跟踪核酸靶标来源以用于核酸测序的方法和试剂盒与流程
本公开大体上涉及用于改进的核酸测序的方法和试剂盒。
背景技术:
本发明属于基因组学的技术领域。更具体来说,本发明属于核酸测序的技术领域。核酸测序可以为多种多样的生物医学应用提供信息,所述生物医学应用包括诊断学、预后学、药物基因组学、以及法医生物学。测序可以利用基本低通量方法,包括马克萨姆-吉尔伯特测序(maxam-gilbertsequencing)(化学修饰的核苷酸)和桑格测序(sangersequencing)(链终止)方法;或高通量下一代方法,包括大规模并行焦磷酸测序、边合成边测序、边连接边测序、半导体测序等。对于大部分的测序方法,在引入测序仪器中之前,需要处理样品,如核酸靶标。举例来说,可以将样品片段化、扩增或与标识物附连。独特的标识物常常用于标识特定样品的来源。大部分的测序方法产生相对短的测序读段(read),长度从数十个碱基到数百个碱基不等,并且由于有限的测序读段长度而不能产生完整的单倍型相信息。
技术实现要素:
本发明提供了用于当核酸靶标被断裂成更小段时通过条形码标记跟踪核酸靶标来源的方法和试剂盒。可以在固体载体(例如珠粒、微粒、载片、平板或流动池)上克隆扩增或克隆合成用作条形码的多个核酸序列。本发明中的条形码序列的设计允许产生数十亿个不同的条形码并且每个条形码序列含有用于提高测序准确度的特征。通过这些克隆定位的核酸条形码模板在固体载体上体外捕获具有或不具有修饰的核酸靶标。使用转座酶和可转座的dna来促进核酸靶标的片段化和条形码标记。可以以大规模并行方式同时处理数百个、数千个或数百万个核酸靶标。所述靶标中的每个可以在开放的本体(bulk)反应中被一组独特的条形码局部捕获而无需另外的分区,如用孔、微孔、洞、管、斑点、纳米通道、液滴、乳液液滴、胶囊、或用于分隔样品的部分的任何其他合适的容器进行分区。可以将这些捕获的靶标断裂成更小的片段,并且靶标特异性条形码序列将被标记到每个片段上作为其原始靶标的标识。这些核酸靶标跟踪方法可以用于全基因组测序和靶向测序这两者中的多种应用。
本文提供的方法和试剂盒相对于现有方法,如illumina公司的合成长读段和10xgenomics公司的连接读段,提供了若干优点。举例来说,本发明提供了数百万个至数十亿个或更多个条形码,这些条形码显著提高标记容量和特异性。条形码设计提供了减少来自长段相同类型的核苷酸,即均聚物序列的测序错误并且滤出低质量读段的特征,以使它提高测序质量。可以使用已知的化学方法(例如乳液pcr方法、桥式pcr)在固体表面上直接克隆合成或克隆或半克隆扩增条形码。基于转座酶的片段化方法简化了样品制备程序。不同于所有的现有方法,本发明中的条形码标记反应可以在开放的本体溶液中进行,而无需用孔、微孔、洞、管、斑点、纳米通道、液滴、乳液液滴、胶囊进行另外的分区。所述程序容易自动化或按比例放大以用于高通量样品制备。本发明提供了条形码标记方法,所述方法不仅用于长核酸样品以用于诸如单倍型定相、结构变异检测以及拷贝数研究的应用,而且还用于短核酸样品以跟踪样品的独特性。
附图说明
图1是示出了条形码序列结构和组成的实例的表。
图2说明了核酸条形码模板。
图3示出了两种不同的可转座的dna设计,(a)具有一体的3'突出端的转座子互补链,(b)具有分开的互补接头寡核苷酸的转座子互补链。
图4说明了通过杂交(a)和连接(b)在固体载体上捕获具有互补的3'突出端的转座子或转座体。
图5说明了通过杂交(a)和连接(b)在固体载体上捕获具有互补接头寡核苷酸设计的转座子或转座体。
图6是示出了核酸靶标(601)由转座体(602)标记,形成连续的转座体-核酸复合物(603),由克隆条形码模板(605)捕获在固体载体(604)上,然后片段化成条形码标记的片段(606)的示意图。
图7是示出了以下的示意图:多个核酸靶标(701)由转座体(702)同时标记,由克隆条形码模板(705)分别捕获在连续固体表面(704,例如流动池表面)上分开的斑点处或单独的固体载体(例如珠粒或微粒,未示出)上,并且在没有另外分区的开放的本体反应中片段化成条形码标记的片段。
图8是示出了通过油包水乳液液滴(805)封装转座体-核酸复合物(801)和带克隆条形码的珠粒(802)以产生条形码(803)标记的核酸片段的示意图。
图9说明了在乳液液滴中产生固定的条形码标记的片段的两种不同的方式。
图10是示出了液滴(1003)中的转座体-核酸复合物(1001)与另一个液滴(1005)中的克隆条形码(1004)池合并成组合液滴(1006)以产生条形码标记的片段(1007)的示意图。
图11说明了在液滴中产生条形码标记的片段的两种不同的方式。
图12说明了以不同形式固定在固体载体上的具有转座酶结合区(tbr)的条形码模板。(a)在一端具有tbr的条形码模板;(b)固体载体上呈双链形式的在游离端具有tbr的固定的条形码模板;(c)固体载体上呈单链形式的在游离端具有tbr的固定的条形码模板,tbr的互补链可以通过引物退火、杂交、和/或引物延伸来引入。
图13是示出了使转座酶(1303)与固体载体(1302)上具有tbr末端的条形码模板(1301)结合,之后捕获并片段化核酸靶标(1305)以用于条形码标记的示意图。
图14是示出了以下的示意图:使转座酶(1403)结合到在固体载体(1402)上的不同分离位置处的具有tbr末端的克隆条形码模板(1401)上,每个位置并行捕获不同的核酸靶标,并且在没有另外分区的开放的本体反应中将它在固体载体上用条形码标签片段化。
图15提供了通过引物延伸(a)和/或pcr扩增(b)释放固定的条形码标记片段的一个或多个拷贝(c)的图示。
图16是illumina公司的由条形码标记片段产生的测序文库和其测序方法的实例。
图17是iontorrent公司的由条形码标记片段产生的测序文库和其测序方法的实例。
图18是使用释放的条形码标记的片段进行靶向扩增的图示。
图19是直接从固体载体上条形码标记的片段富集所关注的基因的图示。
图20示出了条形码以及条形码模板序列和结构的具体实例。
图21示出了通过测序检测的特异性设计的条形码的每个位置处的核苷酸含量。
图22列出了具有用于被条形码模板捕获的可附连的末端的mua可转座的dna设计的三个实例。
图23是在mua标记反应之后片段化的基因组dna的琼脂糖凝胶电泳照片。l:1kbplusdna标准梯(单位:bp),s:片段化的dna样品。
