2-N-甲基氨甲基-5-N-甲基亚氨甲基呋喃的合成的制作方法
本发明涉及有机化合物的合成方法,尤其是涉及一种2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃的合成方法。
背景技术:
5-羟甲基糠醛(hmf)是生物质催化转换中一种极其重要的平台化合物,它的很多下游产品应用广泛,可以转化成各种具有高附加值的呋喃类物质,如2,5-二甲基呋喃(dmf)、2,5-呋喃二甲醇(fdm)、2,5-呋喃二甲醛(dff)、2,5-呋喃二甲酸(fdca)等。hmf进行氨取代反应可合成一大类hmf氨代衍生物,如中国专利cn106349195a报道了一种n,n-二甲基四氢糠胺的合成方法。此方法主要以糠醛为原料,与n,n–二甲基甲酰胺或n,n–二甲基乙酰胺和甲酸混合反应,先生成n,n-二甲基糠胺,然后催化加氢得到n,n-二甲基四氢糠胺产品。文献johnj.roylanceandkyoung-shinchoi,greenchem.,2016,18,5412-5417使用电化学法,以2,5-呋喃二甲醛(dff)、甲胺为原料,水作为还原剂来制备2,5-二(n-甲基氨基甲基)呋喃,此方法中以aggd作为电极。此外,还有不少以金属作为催化剂进行还原加氢的方法被报道。文献ngoc-thucle,areumbyun,yohanhan,kee-inlee,hyungrokkim,greenandsustainablechemistry,2015,5,115-127以2,5-呋喃二甲醛(dff)、氨(nh3,99%)和氢气(h2)为原料,雷尼镍作为催化剂,在乙醇-水,四氢呋喃-水等体系中反应,制备2,5-二氨基甲基呋喃。美国专利us8669383b2报道一种糖类和氢气作为反应物,在烷基酰胺溶剂体系中,同时存在酸催化剂(包括均相的无机酸或基质上的多相酸催化剂)和pt、pd、ni等氢化还原催化剂来制备呋喃和四氢呋喃的各种类型的烷基胺衍生物的方法,产物包括2-(n-烷基氨基甲基)-5-羟甲基呋喃、2-(n,n-二烷基氨基甲基)-5-羟甲基呋喃、2,5-二(n-烷基氨基甲基)呋喃、2,5-二(n,n-二烷基氨基甲基)呋喃、2-(n-烷基氨基甲基)-5-羟甲基四氢呋喃、2-(n,n-二烷基氨基甲基)-5-羟甲基四氢呋喃、2,5-二(n-烷基氨基甲基)四氢呋喃、2,5-二(n,n-二烷基氨基甲基)四氢呋喃等。中国专利cn104277018b报道一种以氨气为胺源,以氢气为氢源,以负载型金属为催化剂,在30-220℃,将2,5-二甲酰基呋喃选择还原胺化为2,5-二甲胺基呋喃的方法。此方法中负载型催化剂的活性组分金属是指:ni、cu、co、cr、sn、al、b1、ce、pt、pd、au、ag、rh、ru、ir、re中的一种或二种以上,以金属计,其含量为催化剂质量的0.1-30wt%,载体为金属氧化物(mxoy)是指:cao、mgo、la2o3、y2o3、ceo2、zro2、al2o3、tio2、nb2o5、sno2、v2o5、mno2、fe2o3、fe3o4、moo3中的至少一种。然而2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃的合成报道很少。
技术实现要素:
本发明的目的在于提供一种工艺简单,成本低廉的2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃的合成方法。
本发明包括以下步骤:
1)将5-羟甲基糠醛(hmf)、有机胺、甲酸水溶液混合于反应容器中,加热反应后,旋蒸去除溶剂,得到溶液a;
在步骤1)中,所述甲酸水溶液的摩尔浓度可为1~10mol/l;所述有机胺可选自甲胺、n-甲基甲酰胺、n-甲基乙酰胺等中的至少一种,所述5-羟甲基糠醛与有机胺的摩尔比可为1︰(5~20);所述反应的温度可为170~230℃,反应的时间可为1~10h。
2)将naoh溶液加入溶液a中,调节溶液ph≥10,得到溶液b;
在步骤2)中,所述naoh溶液的质量百分比浓度超过40%。
3)用乙酸乙酯对溶液b进行萃取,合并萃取剂,减压回收萃取剂乙酸乙酯,得到溶液c;
4)对溶液c进行减压蒸馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃,所得2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃的纯度高于99%。
在步骤4)中,所述蒸馏的温度可为180~230℃,压力为2000pa。
本发明的突出优点在于:
1)本发明以hmf、有机胺作为原料,甲酸水溶液为反应溶液一锅法制备2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。该方法操作简单,反应体系绿色环保,反应溶剂可重复利用,投料浓度大,得率高。
2)本发明以生物质平台化合物hmf为原料制备2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃,原料来源广泛。
3)本发明中有机胺为甲胺、n-甲基甲酰胺、n-甲基乙酰胺,成本低廉。
4)本发明对设备要求低,操作简单。
5)本发明合成产物的分离过程降低,产品纯度和得率高。
本发明所制备的2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃的得率测定按高效气相色谱(gc)外标定量分析法。
附图说明
图1为本发明实施例的gc-ms图。
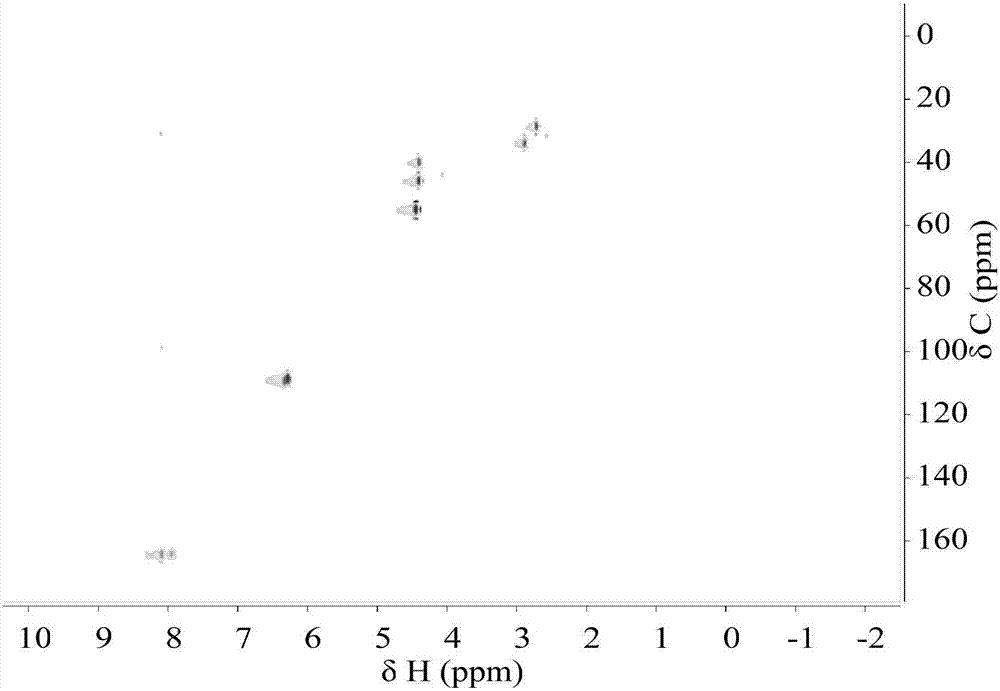
图2为本发明实施例的二维nmr图。
图3为本发明实施例的nmr碳谱图。
图4为本发明实施例的nmr氢谱图。
具体实施方式
下面结合实施例对本发明作进一步的描述。
实施例1
在50ml反应釜中加入1.89ghmf和4.65g甲胺,适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为170℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为31.2%。
实施例2
在50ml反应釜中加入1.89ghmf和9.03gn-甲基甲酰胺溶液(98%),适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为170℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为35.0%。
实施例3
在50ml反应釜中加入1.89ghmf和10.95gn-甲基乙酰胺溶液(>99%),适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为170℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为32.3%。
