一种盐酸表柔比星晶型Ⅲ及其制备方法与流程
本发明属于化学合成领域,具体涉及一种盐酸表柔比星晶型ⅲ及其制备方法。
背景技术:
盐酸表柔比星又名盐酸表阿霉素,属于蒽环类抗生素,化学名:(8s,10s)-10-[(3,-氨基-2,,3,,6,-三脱氧-alpha-l-阿拉伯吡喃糖基)-o-]-6,8,11-三羟基-8-羟乙酰基-1-甲氧基-7,8,9,10-四氢并四苯-5,12-二酮盐酸盐,结构式如下:
美国专利us4861870提供了纯化盐酸表柔比星的方法,在此方法中,借助丙酮使盐酸表柔比星由水溶液中沉淀出来并作为无定型固体获得,但由于其在贮存期间明显的潮解性和化学不稳定性,并伴有苷元多柔比星酮的积累,被认为不适合于药物应用。
现已报道盐酸表柔比星有两种晶型,专利us4861870中所述ⅰ型盐酸表柔比星的xrd图谱显示在24.6左右产生一个单独的强信号。通过实验发现,这种晶型在放置几个月后稳定性降低,导致该晶型在制备制剂产品后降低了其用药安全性。
在专利wo2005004805a1、wo2011118929a2和wo2012163508a1中,虽然盐酸表柔比星的制备方法不同,但x射线衍射图谱显示高度的相似性,在专利wo2005004805a1中描述了盐酸表柔比星晶型ⅱ在长达六个月的贮存期间具备了良好的稳定性,但发明人在后期制剂产品研究中发现其产品在常温不加助溶剂的情况下,溶解度不是很理想。
此外,在上述专利文献中所述晶型的制备方法中需要使用大量的有机溶剂,且易形成溶剂化物,严重影响了其制剂产品在临床上用药的安全性。因此本领域很有必要寻找具备较高稳定性和较高溶解度的盐酸表柔比星的新晶型,从而为制备制剂产品提供更丰富的原料。
技术实现要素:
本发明提供一种盐酸表柔比星晶型ⅲ,本晶型比现有技术报道的晶型ⅱ具有更好的溶解度,比晶型ⅰ具有更高的稳定性。
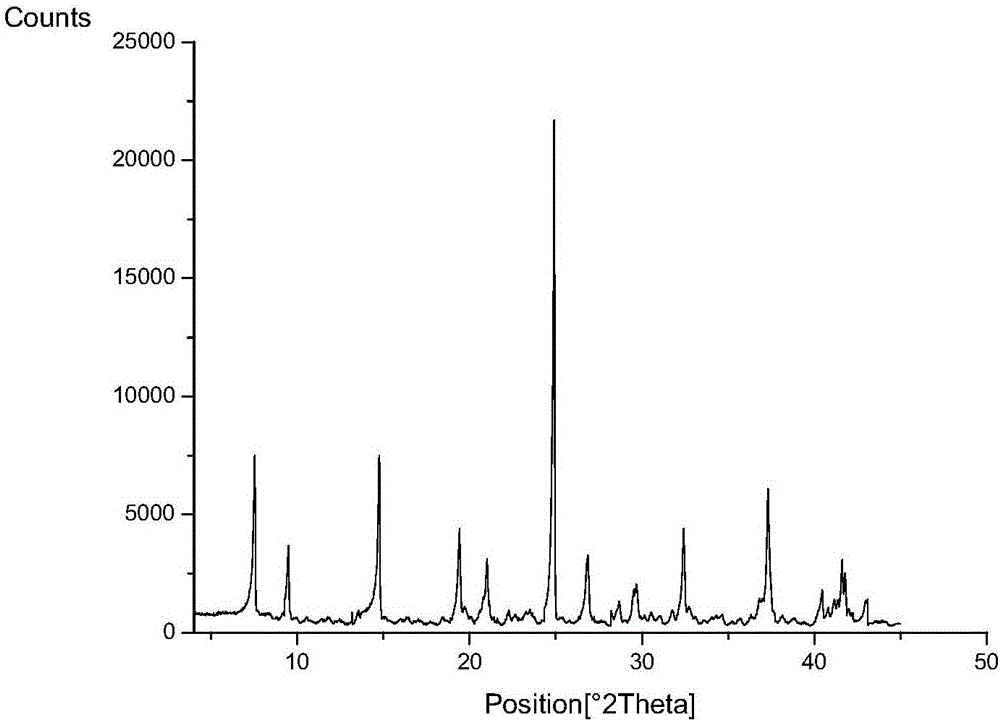
本发明提供一种盐酸表柔比星晶型ⅲ,其x-射线粉末衍射图具有在2θ=7.51±0.2°、9.46±0.2°、14.74±0.2°、19.39±0.2°、24.89±0.2°和37.31±0.2°处有特征衍射峰。
优选地,本发明所述晶型ⅲ,其x射线粉末衍射图具有在2θ=7.51±0.2°、9.46±0.2°、14.74±0.2°、19.39±0.2°、21.00±0.2°、24.89±0.2°、26.85±0.2°、32.41±0.2°和37.31±0.2°处有特征衍射峰。
进一步优选地,本发明所述晶型ⅲ,其x射线粉末衍射图具有在2θ=7.51±0.2°、9.46±0.2°、14.74±0.2°、19.39±0.2°、21.00±0.2°、24.89±0.2°、26.85±0.2°、28.68±0.2°、29.68±0.2°、32.41±0.2°、36.81±0.2°、37.31±0.2°、40.45±0.2°、41.65±0.2°和42.99±0.2°处有特征衍射峰。