图24是在tapestation上的高灵敏度d5000screentape上运行的条形码标记的illumina测序文库的电泳图。
图25是示出了用本发明中所述的条形码标记法构建的illumina测序文库结构的示意图。
图26示出了相同条形码读段1到下一次比对的读段距离的读段1测序读段计数直方图。
所有图中的转座酶在基于mua转座系统的转座体中被示为四聚体。
具体实施方式
大部分可商购获得的测序技术具有有限的测序读段长度。第二代测序技术尤其仅可以对几百个碱基进行测序并且很少达到一千个碱基。然而,基因的核酸序列可以从数千碱基至数十和数百千碱基,这意味着数十千碱基的测序读段长度对于成功确定所有基因的单倍型是必需的。本公开提供了用于将核酸靶标处理成更小段,同时用靶标特异性条形码标签保留其来源信息的方法和试剂盒。经过处理的dna样品可以用于产生用于测序应用的文库。可以将测序数据组装成完整或串联长读段以进行单倍型定相。本文提供的方法和试剂盒相对于现有方法,如illumina公司的合成长读段和10xgenomics公司的连接读段,提供了几个优点。举例来说,本公开提供了数百万个和数十亿个或更多个条形码,这些条形码通过提高标记容量和特异性来提高测序准确度。此外,不同于现有方法,本公开中的条形码标记反应可以在开放的本体溶液中进行,而无需用孔、微孔、洞、管、斑点、纳米通道、液滴、乳液液滴、胶囊等进行进一步分区。所述程序容易自动化或按比例放大以用于高通量样品制备。
用于最大容量、测序准确度以及效用的条形码设计
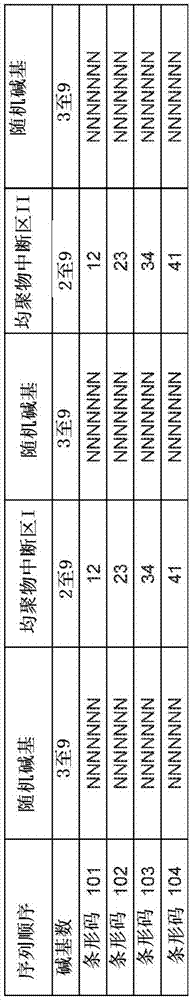
条形编码方法已经被广泛用于高通量测序应用中以用于样品鉴定。具有完全随机或简并核苷酸序列的条形码设计用于对单个核酸和pcr扩增子进行分子标记。“条形码”一般意指可以与靶标缔合(例如附连)并且传递该靶标的信息(例如身份)的标记。“随机”或“简并”意指其中一个或多个位置含有许多可能碱基(例如a、t、g、c、u中的任何2个或3个或4个)的核酸序列。一般来说,条形码可以是具有4个至100个碱基,优选地6个至25个碱基的长度的任何核酸序列,最常见的是6个至8个碱基。本公开中所公开的方法和试剂盒包括改进的条形码设计以能够不仅提供最大的条形编码容量,而且还提供提高的测序准确度,并且同时鉴定分子和样品(例如来自不同患者的不同样品)两者。本公开提供了一种条形码设计,所述条形码设计含有穿插有预定的非均聚物核苷酸区段(被称作均聚物中断区)的两个或更多个随机核苷酸区段。每个随机序列区段可以含有3个至9个简并碱基,优选地3个至7个简并碱基。每个随机区段的长度可以是相同的或不同的。每个均聚物中断区在长度上可以具有2个至9个已知碱基。在一个实施方式中,所述条形码具有两个随机简并核苷酸序列,在这两个核苷酸序列之间具有一个已知的均聚物中断区。在另一个实施方式(图1)中,所述条形码具有三个随机简并核苷酸序列,其中穿插有两个已知的均聚物中断区。在一些情况下,一个中断区可以具有与其他中断区不同的长度。在其他情况下,它可以具有相同的长度。在一些情况下,中断区i与中断区ii具有相同的序列。在其他情况下,每个中断区具有不同的序列。在一个实施方式中,通过设计,中断区序列具有有限的多样性。当中断区中的一个或几个碱基是已知的时,序列的其余部分的身份将是已知的。该条形码设计控制条形码中均聚物碱基的最大长度。它还具有错误检查特征以提高条形码身份的特异性。长段的均聚物碱基(例如8聚体或更高)对于准确测序是非常有挑战性的。使用目前可用的测序技术,非常长的均聚物碱基的测序错误几乎是不可避免的。通过(使用均聚物中断区)缩短条形码中均聚物碱基的最大长度,测序准确度将提高。在条形码中添加可以用作质量过滤器的已知的非均聚物核苷酸序列将进一步提高条形码识别的测序准确度。如果用含有不同已知序列的条形码制备每个样品,那么设计中的已知序列也可以用作高水平鉴定,如样品鉴定。这样的设计的条形码序列的长度最少是8个碱基并且可以多达100个碱基或更多,但是优选的长度在约10个至约50个碱基,并且理想地约12个至约25个碱基的范围内。
对于某些测序技术,如illumina公司的边合成边测序(sbs)技术,如果所有分子的碱基序列在一个特定的测序流程步骤中是相同的,那么它将干扰信号处理流水线并且倾向于导致更高的错误率。在一些情况下,为了避免所有条形码序列具有相同的中断区区段,可以共同使用多于一种条形码序列设计。它们可以具有相同的条形码结构,但是具有不同的中断区序列以使在具体的测序流程步骤中,将有至少两种不同的核苷酸碱基存在。在图1中的一个实施方式中,条形码101具有三个7聚体随机简并区段和两个2聚体均聚物中断区。中断区中的碱基1可以是5个核苷酸碱基a、c、g、t、u中的任一个;中断区中的碱基2可以是5个核苷酸碱基a、c、g、t、u中的任一个,但是必须同时与碱基1不同。为了增加在用illumina公司的sbs技术测序期间均聚物中断区1的碱基1位置处核苷酸的多样性,可以将条形码102、103以及104中的一个、两个或三个与条形码101混合在一起作为最终的条形码设计。具有条形码101、102、103以及104的混合物的条形码设计可以产生最多4×1022种排列(t和u不能通过当前的测序技术区分,因此在排列中考虑总共四个核苷酸碱基,并且由于均聚物中断区是固定序列,因此仅考虑四种可能性),即1.76×1013种不同的条形码序列。
“条形码模板”含有条形码序列,所述条形码序列在一端处侧接有至少一个柄部序列或在两端处侧接有两个柄部序列(图2)。柄部序列可以用作用于杂交或退火的结合位点、用作扩增期间的引发(priming)位点、或用作测序引物或转座酶的结合位点。包括适当设计的条形码和两个柄部的条形码模板可以由从标准合成获得的寡核苷酸或寡核苷酸阵列产生。
