杂合。
在一优选实施方式中,本发明的核酸构建物的序列如seqidno.:1所示。
在另一优选例中,所述的构建物还包括选自下组的元件或其组合:启动子、终止子、poly(a)元件、转运元件、基因靶向元件、筛选标记基因、增强子、抗性基因、转座酶编码基因。
多种选择性标志基因均可应用于本发明,包括但不限于:营养缺陷型标记,抗性标记,报告基因标记。选择性标志的应用对于重组细胞(重组子)的筛选起到作用,使得受体细胞能够与未转化的细胞进行显著区分。营养缺陷型标记是通过转入的标记基因与受体细胞突变基因互补,从而使受体细胞表现野生型生长。抗性标记是指将抗性基因转入受体细胞中,转入的基因使受体细胞在一定的药物浓度下表现抗药性。作为本发明的优选方式,应用抗性标记来实现重组细胞的便捷筛选。
在本发明中,在本发明的酵母体外蛋白质合成体系中应用本发明的核酸构建物,可显著提高蛋白翻译的效率,具体地,应用本发明的核酸构建物所合成的荧光素酶活性的相对光单位值高达6.6×107。
载体,基因工程细胞
本发明还提供了一种载体或载体组合,所述载体含有本发明的核酸构建物。优选地,所述载体选自:细菌质粒、噬菌体、酵母质粒、或动物细胞载体、穿梭载体;所述的载体为转座子载体。用于制备重组载体的方法是本领域普通技术人员所熟知的。只要其能够在宿主体内复制和稳定,任何质粒和载体都是可以被采用的。
本领域普通技术人员可以使用熟知的方法构建含有本发明所述的启动子和/或目的基因序列的表达载体。这些方法包括体外重组dna技术、dna合成技术、体内重组技术等。
本发明还提供了一种基因工程细胞,所述的基因工程细胞含有所述的构建物或载体或载体组合,或所述的基因工程细胞染色体整合有所述的构建物或载体。在另一优选例中,所述的基因工程细胞还包括含有编码转座酶基因的载体或其染色体上整合有转座酶基因。
优选地,所述的基因工程细胞为真核细胞。
在另一优选例中,所述真核细胞,包括(但不限于):酵母细胞(优选,克鲁维酵母细胞,更优选乳酸克鲁维酵母细胞)。
本发明的构建物或载体,可以用于转化适当的基因工程细胞。基因工程细胞可以是原核细胞,如大肠杆菌,链霉菌属、农杆菌:或是低等真核细胞,如酵母细胞;或是高等动物细胞,如昆虫细胞。本领域一般技术人员都清楚如何选择适当的载体和基因工程细胞。用重组dna转化基因工程细胞可用本领域技术人员熟知的常规技术进行。当宿主为原核生物(如大肠杆菌)时,可以用cacl2法处理,也可用电穿孔法进行。当宿主是真核生物,可选用如下的dna转染方法:磷酸钙共沉淀法,常规机械方法(如显微注射、电穿孔、脂质体包装等)。转化植物也可使用农杆菌转化或基因枪转化等方法,例如叶盘法、幼胚转化法、花芽浸泡法等。
体外高通量的蛋白合成方法
本发明提供了一种体外高通量的蛋白合成方法,包括步骤:
(i)在酵母体外蛋白合成体系存在下,提供本发明第一方面或第二方面所述的核酸构建物;
(ii)在适合的条件下,孵育步骤(i)的酵母体外蛋白合成体系一段时间t1,从而合成所述外源蛋白。
在另一优选例中,所述方法还包括:(iii)任选地从所述酵母体外蛋白合成体系中,分离或检测所述外源蛋白。
本发明的主要优点包括:
(1)本发明首次发现,将具有特定启动强度的启动子(如rnr2、adh1、gapdh、tef1、pgk1、sed1等)和rnap(如t7rnap)作为核酸构建物,应用于本发明的酵母体外蛋白质合成体系中,可显著提高蛋白翻译的效率。
(2)本发明首次发现,本发明的核酸构建物所合成的荧光素酶活性的相对光单位值非常高,高达6.6×107。
(3)本发明首次通过crispr/cas9,结合高效的转化技术,将t7rnap基因整合到真核细胞克鲁维酵母基因组中,实现了t7rnap在细胞基因组内的稳定存在和t7rnap蛋白质的持续表达。
(4)本发明将插入t7rnap的克鲁维酵母菌株制备成体外表达系统,实现了外源目标蛋白的高效翻译、大大简化了制备步骤,节约了成本,并增加了蛋白质合成的稳定性。
(5)本发明通过dna重组技术,借助高效的细胞转化平台,将t7rnap基因无标记整合到细胞基因组中,建立其高效内源性表达的稳定细胞,应用之一是在体外蛋白质合成系统中,不需人工添加t7rnap即可高效翻译外源蛋白。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,例如sambrook等人,分子克隆:实验室手册(newyork:coldspringharborlaboratorypress,1989)中所述的条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数是重量百分比和重量份数。
如无特别说明,则本发明实施例中所用的材料和试剂均为市售产品。
实施例1细胞基因组中插入位点的选定
为了克服现有体外翻译系统中需外源手动加入t7rna聚合酶蛋白的缺陷,本发明通过基因编辑技术将t7rnap蛋白整合到细胞基因组内,创造了一个能够稳定适量表达t7rnap蛋白的菌株,进而形成了一个无需外源手动加入t7rnap的简便高效的体外翻译系统。
体外翻译系统对t7rnap的含量有较高要求,过高或过少都会对系统效率产生影响。因此本发明在t7rnap前插入较弱的pscrnr2启动子,并将该结构构建到游离质粒中,验证t7rnap表达盒在体外翻译系统中的作用。
经过游离质粒功能验证以后,本发明通过两种方案将t7rnap整合到细胞基因组中:a将t7rnap通过linker与内源基因连接,形成融合蛋白;b将t7rnap表达盒替换细胞某个基因。本发明中,与t7rnap融合的基因为kltdh3,敲除并进行t7rnap替换的基因为klxrn1。
实施例2pkm-t7rnap1质粒构建、酵母转化及活性测定
2.1pkm-t7rnap1质粒构建