更进一步优选地,本发明所述晶型ⅲ,其x射线粉末衍射图具有在2θ=7.51±0.2°、9.46±0.2°、13.85±0.2°、14.74±0.2°、19.39±0.2°、19.70±0.2°、21.00±0.2°、23.28±0.2°、23.50±0.2°、24.89±0.2°、26.85±0.2°、28.24±0.2°、28.68±0.2°、29.68±0.2°、30.53±0.2°、31.76±0.2°、32.41±0.2°、32.73±0.2°、34.64±0.2°、36.81±0.2°、37.31±0.2°、40.45±0.2°、40.80±0.2°、41.16±0.2°、41.65±0.2°和42.99±0.2°处有特征衍射峰。
经dsc测定该盐酸表柔比星晶型ⅲ的熔点为203℃。
本发明第二方面提供一种盐酸表柔比星晶型ⅲ的制备方法。
本发明提供的盐酸表柔比星晶型ⅲ的制备方法中,所涉及的盐酸表柔比星粗品的来源没有限制,可以化学合成或生物发酵。
一种制备所述晶型ⅲ的方法,该方法包括如下步骤:
(1)将盐酸表柔比星溶在溶剂中,配制成盐酸表柔比星溶液;
(2)设定结晶釜温度、压力等参数,用高压冷冻泵将冷冻的co2以恒定的流速连续通入高压结晶釜中;
(3)用另一高压泵先向高压结晶釜中通入步骤(1)所述溶剂15min,再将盐酸表柔比星溶液喷入高压结晶釜中;
(4)继续通入co240~60min,收集结晶釜底部的晶体即为盐酸表柔比星晶型ⅲ;
其中,步骤(1)所述的溶剂为ch3oh或dmso。
优选地,盐酸表柔比星溶液的浓度为5~25mg/ml。
优选地,结晶釜设定温度为30~45℃,压力为6~30mpa;co2流速为10~30g/min。
优选地,盐酸表柔比星溶液喷入流速为0.2~2.0ml/min。
本发明采用的方法为超临界反溶剂技术。超临界反溶剂技术是制备药物多晶型的一种新方法,同时会使药物微粒化,从而提高药物的溶解度。其原理是将原料药溶于某一溶剂(通常为有机溶剂)形成溶液,选择一种超临界流体(通常为co2)作为反溶剂。这种反溶剂一般不能溶解溶液中的溶质,但能与溶剂互溶,当反溶剂与溶液接触时,反溶剂迅速扩散至该溶液,使其体积迅速膨胀,溶质在溶剂中溶解度大大下降,在极短的时间内形成很大的过饱和度,促使溶质结晶析出。
通过本方法制备的盐酸表柔比星晶型ⅲ粒径小,溶解度大且稳定性较高。本发明所得盐酸表柔比星晶型ⅲ采用马尔文粒度分析仪进行分析,得到平均粒径164nm;采用高效液相色谱法(hplc)测得25℃时其在水中的溶解度为107mg/ml。传统方法所得盐酸表柔比星晶型ⅱ平均粒径为28μm,25℃时在水中的溶解度为19mg/ml。另外通过稳定性实验可知本发明所得盐酸表柔比星晶型ⅲ与盐酸表柔比星晶型ⅰ相比具有更高的稳定性。
另外本发明通过超临界反溶剂技术制备的盐酸表柔比星晶型ⅲ溶剂残留量极其微小,小于700ppm,且不会形成溶剂化物,所使用超临界流体co2可直接循环使用,生产成本低,对环境无污染。工艺流程简单,操作方便,重现性好,易于产业化。
附图说明