可以通过克隆扩增或合成将条形码模板(图2)克隆固定到合适的固体载体上,如珠粒、微粒、载片、平板、或流动池,其中所述固体载体在表面上具有多个相同的条形码模板(“克隆条形码模板”),所述条形码模板具有一个独特的条形码序列。克隆扩增方法可以包括桥式聚合酶链反应(pcr)、乳液pcr扩增、使用模板步移的等温扩增以及重组酶聚合酶扩增(rpa)反应等。为了使用条形码模板在固体载体上进行最佳的克隆扩增,可以使用有限稀释。对于基于乳液的扩增,稀释条形码模板以使大部分的乳液液滴仅含有一个条形码模板。在克隆扩增反应中用于簇或液滴的多于一个条形码模板不是最佳的,但是如果一个克隆反应中不同条形码模板的数量不高,那么它们在一些应用中可以被很好地容忍。这被称为半克隆扩增,其会产生“半克隆条形码模板”。对于乳液pcr扩增,基于泊松分布(poissondistribution),如果以1:1比率混合珠粒和条形码模板,那么在理论上将大致上最后得到36.8%的珠粒具有一个条形码模板,18.4%的珠粒具有两个条形码模板,8%具有三个或更多个条形码模板。在所有阳性珠粒中良好珠粒(具有一个或两个条形码模板)的百分比是87.3%,但是将有约36.8%的珠粒没有条形码模板,它们将被浪费。然而,如果珠粒/条形码模板比率增加到2:1,那么将有约30.3%具有一个条形码,7.6%具有两个条形码,但是只有1.4%具有三个或更多个条形码。在阳性珠粒中良好珠粒比率将增加到96.3%,但是有超过一半的珠粒被浪费。当使用珠粒或微粒作为固体载体时,这些珠粒或微粒可以具有均匀的尺寸或不均匀的尺寸。对于更好的结果,珠粒或微粒的直径应当被控制在200nm至50μm,优选地1μm至15μm,尽管它可以小到40nm或大到100μm。此外,珠粒或微粒可以是磁性的以便于处理。珠粒或微粒也可以是多孔的或无孔的。在一些实施方式中,可以使用另外的程序以仅富集具有扩增的条形码模板的珠粒或微粒。在其他实施方式中,可能不需要富集程序。没有条形码模板的珠粒或微粒可以在下游反应中用作间隔物。对于桥式pcr,稀释条形码模板以使大部分的簇由一个条形码模板形成。对于固体表面,如流动池表面上的条形码簇,簇尺寸应当被控制在50nm至200μm(直径),优选地100nm至100μm,并且簇分隔间距应当至少大于簇尺寸,理想地大于最长核酸靶标的长度,以避免一个核酸靶标被两个或更多个条形码簇捕获。一般规则是核酸靶标越长,则珠粒尺寸或条形码簇尺寸以及珠粒或簇间距越大。簇之间的距离可以通过条形码模板的有限稀释或通过特殊设计的阵列样表面来控制。在任何类型的固体载体上,克隆扩增的条形码模板可以是双链的或被变性为仅单链的。
在一些情况下,单链条形码模板多核苷酸可以直接在固体载体上克隆合成,如使用反向合成以及均分与合并方法(macosko等,2015),而不使用克隆扩增。
在没有另外分区的情况下用带克隆条形码的固体载体捕获核酸-转座体复合物以对核酸进行条形码标记
本公开提供了将由转座体结合的核酸靶标捕获到带克隆条形码的固体载体的方法和试剂盒。然后可以将捕获的核酸靶标片段化并且用带条形码的固体载体上的条形码序列进行标记。
如本文所用的术语“转座酶”指的是作为能够转座的功能性核酸蛋白质复合物的组分并且介导转座的酶。术语“转座酶”还指的是来自逆转录转座子或逆转录病毒来源的整合酶。它还指的是野生型酶和突变型酶以及带有标签,如gst标签、6×his标签等的融合酶。
如本文所用的术语“转座子”指的是由转座酶或整合酶识别并且是能够转座的功能性核酸-蛋白质复合物的必要组分的核酸区段。它指的是野生型和突变型转座子两者。
如本文所用的“转座子末端序列”指的是转座子的远端处的核苷酸序列。转座子末端序列负责鉴定用于转座的转座子;它们是形成转座体并且进行转座反应所需的dna序列。
如本文所用的术语“转座体”指的是与双链核酸(即转座子)非共价结合的转座酶。
如本文所用的“转座反应”指的是其中转座子插入靶核酸中的反应。转座反应中的主要组分是转座子、转座酶或整合酶以及其靶核酸。
如本文所用的术语“转座体-核酸复合物”或“核酸-转座体复合物”指的是转座体和其靶核酸的核酸-蛋白质复合物,在所述靶核酸中插入转座子。
如本文所用的“转座酶结合区”指的是始终处于转座子末端序列内的核苷酸序列,转座酶在介导转座时特异性结合到所述转座子末端序列。转座酶结合区可以包含用于结合转座酶亚基的多于一个位点。
如本文所用的“转座子连接链”意指双链转座子dna的链,所述链通过转座酶在插入位点处连接到靶核酸。
如本文所用的“转座子互补链”意指双链转座子dna中转座子连接链的互补链。
本公开的方法和材料是通过使用体外mua转座(haapa等,1999和savilahti等,1995)来举例说明的。可以使用其他转座系统,例如ty1(devine和boeke,1994)、tn7(craig,1996)、tn10和is10(kleckner等,1996)、mariner转座酶(lampe等,1996)、tc1(vos等,1996,10(6),755-61)、tn5(park等,1992)、p元件(kaufman和rio,1992)和tn3(ichikawa和ohtsubo,1990)、细菌插入序列(ohtsubo和sekine,1996)、逆转录病毒(varmus和brown1989)、以及酵母的逆转录转座子(boeke,1989)。
在本公开中,可转座的dna可以仅包含一个转座子末端序列(图3)。可转座的dna序列中的转座子末端序列因此不通过核苷酸序列与另一个转座子末端序列连接,即可转座的dna仅含有一个转座酶结合区。此外,可转座的dna的连接链的5'末端具有磷酸酯,其可以与任何dna链的具有-oh基团的3'末端连接。转座子互补链的3'末端可以作为部分单链dna突出(图3a)。突出的ssdna序列可以与固体载体上固定的多核苷酸的3'末端互补(图4a)以使它可以退火或杂交在一起。突出末端的长度可以是从1个到固体载体上固定的多核苷酸的长度的任何数量的碱基。转座子连接链的5'末端可以在可转座的dna上存在或不存在转座酶的情况下连接到固体载体上的多核苷酸(图4b)。
在一些情况下,转座子互补链的3'末端可以短于可转座的dna的连接链的5'末端(图3b)。需要与连接链的5'末端结合的接头寡核苷酸(l)。它具有与可转座的dna的连接链的5'末端互补的序列以及与固体载体上固定的多核苷酸的3'末端互补的序列(图5)。转座子连接链的5'末端可以在可转座的dna上存在或不存在转座酶的情况下连接到固体载体上的多核苷酸(图5b)。在一些情况下,接头寡核苷酸可以与可转座的dna结合(图3b)。在一些情况下,接头寡核苷酸可以与固体载体上固定的多核苷酸结合。在一些情况下,可以仅在发生连接反应时添加接头寡核苷酸以使可转座的dna的连接链的5'末端与固体载体上固定的多核苷酸的3'末端连接。