为了验证t7rnap可以在细胞中有效表达且具有转录活性,本发明首先构建t7rnap游离质粒,并转化细胞。游离质粒中t7rnap的启动子为scrnr2promoter,终止子为sccyc1terminator,抗性基因为kan。质粒构建及转化方法如下:
以含有t7rnap基因的质粒为模板,以引物pf1:atgaacacgattaacatcgctaagaacg(seqidno.:2)和pr1:ttacgcgaacgcgaagtccg(seqidno.:3)进行pcr扩增;以乳酸克鲁维酵母游离质粒为模板,以引物pf2:atcttagagtcggacttcgcgttcgcgtaagaagatgcttctgctcatcatc(seqidno.:4)和pr2:agtcgttcttagcgatgttaatcgtgttcatggtaattggacaaataaatacgtgt(seqidno.:5)进行pcr扩增。将两次扩增产物各8.5µl混合,加入1µldpni,2µl10×digestionbuffer,37oc温浴3h。将dpni处理后产物10µl加入100µldh5α感受态细胞中,冰上放置30min,42oc热激45s后,加入1mllb液体培养基37oc振荡培养1h,涂布于kan抗性lb固体培养,37oc倒置培养至单克隆长出。挑取5个单克隆在lb液体培养基中振荡培养,pcr检测阳性并测序确认后,提取质粒保存,命名为pkm-t7rnap1(图1)。
2.2高效乳酸克鲁维酵母感受态制备及转化
将乳酸克鲁维酵母菌液在ypd固体培养基上划线并挑取单克隆,于25ml2×ypd液体培养基中振荡培养过夜,取2ml菌液于50ml液体2×ypd培养基中继续振荡培养2-8h。20oc条件下3000g离心5min收集酵母细胞,加入500µl无菌水重悬,同样条件下离心收集细胞。配制感受态细胞溶液(5%v/v甘油,10%v/vdmso)并将酵母细胞溶解于500µl该溶液中。分装50µl至1.5ml离心管中,-80oc保存。
将感受态细胞置于37oc融化15-30s,13000g离心2min并去除上清。配制转化缓冲液:peg3350(50%(w/v))260µl,liac(1.0m)36µl,carrierdna(5.0mg/ml)20µl,pkm-t7rnap110µl,加入无菌水至最终体积360µl。热激后,13000g离心30s去除上清。加入1mlypd液体培养基,培养2-3h,吸取200µl涂布于固体ypd(200µg/mlg418)培养基,培养2-3天至单菌落出现。
实施例3通过crispr-cas9技术将t7rnap与kltdh3蛋白融合
3.1kltdh3序列检索及crisprgrna序列确定
在酿酒酵母(s.cerevisiae)中,tdh3以四聚体形式存在,参与糖酵解途径中的催化反应。其启动子ptdh3是基因工程中广泛使用的一种持续型强启动子。为了实现乳酸克鲁维酵母中t7rnap的足量表达,并在执行转录功能时形成局部高浓度,本发明将t7rnap基因连接到乳酸克鲁维酵母tdh3基因orf3’端。
i.基于s.cerevisiae酵母中的tdh3基因序列。在ncbi数据库中以tdh3基因进行blast比对分析,确定乳酸克鲁维酵母中tdh3同源基因序列。经比对发现,在乳酸克鲁维酵母基因组中存在两个tdh3同源基因,在本发明中分别命名为kltdh3-1(位于染色体a的1024297...1025292)和kltdh3-2(位于染色体f的1960417...1961406)。在这里以此基因尾部插入一段标记dna为例,其他目标基因或插入位置、序列均可采用类似方法操作。
ii.在kltdh3基因终止密码子附近搜索pam序列(ngg),并确定kltdh3grna序列(kltdh3-1:cttgttgctaagaactaaag(seqidno.:6),位于染色体a的1024272...1024291位点,kltdh3-2:ctctgaaagagttgtcgatt(seqidno.:7)位于染色体f的1960378...1960397位点)。
kltdh3质粒构建
i.对kltdh3-1,使用引物pf3:cttgttgctaagaactaaaggttttagagctagaaatagcaagttaaaat(seqidno.:8),pr3:gctctaaaacctttagttcttagcaacaagaaagtcccattcgccacccg(seqidno.:9),以pcas质粒为模板,进行pcr扩增。将扩增产物17µl混合,加入1µldpni,2µl10×digestionbuffer,37oc温浴3h。将dpni处理后产物10µl加入100µldh5α感受态细胞中,冰上放置30min,42oc热激45s后,加入1mllb液体培养基37oc振荡培养1h,涂布于kan抗性lb固体培养,37oc倒置培养至单克隆长出。挑取5个单克隆在lb液体培养基中振荡培养,pcr检测阳性并测序确认后,提取质粒保存,命名为pkm-cas1.0-kltdh3-1(图2)。
ii.对kltdh3-2,使用引物pf4:ctctgaaagagttgtcgattgttttagagctagaaatagcaagttaaaat(seqidno.:10),pr4:gctctaaaacaatcgacaactctttcagagaaagtcccattcgccacccg(seqidno.:11),以pcas质粒为模板,进行pcr扩增。将扩增产物17µl混合,加入1µldpni,2µl10×digestionbuffer,37oc温浴3h。将dpni处理后产物10µl加入100µldh5α感受态细胞中,冰上放置30min,42oc热激45s后,加入1mllb液体培养基37oc振荡培养1h,涂布于kan抗性lb固体培养,37oc倒置培养至单克隆长出。挑取5个单克隆在lb液体培养基中振荡培养,pcr检测阳性并测序确认后,提取质粒保存,命名为pkm-cas1.0-kltdh3-2(图3)。
