一种盐酸舍曲林杂质的制备方法与流程
本发明涉及一种盐酸舍曲林杂质的制备方法。
背景技术:
盐酸舍曲林(sertraline),化学名为:顺-(1s,4s)-4-(3,4-二氯苯基)-1,2,3,4-四氢-n-甲基-1-萘胺盐酸盐;化学式:c17h17ncl2·hcl,由美国辉瑞公司在20世纪90年代初新开发上市,并获美国fda许可用于经前焦虑障碍(pmdd)的治疗,也是第一个获许可的pmdd治疗物,在国际上广泛用于治疗抑郁性及强迫性精神障碍。舍曲林于1990年12月首先在英国上市,目前为止已经在13个国家陆续上市。舍曲林的结构式如下式i所示:
根据已有文献中报道的盐酸舍曲林的制备方法,在其制备过程中,苯环上的二氯有脱去的风险,脱去其中1个氯会产生如下两个杂质,其结构式如下所示:
通过文献查阅,还未发现有文献公开报道此两种杂质的具体制备方法。
技术实现要素:
本发明所解决的技术问题在于提供一种盐酸舍曲林杂质的制备方法,形成质量标准,对盐酸舍曲林的杂质进行定性和定量检测。
盐酸舍曲林结构式有一个3,4-二氯取代的苯环,分析原料药发现其中存在单氯取代的杂质,如下所示:
式8a化合物和式8b化合物分别为4位一氯取代杂质和3位一氯取代杂质。因此,在杂质制备时我们设计从源头开始进行单氯取代的苯环进行合成。
本发明提供的技术方案如下:
一种盐酸舍曲林杂质的制备方法,其包括下述步骤:将式6化合物经leuckart反应得到式7化合物,再与盐酸经成盐反应得到式8化合物(即杂质8)的顺式异构体:
其中,式6化合物中一氯取代的位置为间位或者对位。
即,所制备得到的式8化合物为下式8a化合物或式8b化合物:
其中,所述leuckart反应的方法和条件可为本领域常规的方法和条件。所述leuckart反应一般在甲酸存在条件下由式6化合物与n-甲基甲酰胺进行反应。所述leuckart反应中水解步骤所用碱可选自氢氧化钠、氢氧化钾和氢氧化锂中的一种或多种。
其中,式7化合物成盐酸盐得到式8化合物的顺式异构体。所述成盐反应的溶剂可选自甲醇、乙醇、丙醇、正丁醇和异丁醇中的一种或多种。
按本领域常识,所述成盐反应后还可进行常规的后处理操作,包括但不限于过滤、洗涤和干燥的步骤。
其中,式6化合物可按本领域常规制备方法制取。在本发明中,所述式6化合物优选由下述路线制得:
1)式1a或1b化合物经缩合对接得到式2化合物;
2)式2化合物还原得到式3化合物;
3)式3化合物经还原得到式4化合物;
4)式4化合物经分子内成酯得到式5化合物;
5)式5化合物经傅克反应得到式6化合物。
具体反应方法和条件优选如下:
1)式1a或式1b化合物在酸性环境下与乙醛酸对接得到式2化合物。所用的酸可选自盐酸、硫酸、醋酸和磷酸中的一种或多种;反应溶剂可选自水、醋酸、盐酸水溶液、磷酸和二氧六环的一种或多种。
2)式2化合物在还原条件下,经还原得到式3化合物。所用的还原剂可选自活泼金属,如铁粉、锌粉和锡粉中的一种或多种,也可选择催化加氢试剂,如钯,铂,镍,钯-碳酸钙和钯-硫酸钡中的一种或多种。反应溶剂可选自乙醇、甲醇、乙酸乙酯、乙醚、四氢呋喃、甲基四氢呋喃、二氧六环、醋酸和水的一种或多种。
3)式3化合物在还原条件下,经还原得到式4化合物。所用的还原剂可选自金属氢化物,如硼氢化钠、硼氢化钾、硼氢化锂、四氢铝锂和硼烷-乙醚中的一种或多种。反应的溶剂可自乙醇、甲醇、乙醚、四氢呋喃、甲基四氢呋喃和水的一种或多种。
4)式4化合物在酸性条件下关环成五元环内酯得到式5化合物。所述酸性条件的酸可选自盐酸、硫酸、醋酸和磷酸的一种或多种。