用于对核酸样品进行片段化和条形编码的方法描述如下(图6和图7)。双链核酸靶标(601)与可转座的dna和转座酶反应以形成转座体-核酸复合物(603)。每一种关键组分,即核酸靶标、可转座的dna以及转座酶可以在同一个反应步骤中添加到反应中而不首先将这三种组分中的任何两种预温育。双链核酸靶标的长度可以在约100bp至约1mb或更多的范围。核酸靶标的长度越长,则定相(phasing)应用的结果越好。可转座的dna可以被设计成图3中的可转座的dna。可转座的dna可以与转座酶一起在一定条件下温育以在与核酸靶标(601)反应之前首先形成转座体(602)。
先前已经描述了tn5转座体和mua转座体这两者在体外同时将dna片段化并且以高频率引入衔接子,从而产生用于下一代dna测序的测序文库(adey等,2010;caruccio等,2011;以及kavanagh等,2013)。由于dna的片段化,因此这些特定的方案去除了任何定相或邻接信息。然而,在这些方案中,在dna与转座体反应之后,需要柱纯化、热处理步骤、蛋白酶处理或与sds溶液一起温育以从转座体-dna复合物中释放转座酶以使dna变成片段。然而,与转座体结合的dna链在自然条件下是非常稳定的(surette等,1987;mizuuchi等,1992;savilahti等,1995;burton和baker2003;au等,2004;amini等,2014),并且因此图6中具有转座体的dna链(603)也是如此。
将具有转座体的dna链与带条形码的固体载体(604)一起温育,如图6中所述。固体载体上的条形码模板(605)可以首先被变性以变成单链或作为单链开始。dna链上来自转座体的转座子互补链的3'突出末端可以通过杂交被固体载体上的单链条形码模板捕获(图6d)。可以使用连接反应,例如使用t4dna连接酶来使转座子互补链与固体载体上的条形码模板的3'末端连接,如图4b中那样。然后可以使用热处理步骤,如在约65℃至约75℃下约5分钟-10分钟,或利用蛋白酶或蛋白质变性剂,例如sds溶液、盐酸胍、尿素等释放捕获的dna链上的转座酶。可以使用dna聚合酶来补平在转座反应期间留下的空位。每个核酸片段(606)在反应之后可以含有条形码序列。
许多双链核酸靶标可以以各种浓度同时与可转座的dna和转座酶反应以产生许多核酸-转座体复合物。当在反应中存在许多核酸-转座体复合物(图7b)时,可以使用有限稀释以使dna链单独地被捕获在带条形码的固体载体上,以使一个克隆条形码区捕获有限数量的dna链。固体载体(704)可以是连续表面,如载片、平板或流动池中的连续表面,其具有分离的克隆或半克隆地固定的条形码模板簇或孔或区域(显示为不同图案,705)。它也可以作为单独的珠粒或微粒而物理分开。每个珠粒或微粒具有多个带有独特序列的条形码模板。本公开中的主要优点是对具有转座体的dna链的捕获可以在开放的本体反应中发生而无需用孔、微孔、斑点、纳米通道、液滴、乳液液滴或胶囊等进行另外的分区。多于一个dna链可以被一个带克隆条形码的珠粒或微粒捕获,或者被载片、平板或流动池上的一个克隆条形码簇捕获。通过控制反应浓度,来自基因组或染色体的相同位置的核酸被同一条形码捕获的机会将是非常有限的。可以使用数百万个和数十亿个或更多个条形码模板来制备带条形码的珠粒或其他固体载体,这将使同一条形码标记来自基因组或染色体的相同位置的核酸靶标的机会进一步最小化。
将核酸-转座体复合物和带克隆条形码的珠粒或微粒封装在油包水乳液液滴中
本公开提供了将与转座体结合的核酸靶标和带克隆条形码的珠粒或微粒封装在油包水乳液液滴中并且进一步产生条形码标记的核酸片段的方法。
如本公开中先前所述(图6b)而产生的具有转座体的dna链,即连续核酸-转座体复合物,在该方法(图8a)中用作起始材料(801)。具有克隆条形码模板(803)的珠粒或微粒(802)是通过如本公开中的前文部分中所述的克隆扩增方法或直接合成方法提供的。在水溶液中提供另外的酶和底物(804),如dna聚合酶、dntp以及引物。在这样的条件下产生油包水乳液液滴(805),所述条件使得一个至几个核酸靶标与一个带条形码的珠粒或微粒在一个液滴中混合。在此可以基于泊松分布来使用核酸链和条形码珠粒或微粒的限制性滴定和/或分区。每个液滴的核酸靶标的数量越少,则用于定相应用的能力越高。在热处理,如在约65℃至约75℃下约5分钟-10分钟之后,转座酶将从转座体-核酸复合物中释放并且核酸靶标断裂成更小的片段(806)。当仍在油包水液滴中时,可以使用dna聚合酶来补平在转座反应期间留下的空位。此外,引物延伸反应或pcr扩增反应可以将核酸片段驱动到带条形码的珠粒或微粒上以产生条形码标记的核酸片段。在一个实施方式中,在图9a中在单链条形码模板(901)的3'末端与核酸片段(903)的一端之间存在重叠核苷酸序列。直接引物延伸或pcr扩增反应能够将核酸片段添加到固体载体(902)上。在另一个实施方式中,在图9b中在单链条形码模板(901)与核酸片段(906)的末端之间没有重叠核苷酸序列。使用具有与条形码模板的3'末端重叠的一端和与核酸片段的一端重叠的另一端的桥接寡核苷酸(905)经由引物延伸或pcr扩增将核酸片段驱动到带条形码的固体载体(902)上。然后可以从乳液液滴中释放具有固定的条形码标记的核酸片段的珠粒或微粒以用于进一步的下游处理。
应当指出的是,分区可以与这些实施方式或其他实施方式结合使用。如本文所用的术语“分区”可以是动词或名词。当用作动词(例如“分区(topartition)”或“分区(partitioning)”)时,所述术语一般是指将物质或样品(例如多核苷酸)在可用于将一个级分(或细分部分)与另一个级分隔离的容器之间进行分级分离(例如细分)。这样的容器使用名词“分区”来指代。可以例如使用微流体、稀释、分配、涡旋、过滤等来进行分区。分区可以是例如孔、微孔、洞、液滴(例如乳液中的液滴)、乳液的连续相、试管、斑点、胶囊、珠粒、稀溶液中珠粒的表面、或用于将样品的一个级分与另一个级分隔离的任何其他合适的容器。分区还可以包括另一个分区。
将核酸-转座体复合物与克隆条形码寡核苷酸池封装在油包水乳液液滴中
本公开提供了将与转座体结合的核酸靶标和克隆条形码寡核苷酸池封装在油包水乳液液滴中并且进一步产生条形码标记的核酸片段的方法。