3.3kltdh3-t7rnap供体dna质粒构建及扩增
为了便于线性供体dna的保存及扩增,本发明首先将供体dna插入到pmd18质粒中,然后通过pcr扩增得到线性供体dna序列。
i.对kltdh3-1,以乳酸克鲁维酵母基因组dna为模板,以引物pf5:gagctcggtacccggggatcctctagagatcatccactccatcaccgctacccaa(seqidno.:12)和pr5:gccaagcttgcatgcctgcaggtcgacgatcaacgtccccatctacaagagc(seqidno.:13)进行pcr扩增;以pmd18质粒为模板,以引物pmd18-f:atcgtcgacctgcaggcatg(seqidno.:14)和pmd18-r:atctctagaggatccccggg(seqidno.:15)进行pcr扩增。将两次扩增产物各8.5µl混合,加入1µldpni,2µl10×消化缓冲液(digestionbuffer),37oc温浴3h。将dpni处理后产物10µl加入100µldh5α感受态细胞中,冰上放置30min,42oc热激45s后,加入1mllb液体培养基37oc振荡培养1h,涂布于kan抗性lb固体培养,37oc倒置培养至单克隆长出。挑取5个单克隆在lb液体培养基中振荡培养,pcr检测阳性并测序确认后,提取质粒保存,命名为pkm-kltdh3-1-dd。
以pkm-kltdh3-1-dd为模板,以引物pf6:atcttagagtcggacttcgcgttcgcgtaaagaggttgatgtaattgatattttcct(seqidno.:16)和pr6:acctccttcgacgtttggtctagatccaccgttcttagcaacaagttcgacc(seqidno.:17)进行扩增;以含有t7rnap序列的质粒为模板,以引物pf7:ggtggatctagaccaaacgtcgaaggaggttctaacacgattaacatcgctaagaac(seqidno.:18)和pr7:ttacgcgaacgcgaagtccg(seqidno.:19)进行扩增。将两次扩增产物各8.5µl,1µldpni,2µl10×digestionbuffer混合,37oc温浴3h。将dpni处理后产物10µl加入100µldh5α感受态细胞中,冰上放置30min,42oc热激45s后,加入1mllb液体培养基37oc振荡培养1h,涂布于amp抗性lb固体培养,37oc倒置培养至单克隆长出。挑取5个单克隆在lb液体培养基中振荡培养,pcr检测阳性并测序确认后,提取质粒保存,命名为pkm-kltdh3-1-t7-dd(图4)。
以pkm-kltdh3-1-t7-dd质粒为模板,以引物m13-f:gtaaaacgacggccagt(seqidno.:20)和m13-r:caggaaacagctatgac(seqidno.:21)进行扩增,得到线性供体dna。
ii.对kltdh3-2,以乳酸克鲁维酵母基因组dna为模板,以引物pf8:gagctcggtacccggggatcctctagagatgaagctttgatgactaccgttc(seqidno.:22)和pr8:gccaagcttgcatgcctgcaggtcgacgatgtctattgtatcggaagaactgtca(seqidno.:23)进行pcr扩增;以pmd18质粒为模板,以引物pmd18-f:atcgtcgacctgcaggcatg(seqidno.:24)和pmd18-r:atctctagaggatccccggg(seqidno.:25)进行pcr扩增。将两次扩增产物各8.5µl混合,加入1µldpni,2µl10×digestionbuffer,37oc温浴3h。将dpni处理后产物10µl加入100µldh5α感受态细胞中,冰上放置30min,42oc热激45s后,加入1mllb液体培养基37oc振荡培养1h,涂布于kan抗性lb固体培养,37oc倒置培养至单克隆长出。挑取5个单克隆在lb液体培养基中振荡培养,pcr检测阳性并测序确认后,提取质粒保存,命名为pkm-kltdh3-2-dd。
以pkm-kltdh3-2-dd1为模板,以引物pf9:atcttagagtcggacttcgcgttcgcgtaaattactcttttaagttaacgaacgct(seqidno.:26)和pr9:acctccttcgacgtttggtctagatccaccagcaacgtgctcaactaagtcaacgaccctttcagagtaaccgtat(seqidno.:27)进行扩增;以含有t7rnap序列的质粒为模板,以引物pf10:ggtggatctagaccaaacgtcgaaggaggttctaacacgattaacatcgctaagaac(seqidno.:28)和pr10:ttacgcgaacgcgaagtccg(seqidno.:29)进行扩增。将两次扩增产物各8.5µl,1µldpni,2µl10×digestionbuffer混合,37oc温浴3h。将dpni处理后产物10µl加入100µldh5α感受态细胞中,冰上放置30min,42oc热激45s后,加入1mllb液体培养基37oc振荡培养1h,涂布于amp抗性lb固体培养,37oc倒置培养至单克隆长出。挑取5个单克隆在lb液体培养基中振荡培养,pcr检测阳性并测序确认后,提取质粒保存,命名为pkm-kltdh3-2-t7-dd(图5)。
以pkm-kltdh3-2-t7-dd质粒为模板,以引物m13-f:gtaaaacgacggccagt(seqidno.:30)和m13-r:caggaaacagctatgac(seqidno.:31)进行扩增,得到线性供体dna。
3.4乳酸克鲁维酵母转化及阳性鉴定