实施例4
在50ml反应釜中加入1.89ghmf和2.325g甲胺,适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为170℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为29.7%。
实施例5
在50ml反应釜中加入1.89ghmf和4.51gn-甲基甲酰胺溶液(98%),适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为170℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为30.6%。
实施例6
在50ml反应釜中加入1.89ghmf和5.47gn-甲基乙酰胺溶液(>99%),适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为170℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为27.9%。
实施例7
在50ml反应釜中加入1.89ghmf和9.3g甲胺,适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为170℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为32.1%。
实施例8
在50ml反应釜中加入1.89ghmf和18.06gn-甲基甲酰胺溶液(98%),适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为170℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为37.9%。
实施例9
在50ml反应釜中加入1.89ghmf和21.9gn-甲基乙酰胺溶液(>99%),适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为170℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为34.7%。
实施例10
在50ml反应釜中加入1.89ghmf和9.03gn-甲基甲酰胺溶液(98%),适当振荡混匀。再加入25ml甲酸溶液(1mol/l)。加热反应,温度为170℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为33.0%。
实施例11
在50ml反应釜中加入1.89ghmf和9.03gn-甲基甲酰胺溶液(98%),适当振荡混匀。再加入25ml甲酸溶液(10mol/l)。加热反应,温度为170℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为36.5%。
实施例12
在50ml反应釜中加入1.89ghmf和9.03gn-甲基甲酰胺溶液(98%),适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为200℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为36.9%。
实施例13
在50ml反应釜中加入1.89ghmf和9.03gn-甲基甲酰胺溶液(98%),适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为230℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为35.8%。
实施例14
在50ml反应釜中加入1.89ghmf和9.03gn-甲基甲酰胺溶液(98%),适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为200℃,反应时间为1h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为34.1%。
实施例15
在50ml反应釜中加入1.89ghmf和9.03gn-甲基甲酰胺溶液(98%),适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为230℃,反应时间为10h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为38.2%。
实施例16
在50ml反应釜中加入1.89ghmf和9.03gn-甲基甲酰胺溶液(98%),适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为200℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在160℃,压力为2000pa条件下精馏。经gc测定,未得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。
实施例17
在50ml反应釜中加入1.89ghmf和9.03gn-甲基甲酰胺溶液(98%),适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为200℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=12,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在230℃,压力为2000pa条件下精馏。,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为36.7%。
实施例18
在50ml反应釜中加入1.89ghmf和9.03gn-甲基甲酰胺溶液(98%),适当振荡混匀。再加入25ml甲酸溶液(7mol/l)。加热反应,温度为170℃,反应时间为3h,旋转混匀,转速300r/min。反应完毕后,放入水中冷却。取出反应液,在80℃下旋蒸去除溶剂,直到溶液质量不再减少。旋蒸结束后,向溶液中加入浓naoh溶液调节酸碱度至ph=8,连续使用等体积乙酸乙酯萃取残余物数次,合并萃取液,减压蒸馏回收萃取剂。在200℃,压力为2000pa条件下精馏,得到2-n-甲基氨甲基-5-n-甲基亚氨甲基呋喃。经gc测定,其纯度达到99%以上,产品摩尔得率为30.5%。
本发明实施例的gc-ms图参见图1,二维nmr图参见图2,nmr碳谱图参见图3,nmr氢谱图参见图4。
- 还没有人留言评论。精彩留言会获得点赞!