如本公开中先前所述(图6b)而产生的具有转座体的dna链,即连续核酸-转座体复合物,在该方法(图10a)中用作起始材料(1001)。在水溶液中提供另外的酶和底物(1002),如dna聚合酶、dntp以及引物。在这样的条件下产生油包水乳液靶标液滴(1003),所述条件使得在大部分的液滴中存在一个至几个dna链(例如基于泊松分布通过限制滴定或分区)。每个液滴的dna链的数量越少,则用于定相应用的能力越高。克隆条形码模板(1004)可以作为油包水液滴(1005)中的克隆条形码寡核苷酸池提供。在一个实施方式中,使用t形阀或y形阀或其他装置将一个靶标液滴与一个条形码液滴合并以产生新的组合的油包水乳液液滴(1006),其含有一个dna链和一个条形码池。在热处理,如在约65℃至约75℃下约5分钟-10分钟之后,转座酶将从转座体-核酸复合物中释放并且核酸靶标断裂成更小的片段(1007)。当仍在油包水液滴中时,可以使用dna聚合酶来补平在转座反应期间留下的空位。此外,引物延伸反应或pcr扩增反应可以使条形码序列附连到核酸片段上以产生条形码标记的核酸片段。在一个实施方式中,在图11a中在单链条形码模板(1101)的3'末端与核酸片段(1102)的一端之间存在重叠核苷酸序列。直接引物延伸或pcr扩增反应能够使条形码序列与核酸片段附连。在另一个实施方式中,在图11b中在单链条形码模板(1101)与核酸片段(1105)的末端之间没有重叠核苷酸序列。使用具有与条形码模板的3'末端重叠的一端和与核酸片段的一端重叠的另一端的桥接寡核苷酸(1104)经由引物延伸或pcr扩增使条形码模板与核酸片段连接。然后可以从乳液液滴中释放条形码标记的核酸片段以用于下游应用。
在没有另外分区的情况下用固定的带克隆条形码的转座体捕获核酸以对核酸进行条形码标记
本公开提供了用固定的带克隆条形码的转座体复合物捕获核酸靶标,将捕获的核酸片段化并且使条形码序列与片段附连而无需另外分区的方法。
用于该应用的条形码模板含有条形码序列和转座酶结合区这两者。在一个实施方式中,条形码模板可以具有如图12a所示的结构。条形码序列侧接有两个柄部,即柄部1和柄部2。柄部2含有转座酶结合区。具有克隆条形码模板的固体载体(图12b)可以如本公开中先前所述使用克隆扩增方法或直接合成方法制备。固体载体上的双链条形码模板可以使用克隆扩增方法产生。在必要时,可以用引物退火或引物延伸反应,使用柄部2的部分或全部序列作为引物将固体载体上的单链条形码模板转化成部分或完全双链的条形码模板(图12c)。
用于对核酸样品进行克隆条形码标记和片段化的方法描述如下。具有包括转座酶结合区的双链条形码模板(1301)的固体载体(1302)可以同时或分开地与转座酶(1303)和核酸靶标(1305)一起温育。在一个实施方式中,转座酶(1303)可以与条形码固体载体(1302)一起温育,与条形码模板的tbr结合并且在固体载体上形成转座体(1304)。核酸靶标可以被固定的转座体捕获。在热处理步骤,如在约65℃至约75℃下约5分钟-10分钟,蛋白酶处理,或与蛋白质变性剂,例如sds溶液、盐酸胍、尿素等一起温育之后,转座酶将从固体载体释放并且片段化的核酸靶标暴露(1307)。可以用dna聚合酶进行另外的反应以补平在转座反应期间产生的空位。
在开放的本体反应中,当存在许多不同的核酸靶标时,将使用根据本公开中先前所述的程序制备的具有许多不同克隆条形码模板的一个固体载体或多个固体载体来克隆地捕获每个核酸靶标。可以使用核酸靶标的有限稀释。然而,不需要用孔、微孔、斑点、纳米通道、液滴、乳液液滴或胶囊进行另外的分区。固体载体(1402)可以是分开的,作为单独的珠粒或微粒(图14)。每个珠粒或微粒具有多个带有独特序列的条形码模板(显示为1401的不同图案)。固体载体也可以是连续的,如同载片、平板或流动池一样。使不同的条形码模板在同一载片、平板或流动池表面的不同位置处克隆扩增。多于一个核酸靶标可以被一个带条形码模板的珠粒或微粒捕获,或被载片、平板或流动池上的一个条形码模板区捕获。通过控制反应浓度,来自基因组或染色体的相同位置的核酸靶标被同一条形码捕获的机会将是非常有限的。可以使用数百万个和数十亿个条形码模板来制备带条形码的固体载体,这将使同一条形码标记来自基因组或染色体的相同位置的核酸靶标的机会进一步最小化。
转座酶可以在图13和图14所描绘的方法中被预负载到带条形码的固体载体上。然而,固体载体上的条形码密度对转座反应效率和条形码标记的核酸靶标的片段尺寸可能具有显著的影响。与图6和图7中所描绘的方法相比,对于产生相同量的产物来说,可能浪费更多的转座酶。
在没有另外分区的情况下用固定的克隆条形码捕获核酸以对核酸进行条形码标记
前一部分中的方法使用转座反应将核酸靶标捕获到带克隆条形码的固体载体上而无需另外的分区。可替选地,可以经由引物延伸反应将核酸靶标捕获到带克隆条形码的固体载体上,所述引物延伸反应发生或不发生链置换。固定的条形码模板的远端可以含有在6个碱基至20个碱基的范围的一串简并核苷酸,其可以用作随机引物并且与核酸靶标退火以进行靶标捕获。使用具有或没有链置换功能的dna聚合酶的进一步引物延伸反应将产生附连有条形码的靶核酸的部分的一个或多个拷贝。
释放克隆条形码标记的核酸片段以产生测序文库
条形码标记的片段(706、1407)固定在固体载体上。它们可以通过许多方式从固体载体释放。在一个实施方式中,可切割的接头或罕见的限制性酶切位点可以被包括在与固体载体附连的寡核苷酸序列中。使用切割反应或限制性内切酶消化,条形码标记的片段可以从固体载体释放。在一些情况下,可以进行引物延伸以制备条形码标记的片段的一个或多个拷贝(图15)。使用对任何测序平台具有特异性的引物的进一步pcr扩增可以产生用于特定的测序平台的即可测序(sequencingready)文库,所述引物例如用于illumina公司的sbs文库的p5和p7引物、或用于iontorrent公司的文库的p1和a引物。当在通过从固体载体释放条形码标记的片段来制备文库时,可以使用具有样品特异性索引(index)的引物。在一些情况下,条形码模板中的已知序列可以用作样品特异性索引。释放的具有样品特异性索引的条形码标记的片段可以与来自其他样品的具有它们自身的样品特异性索引的标记片段混合在一起以用于进一步的下游工作流程以增加样品制备通量并且简化工序。可以对构建的文库进行测序以产生条形码和核酸靶标这两者的序列。在一个实施方式中,由条形码标记的核酸片段产生用于illumina公司的sbs测序化学方法的文库(图16)。条形码序列可以在illumina公司的测序平台上被检测为长定制索引读段。核酸靶标序列可以根据需要作为单端读段或双端读段产生。在另一个实施方式中,由条形码标记的核酸片段产生用于iontorrent公司的测序化学方法的文库(图17)。在一个实施方式中,条形码序列和核酸靶标序列可以由单一长读段产生(图17a)。在另一个实施方式中,可以首先用测序引物a产生条形码序列。可以从仪器中取出芯片。可以用低浓度氢氧化钠溶液使芯片上的双链dna变性并且仅留下与ionsphere粒子附连的单链dna。可以在同一芯片上与测序聚合酶一起使用第二测序引物以产生核酸靶标的测序读段(图17b)。