i.将乳酸克鲁维酵母菌液在ypd固体培养基上划线并挑取单克隆,于25ml2×ypd液体培养基中振荡培养过夜,取2ml菌液于50ml液体2×ypd培养基中继续振荡培养2-8h。20oc条件下3000g离心5min收集酵母细胞,加入500µl无菌水重悬,同样条件下离心收集细胞。配制感受态细胞溶液(5%v/v甘油,10%v/vdmso)并将酵母细胞溶解于500µl该溶液中。分装50µl至1.5ml离心管中,-80oc保存。
将感受态细胞置于37oc融化15-30s,13000g离心2min并去除上清。配制转化缓冲液:peg3350(50%(w/v))260µl,liac(1.0m)36µl,carrierdna(5.0mg/ml)20µl,cas9/grna质粒15µl,供体dna10µl,加入无菌水至最终体积360µl。热激后,13000g离心30s去除上清。加入1mlypd液体培养基,培养2-3h,吸取200µl涂布于固体ypd(200µg/mlg418)培养基,培养2-3天至单菌落出现。
ii.在乳酸克鲁维酵母转化后的平板上挑取10-20个单克隆,置于1mlypd(200µg/mlg418)液体培养基中振荡培养过夜,以菌液为模板,分别以引物pf11(t7rnap序列内引物):attggacaaaatgccagcacttccg(seqidno.:32)/pr11(kltdh3-1供体dna5’外侧引物):cttctactgctccaatgttcgtcgtt(seqidno.:33)和引物pf12(t7rnap序列内引物):attggacaaaatgccagcacttccg(seqidno.:34)/pr12(kltdh3-2供体dna5’外侧引物):ttaacgaagacaagtacaacggtga(seqidno.:35)进行pcr扩增,对kltdh3位点的crispr插入进行检测,有阳性条带表明t7rnap序列插入靶位点成功。
实施例4通过crispr-cas9技术将klxrn1敲除并替换为t7rnap表达盒4.1klxrn1序列检索及crisprgrna序列确定
xrn1是一种针对无帽结构mrna的5′-3′核酸外切酶。为了实现,在t7rnap体内表达的同时,酵母的体外翻译活性也得到提高,本发明将klxrn1基因完全敲除,并替换为t7rnap序列及相关启动子和终止子。
i.基于酿酒酵母中的xrn1基因序列。在ncbi数据库中以xrn1基因进行blast比对分析,确定乳酸克鲁维酵母中xrn1同源基因序列klxrn1(位于染色体f的2091235...2095596)。在这里以此基因尾部插入一段标记dna为例,其他目标基因或插入位置、序列均可采用类似方法操作。
ii.在klxrn1基因两端搜索pam序列(ngg),并确定grna序列。grna选择的原则为:gc含量适中,本发明的标准为gc含量为40%-60%;避免polyt结构的存在。最终,本发明确定的klxrn1grna序列为agagttcgacaatttgtact(seqidno.:36)和cgtcgtggccgtagtaatcg(seqidno.:37)。
klxrn1质粒构建
使用引物pf13:agagttcgacaatttgtactgttttagagctagaaatagcaagttaaaataaggctagtc(seqidno.:38),pr13:gctctaaaacagtacaaattgtcgaactctaaagtcccattcgccacccg(seqidno.:39),以pcas质粒为模板,进行pcr扩增,将grna1序列整合到crispr质粒中。将扩增产物17µl混合,加入1µldpni,2µl10×digestionbuffer,37oc温浴3h。将dpni处理后产物10µl加入100µldh5α感受态细胞中,冰上放置30min,42oc热激45s后,加入1mllb液体培养基37oc振荡培养1h,涂布于kan抗性lb固体培养,37oc倒置培养至单克隆长出。挑取5个单克隆在lb液体培养基中振荡培养,pcr检测阳性并测序确认后,提取质粒保存。使用同样的方法,用引物pf14:cgtcgtggccgtagtaatcggttttagagctagaaatagcaagttaaaataaggctagtc(seqidno.:40),pr14:gctctaaaaccgattactacggccacgacgaaagtcccattcgccacccg(seqidno.:41),将grna2整合到质粒中,命名为pkm-cas1.0-klxrn1(图6)。
4.3klxrn1-t7rnap供体dna质粒构建及扩增
为了便于线性供体dna的保存及扩增,本发明首先将供体dna插入到pmd18质粒中,然后通过pcr扩增得到线性供体dna序列。
以乳酸克鲁维基因组dna为模板,以引物pf15:gagctcggtacccggggatcctctagagataaaagcttgaacttatggatccgggta(seqidno.:42)和pr15:gccaagcttgcatgcctgcaggtcgacgatgtaatcctcttgtgctctaattgct(seqidno.:43)进行pcr扩增;以pmd18质粒为模板,以引物pmd18-f:atcgtcgacctgcaggcatg(seqidno.:44)和pmd18-r:atctctagaggatccccggg(seqidno.:45)进行pcr扩增。将两次扩增产物各8.5µl混合,加入1µldpni,2µl10×digestionbuffer,37oc温浴3h。将dpni处理后产物10µl加入100µldh5α感受态细胞中,冰上放置30min,42oc热激45s后,加入1mllb液体培养基37oc振荡培养1h,涂布于kan抗性lb固体培养,37oc倒置培养至单克隆长出。挑取5个单克隆在lb液体培养基中振荡培养,pcr检测阳性并测序确认后,提取质粒保存,命名为pkm-klxrn1-dd。