图1:盐酸表柔比星晶型ⅲ的x射线粉末衍射图。
图2:盐酸表柔比星晶型ⅲ的dsc图。
图3:对比实施例1所得盐酸表柔比星晶型ⅱ的x射线粉末衍射图。
具体实施方式
下面通过实施例来进一步说明本发明。应该正确理解的是,本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属于本发明要求保护的范围。
实施例1
准确称取3.06g盐酸表柔比星溶于170mlch3oh中,过滤,滤液即为盐酸表柔比星溶液备用;在控制软件中将高压结晶釜设置温度为35℃,压力为12mpa,co2注入结晶釜中的流速为20g/min;打开钢瓶,用高压泵将co2通入结晶釜,待结晶釜中温度、压力达到设定值时,用另一高压泵将ch3oh以0.8ml/min的速率通入结晶釜,15min后停止通入ch3oh,以同样的速率将上述盐酸表柔比星溶液注入;进样完毕后,继续通入co250min,停止控制系统,将结晶釜压力降为大气压时取出,在结晶釜底部即得盐酸表柔比星晶型ⅲ。
对盐酸表柔比星晶型ⅲ经gc测定ch3oh残留量为652ppm。
实施例2
准确称取3.00g盐酸表柔比星溶于600mlch3oh中,过滤,滤液即为盐酸表柔比星溶液备用;在控制软件中将高压结晶釜设置温度为38℃,压力为6mpa,co2注入结晶釜中的流速为30g/min;打开钢瓶,用高压泵将co2通入结晶釜,待结晶釜中温度、压力达到设定值时,用另一高压泵将ch3oh以2.0ml/min的速率通入结晶釜,15min后停止通入ch3oh,以同样的速率将上述盐酸表柔比星溶液注入;进样完毕后,继续通入co260min,停止控制系统,将结晶釜压力降为大气压时取出,在结晶釜底部即得盐酸表柔比星晶型ⅲ。
对盐酸表柔比星晶型ⅲ经gc测定ch3oh残留量为665ppm。
实施例3
准确称取3.00g盐酸表柔比星溶于120mlch3oh中,过滤,滤液即为盐酸表柔比星溶液备用;在控制软件中将高压结晶釜设置温度为45℃,压力为30mpa,co2注入结晶釜中的流速为10g/min;打开钢瓶,用高压泵将co2通入结晶釜,待结晶釜中温度、压力达到设定值时,用另一高压泵将ch3oh以0.2ml/min的速率通入结晶釜,15min后停止通入ch3oh,以同样的速率将上述盐酸表柔比星溶液注入;进样完毕后,继续通入co240min,停止控制系统,将结晶釜压力降为大气压时取出,在结晶釜底部即得盐酸表柔比星晶型ⅲ。
对盐酸表柔比星晶型ⅲ经gc测定ch3oh残留量为674ppm。
实施例4
准确称取3.00g盐酸表柔比星溶于140mldmso中,过滤,滤液即为盐酸表柔比星溶液备用;在控制软件中将高压结晶釜设置温度为30℃,压力为8mpa,co2注入结晶釜中的流速为20g/min;打开钢瓶,用高压泵将co2通入结晶釜,待结晶釜中温度、压力达到设定值时,用另一高压泵将dmso以0.5ml/min的速率通入结晶釜,15min后停止通入dmso,以同样的速率将上述盐酸表柔比星溶液注入;进样完毕后,继续通入co240min,停止控制系统,将结晶釜压力降为大气压时取出,在结晶釜底部即得盐酸表柔比星晶型ⅲ。