将条形码测序读段组装成长读段
本公开提供了在开放的本体反应中对核酸样品进行克隆条形码标记而无需如其他方法中的复杂隔室化或分区方案的方法和试剂盒。条形码标记的片段可以来自全基因组样品。由这些条形码标记的片段产生的测序读段可以用于组装全基因组,从而作为单倍体测序方法。
由这些条形码标记的片段产生的测序读段含有条形码信息,所述条形码信息可以用于鉴定这些片段的靶标来源。具有相同条形码的这些短测序读段可以被集合在一起并且包含沿原始核酸靶标延伸的许多短的串联读段。它们提供了用于单倍型定相的有用的长距离连接信息。原始核酸靶标越长,则串联读段将越长,它们对于定相应用就越有用。可以开发分析流水线用于使用这些条形码读段进行全基因组组装或结构变异分析以用于从头测序和重新测序两者。在一种情况下,所有测序读段都可以用于标准鸟枪法组装分析以首先建立许多初始重叠群。然后可以使用条形码信息来将初始重叠群定相成长得多的重叠群。本公开中的实施方式之一是使用克隆扩增产生条形码固体载体。即使使用有限稀释方法,也可能在相同的珠粒或微粒上或在载片或流动池上的靠近位置处克隆扩增多于一个条形码模板。还可能的是,可以在多于一个固体载体或固体载体表面区域上克隆扩增一个条形码模板以产生复制的条形码固体载体。然而,在本公开中设计的条形码模板可以产生数百万个和数十亿个或更多个不同的条形码,在该过程中产生的多克隆条形码固体载体和复制的条形码固体载体的水平将不会显著干扰条形码标记的读段的整体组装。
使用条形码标记的片段进行靶向测序
本公开还提供了根据下文使用这些条形码标记的片段用于靶向测序应用的方法。
在一种情况下,所关注的区域,如hla基因或cyp2d6基因可以被扩增为长距离pcr产物。使用本公开中所述的条形码标记方法,这些长距离pcr产物可以直接用作dna靶标。由所述方法产生的串联长读段可以相应地定相回到这些长距离pcr片段。
在一些情况下,可以首先使用本公开中所述的方法对全基因组dna样品进行条形码标记。在一个实施方式中,这些条形码标记的基因组dna片段可以作为引物延伸产物从固体载体释放或通过生物化学方法从固体载体切割(图18a)。所关注的基因的第一组基因特异性引物(gsp1)可以用于一轮引物延伸或与条形码标记的片段上的通用引物一起进行几轮扩增。gsp1引物的数量可以是约3个至约40,000个。嵌套在gsp1引发产物内部的第二组基因特异性引物(gsp2)可以与条形码标记的片段上的通用引物一起用于进一步扩增所关注的基因。gsp2引物的数量可以是约3个至约40,000个,与gsp1引物的数量一样。gsp2引物的使用可以显著提高中靶率。当需要处理多个样品时,可以包括样品特异性索引作为gsp2引物或条形码标记的片段上的通用引物的尾部,以便随后可以将来自不同样品的扩增产物混合在一起用于进一步的下游程序,如测序。
在另一个实施方式中,这些条形码标记的基因组dna片段留在固体载体上(图19a)。所关注的基因的第一组基因特异性引物(gsp1)可以用于直接用固体载体上的片段进行一轮引物延伸。gsp1引物的数量可以是约3个至约40,000个。可以将靶基因的引物延伸拷贝变性并且从固体载体释放。可以用gsp2引物和条形码标记的片段上的通用引物进行另外的扩增以富集具有所关注的基因的片段。gsp2引物的数量可以是约3个至约40,000个,与gsp1引物的数量一样。当需要处理多个样品时,可以包括样品特异性索引作为gsp2引物或条形码标记的片段上的通用引物的尾部,以便随后可以将来自不同样品的扩增产物混合在一起用于进一步的下游程序,如测序。
这些条形码标记方法可以用于对靶基因、基因或外显子组进行定相。这些条形码标记方法也可以用作在靶向测序应用中区分重复读段的工具。这种方法提高了对异质样品的测序分析检测限度,例如在癌症活检样品或循环肿瘤细胞/dna中的体细胞突变检测。
本公开的一个实施方式是条形码模板,所述条形码模板包含条形码序列和侧接所述条形码序列的两个柄部序列。所述条形码序列包含一个或多个随机核苷酸序列区段和一个或多个已知核苷酸序列区段。在一些实施方式中,每个柄部序列大致具有约10个核苷酸至约100个核苷酸的长度。在其他实施方式中,柄部序列包含用于引发和/或杂交的序列。此外,柄部序列可以包含转座子末端序列。在一些情况下,条形码序列具有约6个核苷酸至约100个核苷酸的长度。条形码序列中的已知序列具有约2个核苷酸至约50个核苷酸的长度。条形码序列中的已知序列可以用作质量过滤器以去除易错的测序读段。
本公开的另一个实施方式是一种对核酸靶标进行克隆条形码标记的方法,所述方法包括:提供上面固定有克隆条形码模板的固体载体;提供可转座的dna,其中所述可转座的dna的转座子连接链的5'末端可与所述固定的条形码模板的3'末端连接;向所述可转座的dna和转座酶施加核酸靶标以在溶液中形成dna-转座体链;使所述dna-转座体链与所述具有条形码模板的固体载体杂交,其中所述可转座的dna的连接链的5'末端与条形码模板连接,而无需任何另外的隔室化;以及施加热处理、蛋白酶或蛋白质变性剂,例如sds溶液、盐酸胍、尿素等以从所述转座体中释放所述转座酶。在一些实施方式中,所述可转座的dna具有来自野生型或突变型tn5或mua转座子dna的一个转座子末端序列;其中所述转座酶是野生型或突变型tn5或mua转座酶之一。所述可转座的dna的转座子连接链的5'末端具有适用于连接的磷酸酯。所述可转座的dna的转座子互补链的3'末端具有突出末端并且所述突出末端包含固体载体上的所述条形码模板的互补核苷酸序列;并且所述可转座的dna可以与所述条形码模板杂交;并且条形码模板的3'末端可直接或在用酶修饰之后与转座子连接链的5'末端连接。所述突出末端的长度是约1个碱基、约3个碱基、约5个碱基、约10个碱基、约15个碱基、约20个碱基、约25个碱基、约30个碱基或长达固体载体上固定的寡核苷酸的长度。所述核酸分子的数量是至少约102个、103个、104个、105个、106个,其中在与所述固体载体杂交之前将所述dna-转座体链在反应溶液中稀释。可以在平板、微孔或纳米通道中进一步隔室化而进行杂交反应。