以pkm-klxrn1-dd为模板,以引物pf16:atgaattctattttgcataattgtacgcatggtg(seqidno.:46)和pr16:ttacaagtacaaattgtcgaactctggaatctg(seqidno.:47)进行扩增;以pkm-t7rnap1质粒为模板,以引物pf17:attccagagttcgacaatttgtacttgtaaagtcgaacaagaagcaggcaaag(seqidno.:48)和pr17:atgcgtacaattatgcaaaatagaattcatcttcgagcgtcccaaaaccttc(seqidno.:49)进行扩增。将两次扩增产物各8.5µl,1µldpni,2µl10×digestionbuffer混合,37oc温浴3h。将dpni处理后产物10µl加入100µldh5α感受态细胞中,冰上放置30min,42oc热激45s后,加入1mllb液体培养基37oc振荡培养1h,涂布于amp抗性lb固体培养,37oc倒置培养至单克隆长出。挑取5个单克隆在lb液体培养基中振荡培养,pcr检测阳性并测序确认后,提取质粒保存,命名为pkm-klxrn1-t7-dd(图7)。
以pkm-klxrn1-t7-dd质粒为模板,以引物m13-f:gtaaaacgacggccagt(seqidno.:50)和m13-r:caggaaacagctatgac(seqidno.:51)进行扩增,得到线性供体dna。
4.4乳酸克鲁维酵母转化及阳性鉴定
iii.将乳酸克鲁维酵母菌液在ypd固体培养基上划线并挑取单克隆,于25ml2×ypd液体培养基中振荡培养过夜,取2ml菌液于50ml液体2×ypd培养基中继续振荡培养2-8h。20oc条件下3000g离心5min收集酵母细胞,加入500µl无菌水重悬,同样条件下离心收集细胞。配制感受态细胞溶液(5%v/v甘油,10%v/vdmso)并将酵母细胞溶解于500µl该溶液中。分装50µl至1.5ml离心管中,-80oc保存。
将感受态细胞置于37oc融化15-30s,13000g离心2min并去除上清。配制转化缓冲液:peg3350(50%(w/v))260µl,liac(1.0m)36µl,carrierdna(5.0mg/ml)20µl,cas9/grna质粒15µl,供体dna10µl,加入无菌水至最终体积360µl。热激后,13000g离心30s去除上清。加入1mlypd液体培养基,培养2-3h,吸取200µl涂布于固体ypd(200µg/mlg418)培养基,培养2-3天至单菌落出现。
在乳酸克鲁维酵母转化后的平板上挑取10-20个单克隆,置于1mlypd(200µg/mlg418)液体培养基中振荡培养过夜,以菌液为模板,以引物pf18(t7rnap序列内引物):attggacaaaatgccagcacttccg(seqidno.:52)和引物pr18(供体dna5’外侧引物):tttgctggttgcccgtattccc(seqidno.:53)进行pcr扩增,对klxrn1位点的crispr插入进行检测,有阳性条带表明t7rnap序列插入靶位点成功。
实施例4活性测定
将基因改造后的乳酸克鲁维酵母菌株制备成体外蛋白质合成系统,并加入萤火虫荧光素酶(fireflyluciferase,fluc)基因dna模板以测定改造菌株的蛋白翻译能力。将上述反应体系置于25-30oc的环境中,静置孵育约2-6h。反应结束后,在96孔白板或者384孔白板中加入等体积的fluc底物荧光素(luciferin),立即放置于envision2120多功能酶标仪(perkinelmer),读数,检测fluc活性,相对光单位值(relativelightunit,rlu)作为活性单位。
转入游离质粒pkm-t7rnap1的菌株,在外加t7rnap浓度为0时表现出最高活性且处于正常范围内(107),随着外加t7rnap浓度增加,对活性产生抑制作用,说明pkm-t7rnap1质粒中的t7rnap结构(pscrnr2+t7rnap)能够正常发挥功能,其rlu值为1.4×107(图8)。
在klxrn1δ-pscrnr2-t7rnap结构中,在外加t7rnap浓度为0时表现出最高活性且处于正常范围内(107),表面该结构中t7rnap可以适量表达,能够满足体外翻译系统的要求,并且该结构的核酸构建物能够显著增强酵母体外蛋白质合成体系产生蛋白质的效率,其rlu值为6.6×107(图11)。
对比例
kltdh3-1与t7rnap融合蛋白的结构中,随着外加t7rnap浓度增加,体外翻译活性增强,表明该结构中t7rnap的活力不足以支持体外翻译系统的运行(图9),其rlu值仅为4.6×105。
在kltdh3-2与t7rnap融合蛋白的结构中,随着外加t7rnap浓度增加,体外翻译活性减弱,且整体处于受抑制状态(106),说明该结构中t7rnap表达量过高,对系统产生了抑制作用,其rlu值仅为1.4×106。(图10)。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
序列表
<110>康码(上海)生物科技有限公司
<120>一种细胞中内源性表达rna聚合酶的核酸构建物
<130>2017-1564
<141>2017-08-31
<160>53
<170>siposequencelisting1.0
<210>1
<211>3542
<212>dna
<213>人工序列(artificialsequence)
<400>1
agtcgaacaagaagcaggcaaagtttagagcactgcccctccgcactcaaaaaagaaaaa60
actaggaggaaaataaaattctcaaccacacaaacacataaacacatacaaatacaaata120