对盐酸表柔比星晶型ⅲ经gc测定dmso残留量为691ppm。
实施例5
准确称取3.00g盐酸表柔比星溶于375mldmso中,过滤,滤液即为盐酸表柔比星溶液备用;在控制软件中将高压结晶釜设置温度为40℃,压力为25mpa,co2注入结晶釜中的流速为25g/min;打开钢瓶,用高压泵将co2通入结晶釜,待结晶釜中温度、压力达到设定值时,用另一高压泵将dmso以1.5ml/min的速率通入结晶釜,15min后停止通入dmso,以同样的速率将上述盐酸表柔比星溶液注入;进样完毕后,继续通入co260min,停止控制系统,将结晶釜压力降为大气压时取出,在结晶釜底部即得盐酸表柔比星晶型ⅲ。
对盐酸表柔比星晶型ⅲ经gc测定dmso残留量为670ppm。
对比实施例1
将3.00g盐酸表柔比星加入到ph3~4的乙醇-水的混合溶液中,在40℃下低压蒸发至溶液变为凝胶状态,向剩余溶液中加入12倍体积的1-丙醇并搅拌3h,过滤收集固体,用10ml丙酮洗涤并在35℃真空下干燥16h,所得晶体即为盐酸表柔比星晶型ⅱ,其xrd图如图3所示。
对盐酸表柔比星晶型ⅱ经gc测定乙醇残留含量为0.45%,1-丙醇残留量为0.68%,丙酮残留量为0.37%。
对比实施例2
将3.00g盐酸表柔比星加入到ph3.5的水中,40℃下低压蒸发至溶液变为凝胶状态,向上述体系中加入90ml丙酮,过滤收集固体,用20ml丙酮洗涤,并在35℃下真空干燥16h,所得晶体即为盐酸表柔比星晶型ⅰ。
对盐酸表柔比星晶型ⅰ经gc测定丙酮残留量为0.40%。
稳定性试验
称取两份实施例1制备得到的盐酸表柔比星晶型ⅲ样品,一份标为样品a,另一份经1-丙醇淋洗抽滤,并置于真空干燥箱中干燥,控制1-丙醇残留量为0.60%~0.70%时取出,标为样品b,将样品a与样品b放置在40℃的条件下,考察放置1个月、2个月、3个月、6个月的稳定性。并将样品a与样品b同现有技术中得到的晶体ⅰ型和ⅱ型样品进行稳定性试验的结果比较,试验结果详见表1。
具体的稳定性考察的方法可以按照中国药典2010版第二部附录xixc的方法;纯度检测用hplc法,可以参照中国药典2010版第二部附录vd的方法。
表1稳定性考察结果
溶解度实验
向过量的待测实施例1及对比实施例1样品中加入一定体积的水,置于25℃的恒温水浴中持续搅拌24h,每3h取样,滤膜过滤,适当稀释后经hplc分析,直到溶液达到平衡,并通过外标法测得该样品的溶解度。
表2溶解度测定结果
由上述结果可知,本发明采用超临界反溶剂技术可以制备出稳定性较晶型ⅰ高而溶解度较晶型ⅱ更好的盐酸表柔比星晶型ⅲ,而晶型ⅲ稳定性的提高并非是溶剂残留量较少所致。与现有技术中制备盐酸表柔比星晶型ⅰ和ⅱ的方法相比,本发明超临界反溶剂技术可一步完成药物晶型制备及微粒化过程,使生产过程大幅简化。此工艺简单,条件温和,重现性好,且溶剂残留量及其微小,适宜规模化生产。
- 还没有人留言评论。精彩留言会获得点赞!