本公开的另一个实施方式是一种对核酸靶标进行克隆条形码标记的方法,所述方法包括提供上面固定有克隆条形码模板的固体载体;使得所述条形码模板的远离固体载体的远端具有转座子结合区;使得所述固体载体上的所述条形码模板与可转座的dna末端形成双链;向所述具有固定的条形码模板的固体载体施加转座酶和核酸靶标以在固体载体的表面上形成dna-转座体链而无需任何另外的隔室化;以及施加热处理、蛋白酶或蛋白质变性剂以从所述转座体中释放所述转座酶。所述转座子结合区来自野生型或突变型tn5或mua转座子dna;其中所述转座酶是野生型或突变型tn5或mua转座酶之一。核酸靶标的数量在约至少102个、103个、104个、105个或106个的范围。在与所述固定的条形码模板和转座酶反应之前,将核酸靶标在反应溶液中稀释。
本公开的另一个实施方式是一种产生条形码标记的dna片段的文库的方法,所述方法包括在固体载体上提供所述克隆条形码标记的核酸靶标;在加热、蛋白酶或蛋白质变性剂处理之后,用dna聚合酶处理所述固定的条形码标记的片段以补平在转座反应中产生的空位;用引物延伸反应释放条形码标记的dna片段。在一些实施方式中,引物具有与所述转座体中的转座子连接链序列的一部分或全部相同的核苷酸序列。当在所述引物序列和所述条形码模板序列中包括测序文库衔接头序列时,释放的条形码标记的片段是即可测序文库。用含有文库衔接头序列的引物进一步扩增释放的条形码标记的片段以产生即可测序文库。所述文库含有在所述引物延伸反应或所述扩增反应中引入的样品特异性索引;因此,可以将来自不同样品的文库汇集在一起以进行测序。将所述条形码标记的核酸片段的测序读段分组成来自相同的核酸靶标的一串串联读段;这能够用于单倍型定相。进行切割反应以从固体载体释放固定的条形码模板是另一个实施方式。
本公开的另一个实施方式是一种用条形码标记的核酸片段产生靶基因、基因或外显子组的文库的方法,所述方法包括提供所述释放的条形码标记的核酸片段;用针对靶基因、基因或外显子组的第一组引物进行引物延伸反应;以及用含有所述条形码模板序列的一部分的通用引物和针对靶基因、基因或外显子组的第二组引物进行扩增反应;其中所述第二组引物被嵌套在所述第一组引物的产物中。在扩增步骤期间添加用于测序文库的衔接头序列。
本公开的另一个实施方式是一种用条形码标记的核酸片段产生靶基因、基因或外显子组的文库的方法,所述方法包括提供所述释放的条形码标记的核酸片段;用含有所述条形码模板序列的一部分的通用引物和针对靶基因、基因或外显子组的第一组引物进行扩增;用含有所述条形码模板序列的一部分的通用引物和针对靶基因、基因、或外显子组的第二组引物进行扩增;并且所述第二组引物被嵌套在所述第一组引物的产物中。在扩增步骤期间添加用于测序文库的衔接头序列。
本公开的另一个实施方式是一种用条形码标记的核酸片段产生靶基因、基因或外显子组的文库的方法,所述方法包括在固体载体上提供所述克隆条形码标记的核酸靶标;用针对靶基因、基因或外显子组的第一组引物进行引物延伸反应;用含有所述条形码模板序列的一部分的通用引物和针对靶基因、基因或外显子组的第二组引物进行扩增;并且所述第二组引物被嵌套在所述第一组引物的产物中。在扩增步骤期间添加用于测序文库的衔接头序列。
本公开的另一个实施方式是一种用条形码标记的核酸片段产生靶基因、基因或外显子组的文库的方法,所述方法包括在固体载体上提供所述克隆条形码标记的核酸靶标;用含有所述条形码模板序列的一部分的通用引物和针对靶基因、基因或外显子组的第一组引物进行扩增;用含有所述条形码模板序列的一部分的通用引物和针对靶基因、基因、或外显子组的第二组引物进行扩增;并且所述第二组引物被嵌套在所述第一组引物的产物中。在所述扩增步骤期间添加用于测序文库的衔接头序列。在一些实施方式中,所述文库含有在引物延伸反应或所述扩增反应中引入的样品特异性索引;因此,可以将来自不同样品的文库汇集在一起以进行测序。
本公开的另一个实施方式是一种对核酸靶标进行克隆条形码标记的方法,所述方法包括提供上面固定有克隆条形码模板的珠粒或微粒;提供可转座的dna;向所述可转座的dna和转座酶施用核酸靶标以在溶液中形成dna-转座体链;将所述dna-转座体链、所述具有条形码模板的珠粒或微粒以及水性反应试剂封装到油包水乳液液滴中;施加热处理以从所述转座体中释放所述转座酶以在乳液液滴中将所述核酸靶标断裂成片段;以及将所述核酸片段驱动到所述珠粒或微粒上的所述条形码模板上。
本公开的另一个实施方式是一种对核酸靶标进行克隆条形码标记的方法,所述方法包括提供可转座的dna;向所述可转座的dna和转座酶施用核酸靶标以在溶液中形成核酸-转座体复合物;将所述核酸-转座体复合物和水性反应试剂封装到油包水乳液液滴中作为靶标液滴;提供油包水液滴中的克隆条形码模板作为条形码液滴;将所述靶标液滴与条形码液滴逐个合并;向所述合并的液滴施加热处理以从所述转座体中释放所述转座酶以在所述乳液液滴内将所述dna靶标断裂成片段;以及使所述条形码与所述dna片段在所述液滴中附连。
尽管已经关于实施方式解释了本发明,但是应当了解的是,可以作出许多其他可能的修改和变化而不脱离如本文所述的本发明的精神和范围。
此外,一般来说,关于本文所述的工艺、系统、方法等,应当了解的是,尽管所述工艺等的步骤已经被描述为根据某个有序序列发生,但是所述工艺可以通过按照与本文所述的顺序不同的顺序执行所述步骤来实施。还应当了解的是,可以同时执行某些步骤,可以增添其他步骤,或可以省去本文所述的某些步骤。换句话说,提供本文的工艺的说明是为了说明某些实施方式的目的,而决不应当被解释为限制要求保护的发明。
此外,应当了解的是,上述说明旨在是说明性的而非限制性的。在阅读上述说明之后,除所提供的实例以外的许多实施方式和应用对本领域技术人员将是显而易见的。本发明的范围不应当参考上述说明来确定,而是应当参考权利要求书连同权利要求书有权利具有的等同方案的全部范围一起来确定。预计并且预期,在本文论述的领域中将发生未来的发展,并且所公开的系统和方法将被并入这样的未来的实施方式中。总之,应当了解的是,本发明能够进行修改和变化并且仅由权利要求书限制。