caagcttatttacttgacatcgcgcgatcttccactattcagcgccgtccgccctctctc180
gtgttttttgtttacgcgacaactatgcgaaatccggagcaacgggcaaccgtttgggga240
aagaccacacccacgcgcgatcgccatggcaacgaggtcgcacacgccccacacccagac300
ctccctgcgagcgggcatgggtacaatgtccccgttgccacagacaccacttcgtagcac360
agcgcagagcgtagcgtgttgttgctgctgacaaaagaaaatttttcttagcaaagcaaa420
ggaggggaagcacgggcagatagcaccgtaccatacccttggaaactcgaaatgaacgaa480
gcaggaaatgagagaatgagagttttgtaggtatatatagcggtagtgtttgcgcgttac540
catcatcttctggatctatctattgttcttttcctcatcactttcccctttttcgctctt600
cttcttgtcttttatttctttcttttttttaattgttccctcgattggctatctaccaaa660
gaatccaaacttaatacacgtatttatttgtccaattaccatgaacacgattaacatcgc720
taagaacgacttctctgacatcgaactggctgctatcccgttcaacactctggctgacca780
ttacggtgagcgtttagctcgcgaacagttggcccttgagcatgagtcttacgagatggg840
tgaagcacgcttccgcaagatgtttgagcgtcaacttaaagctggtgaggttgcggataa900
cgctgccgccaagcctctcatcactaccctactccctaagatgattgcacgcatcaacga960
ctggtttgaggaagtgaaagctaagcgcggcaagcgcccgacagccttccagttcctgca1020
agaaatcaagccggaagccgtagcgtacatcaccattaagaccactctggcttgcctaac1080
cagtgctgacaatacaaccgttcaggctgtagcaagcgcaatcggtcgggccattgagga1140
cgaggctcgcttcggtcgtatccgtgaccttgaagctaagcacttcaagaaaaacgttga1200
ggaacaactcaacaagcgcgtagggcacgtctacaagaaagcatttatgcaagttgtcga1260
ggctgacatgctctctaagggtctactcggtggcgaggcgtggtcttcgtggcataagga1320
agactctattcatgtaggagtacgctgcatcgagatgctcattgagtcaaccggaatggt1380
tagcttacaccgccaaaatgctggcgtagtaggtcaagactctgagactatcgaactcgc1440
acctgaatacgctgaggctatcgcaacccgtgcaggtgcgctggctggcatctctccgat1500
gttccaaccttgcgtagttcctcctaagccgtggactggcattactggtggtggctattg1560
ggctaacggtcgtcgtcctctggcgctggtgcgtactcacagtaagaaagcactgatgcg1620
ctacgaagacgtttacatgcctgaggtgtacaaagcgattaacattgcgcaaaacaccgc1680
atggaaaatcaacaagaaagtcctagcggtcgccaacgtaatcaccaagtggaagcattg1740
tccggtcgaggacatccctgcgattgagcgtgaagaactcccgatgaaaccggaagacat1800
cgacatgaatcctgaggctctcaccgcgtggaaacgtgctgccgctgctgtgtaccgcaa1860
ggacaaggctcgcaagtctcgccgtatcagccttgagttcatgcttgagcaagccaataa1920
gtttgctaaccataaggccatctggttcccttacaacatggactggcgcggtcgtgttta1980
cgctgtgtcaatgttcaacccgcaaggtaacgatatgaccaaaggactgcttacgctggc2040
gaaaggtaaaccaatcggtaaggaaggttactactggctgaaaatccacggtgcaaactg2100
tgcgggtgtcgataaggttccgttccctgagcgcatcaagttcattgaggaaaaccacga2160
gaacatcatggcttgcgctaagtctccactggagaacacttggtgggctgagcaagattc2220
tccgttctgcttccttgcgttctgctttgagtacgctggggtacagcaccacggcctgag2280
ctataactgctcccttccgctggcgtttgacgggtcttgctctggcatccagcacttctc2340
cgcgatgctccgagatgaggtaggtggtcgcgcggttaacttgcttcctagtgaaaccgt2400
tcaggacatctacgggattgttgctaagaaagtcaacgagattctacaagcagacgcaat2460