5)式5化合物与苯环经傅-克反应得到式6化合物。所述傅-克反应的路易斯酸可选择氯化锌、氯化铝、氯化铁、四氯化锡、四氯化钛、甲磺酸和三氟甲磺酸中的一种或多种。反应的溶剂可自苯、二氯甲烷和三氯甲烷中的一种或多种。
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明所用试剂和原料均市售可得。
本发明的积极进步效果在于:
本发明的盐酸舍曲林杂质的制备方法简单,得到的杂质8a和杂质8b纯度高,可形成质量标准,对盐酸舍曲林的杂质进行定性和定量检测。
附图说明
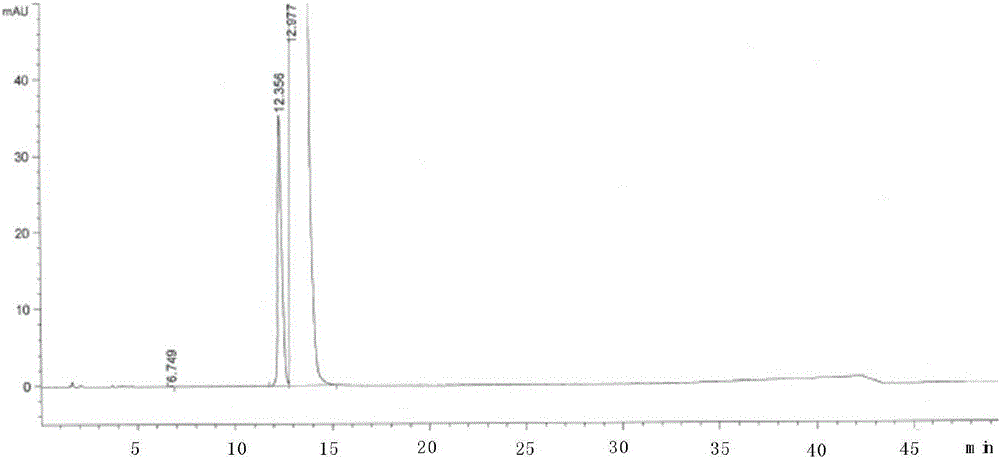
图1是杂质8a的hplc图谱。
图2是杂质8b的hplc图谱。
具体实施方式
实施例1:杂质8a的制备
将200g式1a化合物、260g50%乙醛酸和400ml乙酸混匀,搅拌。升温至回流反应过夜,tlc检测原料消失后降温到70-80度,减压浓缩蒸出乙酸,残余物用乙醇:水=1:10搅拌析晶1小时,抽滤,滤饼烘干得式2化合物138克黄色固体。
将138克式2化合物、50克锌粉、700ml乙酸、150ml水混匀,机械搅拌,室温反应过夜,tlc检测原料反应完后,抽滤,滤饼用乙酸乙酯洗涤。收集滤液,减压浓缩至干。残余物用乙酸乙酯:正己烷=1:12打浆析晶,抽滤,滤饼烘干得120克式3化合物。
将120克式3化合物溶于350毫升水中,升温至80℃,加入1.5n氢氧化钠调节ph=11,搅拌溶清。滴加25克硼氢化钠水溶液160ml,滴加时要慢,体系会放出大量气泡。滴加结束,反应4小时,原料过滤,滤去白的不溶物,得式4化合物水溶液。
将式4化合物水溶液,用乙酸乙酯萃取除杂,收集水层,水层升至50度,用6nhcl调节反应体系ph=1-2,保温反应3小时。tlc检测原料消失后,降温,加入二氯甲烷萃取,收集有机层,无水硫酸钠干燥、过滤、滤液减压浓缩得80克式5化合物。
将80克式5化合物加入到200ml苯中,缓慢滴加170ml三氟甲磺酸,加完后60度反应2小时。结束后降温,体系缓慢加入到冰水中,用4n氢氧化钠调节ph=9,加入二氯甲烷萃取分层,收集有机层,干燥、过滤、浓缩得95克式6化合物,不进一步纯化,直接下一步反应。
将上一步中式6化合物溶于n-甲基甲酰胺中,升温至140度。缓慢加入20克甲酸,加完后,升至190度反应约10小时,tlc监测原料消失后,降温至160摄氏度,减压蒸去n-甲基甲酰胺。冷却到110℃加入400克正丁醇,缓慢加入57.3克氢氧化钾,毕,升至130度反应10小时,反应结束后,降至室温加入水分层。收集有机层,无水硫酸钠干燥、过滤得式7化合物溶液,不进一步后处理,直接下一步。
将上一步中的式7化合物溶液,用浓盐酸调节ph=1,常温搅拌析晶过夜,抽滤,滤饼用正丁醇洗两次,固体重新用饱和碳酸氢钠水溶液和二氯甲烷游离,收集二氯甲烷层,无水硫酸钠干燥、过滤、浓缩得到的残余物加入70克正丁醇,再用浓盐酸调节ph=1成盐,析出白色固体,抽滤,滤饼用正丁醇洗涤,烘干得8克杂质8a。1h-nmr(500mhz,dmso-d6):δ7.81-7.67(m,1h),7.47-7.34(m,4h),7.33-7.19(m,2h),6.75(d,j=7.5hz,1h),4.43(s,1h),4.11(dd,j=9.2,5.6hz,1h),2.63(t,j=5.3hz,3h),2.30-2.10(m,2h),2.09-1.93(m,2h).esi[m+h]+:238.15