caatgggaccgataacgaagtagttaccgtgaccgatgagaacactggtgaaatctctga2520
gaaagtcaagctgggcactaaggcactggctggtcaatggctggcttacggtgttactcg2580
cagtgtgactaagcgttcagtcatgacgctggcttacgggtccaaagagttcggcttccg2640
tcaacaagtgctggaagataccattcagccagctattgattccggcaagggtctgatgtt2700
cactcagccgaatcaggctgctggatacatggctaagctgatttgggaatctgtgagcgt2760
gacggtggtagctgcggttgaagcaatgaactggcttaagtctgctgctaagctgctggc2820
tgctgaggtcaaagataagaagactggagagattcttcgcaagcgttgcgctgtgcattg2880
ggtaactcctgatggtttccctgtgtggcaggaatacaagaagcctattcagacgcgctt2940
gaacctgatgttcctcggtcagttccgcttacagcctaccattaacaccaacaaagatag3000
cgagattgatgcacacaaacaggagtctggtatcgctcctaactttgtacacagccaaga3060
cggtagccaccttcgtaagactgtagtgtgggcacacgagaagtacggaatcgaatcttt3120
tgcactgattcacgactccttcggtaccattccggctgacgctgcgaacctgttcaaagc3180
agtgcgcgaaactatggttgacacatatgagtcttgtgatgtactggctgatttctacga3240
ccagttcgctgaccagttgcacgagtctcaattggacaaaatgccagcacttccggctaa3300
aggtaacttgaacctccgtgacatcttagagtcggacttcgcgttcgcgtaaatccgctc3360
taaccgaaaaggaaggagttagacaacctgaagtctaggtccctatttatttttttatag3420
ttatgttagtattaagaacgttatttatatttcaaatttttcttttttttctgtacagac3480
gcgtgtacgcatgtaacattatactgaaaaccttgcttgagaaggttttgggacgctcga3540
ag3542
<210>2
<211>28
<212>dna
<213>人工序列(artificialsequence)
<400>2
atgaacacgattaacatcgctaagaacg28
<210>3
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>3
ttacgcgaacgcgaagtccg20
<210>4
<211>52
<212>dna
<213>人工序列(artificialsequence)
<400>4
<210>5
<211>56
<212>dna
<213>人工序列(artificialsequence)
<400>5
<210>6
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>6
<210>7
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>7
<210>8
<211>50
<212>dna
<213>人工序列(artificialsequence)
<400>8
<210>9
<211>50
<212>dna
<213>人工序列(artificialsequence)
<400>9
gctctaaaacctttagttcttagcaacaagaaagtcccattcgccacccg50
<210>10
<211>50
<212>dna
<213>人工序列(artificialsequence)
<400>10
<210>11
<211>50
<212>dna
<213>人工序列(artificialsequence)
<400>11
gctctaaaacaatcgacaactctttcagagaaagtcccattcgccacccg50
<210>12
<211>55
<212>dna
<213>人工序列(artificialsequence)
<400>12
<210>13
<211>52
<212>dna
<213>人工序列(artificialsequence)
<400>13
<210>14
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>14
<210>15
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>15
<210>16
<211>57
<212>dna
<213>人工序列(artificialsequence)
<400>16
<210>17
<211>52
<212>dna
<213>人工序列(artificialsequence)
<400>17
<210>18
<211>57
<212>dna
<213>人工序列(artificialsequence)
<400>18
<210>19
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>19
<210>20
<211>17
<212>dna
<213>人工序列(artificialsequence)
<400>20
<210>21
<211>17
<212>dna
<213>人工序列(artificialsequence)
<400>21
<210>22
<211>52
<212>dna
<213>人工序列(artificialsequence)
<400>22
<210>23
<211>55
<212>dna
<213>人工序列(artificialsequence)
<400>23
<210>24
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>24
<210>25
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>25
<210>26
<211>56
<212>dna
<213>人工序列(artificialsequence)
<400>26
atcttagagtcggacttcgcgttcgcgtaaattactcttttaagttaacgaacgct56
<210>27
<211>76
<212>dna
<213>人工序列(artificialsequence)
<400>27
acctccttcgacgtttggtctagatccaccagcaacgtgctcaactaagtcaacgaccct60
ttcagagtaaccgtat76
<210>28
<211>57
<212>dna
<213>人工序列(artificialsequence)
<400>28
<210>29
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>29
<210>30
<211>17
<212>dna
<213>人工序列(artificialsequence)
<400>30
<210>31
<211>17
<212>dna
<213>人工序列(artificialsequence)
<400>31
<210>32
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>32
<210>33
<211>26
<212>dna
<213>人工序列(artificialsequence)
<400>33
<210>34
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>34
<210>35
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>35
<210>36
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>36
agagttcgacaatttgtact20
<210>37
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>37
<210>38
<211>60
<212>dna
<213>人工序列(artificialsequence)
<400>38
agagttcgacaatttgtactgttttagagctagaaatagcaagttaaaataaggctagtc60
<210>39
<211>50
<212>dna
<213>人工序列(artificialsequence)
<400>39
<210>40
<211>60
<212>dna
<213>人工序列(artificialsequence)
<400>40
<210>41
<211>50
<212>dna
<213>人工序列(artificialsequence)
<400>41
<210>42
<211>57
<212>dna
<213>人工序列(artificialsequence)
<400>42
<210>43
<211>55
<212>dna
<213>人工序列(artificialsequence)
<400>43
gccaagcttgcatgcctgcaggtcgacgatgtaatcctcttgtgctctaattgct55
<210>44
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>44
atcgtcgacctgcaggcatg20
<210>45
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>45
<210>46
<211>34
<212>dna
<213>人工序列(artificialsequence)
<400>46
<210>47
<211>33
<212>dna
<213>人工序列(artificialsequence)
<400>47
<210>48
<211>53
<212>dna
<213>人工序列(artificialsequence)
<400>48
<210>49
<211>52
<212>dna
<213>人工序列(artificialsequence)
<400>49
<210>50
<211>17
<212>dna
<213>人工序列(artificialsequence)
<400>50
<210>51
<211>17
<212>dna
<213>人工序列(artificialsequence)
<400>51
<210>52
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>52
<210>53
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>53
- 还没有人留言评论。精彩留言会获得点赞!