实施例2:杂质8b的合成
将100g式1b化合物、1300g50%乙醛酸和200ml乙酸混匀,搅拌。升温至回流反应过夜,tlc检测原料消失后降温到70-80度,减压浓缩蒸出乙酸,残余物用乙醇:水=1:7搅拌析晶1小时,抽滤,滤饼烘干得式2化合物70克黄色固体。
将70克式2化合物、24克锌粉、300ml乙酸、80ml水混匀,机械搅拌,室温反应过夜,tlc检测原料反应完后,抽滤,滤饼用乙酸乙酯洗涤。收集滤液,减压浓缩至干。残余物用乙酸乙酯:正己烷=1:10打浆析晶,抽滤,滤饼烘干得55克式3化合物。
将55克式3化合物溶于120毫升水中,升温至80℃,加入1.5n氢氧化钠调节ph=11,搅拌溶清。滴加12克硼氢化钠水溶液80ml,滴加时要慢,体系会放出大量气泡。滴加结束,反应4小时,原料过滤,滤去白的不溶物,得式4化合物水溶液。
将式4化合物水溶液,用乙酸乙酯萃取除杂,收集水层,水层升至50度,用6nhcl调节反应体系ph=1-2,保温反应3小时。tlc检测原料消失后,降温,加入二氯甲烷萃取,收集有机层,无水硫酸钠干燥、过滤、滤液减压浓缩得35克式5化合物。
将35克式5化合物加入到100ml苯中,缓慢滴加70ml三氟甲磺酸,加完后60度反应2小时。结束后降温,体系缓慢加入到冰水中,用4n氢氧化钠调节ph=9,加入二氯甲烷萃取分层,收集有机层,干燥、过滤、浓缩得46克式6化合物,不进一步纯化,直接下一步反应。
将上一步中式6化合物溶于n-甲基甲酰胺中,升温至140度。缓慢加入10克甲酸,加完后,升至190度反应约10小时,tlc监测原料消失后,降温至160摄氏度,减压蒸去n-甲基甲酰胺。冷却到110℃加入170克正丁醇,缓慢加入28克氢氧化钾,毕,升至130度反应10小时,反应结束后,降至室温加入水分层。收集有机层,无水硫酸钠干燥、过滤得式7化合物溶液,不进一步后处理,直接下一步。
将上一步中的式7化合物溶液,用浓盐酸调节ph=1,常温搅拌析晶过夜,抽滤,滤饼用正丁醇洗两次,固体重新用饱和碳酸氢钠水溶液和二氯甲烷游离,收集二氯甲烷层,无水硫酸钠干燥、过滤、浓缩得到的残余物加入40克正丁醇,再用浓盐酸调节ph=1成盐,析出白色固体,抽滤,滤饼用正丁醇洗涤,烘干得3.5克杂质8b。1h-nmr(500mhz,dmso-d6):δ7.73(d,j=7.3hz,1h),7.44(s,1h),7.40-7.23(m,5h),6.75(d,j=7.3hz,1h),4.53-4.36(m,1h),4.13(dd,j=9.7,5.5hz,1h),2.64(t,j=5.3hz,3h),2.32-2.09(m,2h),2.09-1.93(m,2h).esi[m+h]+:238.15
效果实施例
按照高效液相色谱法(中国药典2015年版四部通则0512)测定实施例1和2的产物,用十八烷基硅烷键合硅胶为填充剂(xbridgec18,4.6×250mm,5μm),以0.272%磷酸二氢钾溶液(用磷酸调节ph值至3.0)为流动相a,以乙腈为流动相b,按下表1进行线性梯度洗脱;检测波长为210nm;柱温为30℃。杂质8a和杂质8b的hplc测试结果见图1和图2。
表1
根据图1和图2可看出由本发明的盐酸舍曲林杂质的制备方法得到的杂质8a和杂质8b的纯度分别为96.38%和99.42%,纯度较高,副反应少,含量可控,可形成质量标准,对盐酸舍曲林的杂质进行定性和定量检测。
- 还没有人留言评论。精彩留言会获得点赞!