最后,本申请中所用的所有定义的术语都旨在被给予它们与本文提供的定义一致的最宽泛的合理解释。除非在本文作出明确相反的指示,否则权利要求书中所用的所有未定义的术语都旨在被给予它们与它们如本领域技术人员所了解的普通含义一致的最宽泛的合理解释。特别地,除非权利要求叙述明确相反的限制,否则使用诸如“一个/种(a)”、“所述(the)”、“所述(said)”等单数形式冠词时,应当被解读成叙述所指要素中的一个或多个。
实施例1
本实施例描述了基于图1中所述的构思的特定条形码设计。图20a中的条形码201具有五个简并碱基,之后是c和t,之后是另外五个简并碱基,之后是c和t,并且最后是四个简并碱基。条形码202和条形码203序列与条形码201非常相似,除了已知序列分别是tg和ac之外。这些条形码可以侧接有illumina测序平台所用的p5和p7'衔接头序列以形成条形码模板(图20b)。图20c示出了如图20b所示的形式的条形码模板的详细序列。在一些情况下,可以在随后的阶段引入测序平台特异性衔接头(例如用于illumina测序平台的p5序列和p7序列),如图20d所示。图20d中的条形码模板设计具有非平台依赖性通用柄部1和柄部2。可以通过pcr扩增添加illumina平台特异性p5和p7衔接头序列以产生用于测序检测的测序文库。
合成p7寡核苷酸(5'-caagcagaagacggcatacgagat-3'),其具有5'末端处的胺基和所述胺与其他核苷酸之间的六碳接头(c6)(爱荷华州科拉尔维尔的integrateddnatechnologies公司(integrateddnatechnologies,coralville,ia))。根据制造商的方案将该寡核苷酸与
图21a示出了所有经测序的条形码读段的每个位置处的核苷酸含量。在所有简并碱基位置处g碱基含量是42%至47%,而非如果在寡核苷酸合成期间简并位置中a、c、g以及t的分布是真正随机的情况下所预期的25%。经由机器混合方法合成简并核苷酸,已知所述机器混合方法在四种核苷酸中产生一定的偏好表示,特别是g过度表示。然而,经测序的条形码中的g碱基含量似乎比预期的g偏好高得多。条形码位置6和13应当没有任何g;条形码位置7和14应当没有任何a。测序数据显示在位置6和13处各自约6.7%的g以及在位置7和14处各自约0.7%的a(图21a)。这些g和a可能部分归因于寡核苷酸合成错误。但是我们并不预期,在合成期间g的错误掺入率比a的错误掺入率高得多。更大程度过度调用的g(约6%)的可能来源是来自于测序相关错误。图21b示出了当在位置6、7、13以及14处显示任何错误序列的所有条形码读段被滤出时,仅有的在位置6、7、13以及14处具有正确碱基的条形码的核苷酸含量。在所有简并碱基位置处,g碱基含量降低并且是38%至44%。这表明滤出的条形码读段在所有简并位置上具有高得多的g表示,它们可能是由测序错误产生的易错读段。
实施例2
本实施例描述了特别设计的mua可转座dna和它在c末端his标记的mua转座酶存在下的转座功能。mua可转座dna设计之一(图22a)具有各自呈单段的连接链(2201)和互补链(2202)。互补链具有3'末端突出端,它可以用作接头以被固定的固体载体上的条形码模板捕获。第二种设计(图22b)的互补链具有两段(2203和2204),这将提高用于捕获的突出尾部的柔性。第三种设计(图22c)的互补链具有3段(2203、2205、2206),这将简化寡核苷酸合成。使c末端6×his标签mua转座酶在大肠杆菌bl21中表达并且纯化至均一。在37℃将1ng大肠杆菌基因组dna与0.05μm的mua可转座dna和0.3ng的mua转座酶在缓冲液中温育30分钟至60分钟,所述缓冲液含有tris-hcl、ph8.0、nacl、mgcl2、dmso以及peg-8000。在温育之后,添加sds达到最终0.05%。使用十分之一的反应混合物与phusionhotstartii高保真pcr主混合物建立30μl的pcr反应以进行18个循环的扩增。将10μl的pcr产物上样到2%e-gelex上。对于mua可转座dna的所有三种设计都明显观测到150bp至2kbp范围的来自标记及片段化的dna片段(图23)。
在一些情况下,接头寡核苷酸(2203)在转座反应期间可能不与可转座的dna退火。它可以只在可转座的dna末端需要与条形码模板附连时才用于捕获反应中。
实施例3
本实施例描述了在开放的本体反应中用带条形码的珠粒对dna进行条形码标记而无需另外分区的方法。将对于柄部1使用不同通用序列的修饰的条形码模板301、302以及303以相等的摩尔浓度汇集,并且根据beaming方案(diehl等,2005)克隆扩增到与p7寡核苷酸缀合的珠粒上。在beaming反应之后直接收集具有单链dna的珠粒作为带条形码的珠粒。将1ng的大肠杆菌基因组dna通过与0.05μm的mua可转座dna(呈图22中的设计b)和0.3ng的mua转座酶在缓冲液中在37℃温育30分钟来标记,所述缓冲液含有tris-hcl、ph8.0、nacl、mgcl2、dmso以及peg-8000。然后在37℃使用t4dna连接酶(马萨诸塞州贝弗利的enzymatics公司(enzymatics,beverly,ma))将标记反应混合物与2000万个带条形码的珠粒在快速连接缓冲液中温育15分钟以将标记的dna捕获到带条形码的珠粒上。添加edta以使捕获反应失活。将反应混合物在72℃加热5分钟以从标记的dna释放转座酶。用核酸外切酶i处理洗涤过的珠粒以去除单链多核苷酸,然后用于20个循环的pcr扩增以释放固定的条形码标记的dna片段。用0.8×ampurexp珠粒纯化pcr产物以去除小的引物二聚体并且在tapestation上使用高灵敏度d5000screentape检查(图24)。使用纯化的pcr产物作为illumina测序文库(图25),可以对其进行测序以使用测序引物1确定基因组dna插入序列并且使用测序引物2确定条形码序列。
实施例4
本实施例证实了条形码标记的dna测序读段的邻接性。在nextseq500仪器上对实施例3中所述的条形码标记的大肠杆菌dna文库进行测序,对于基因组dna插入序列进行73个循环的读段1测序并且对于条形码序列进行18个循环的索引1测序(图25)。将输出的bcl文件转换为fastq文件,其用于进一步分析。基于条形码模板301、302以及303中的已知核苷酸序列,约10%的条形码读段在所述四个位置中含有多于一个错误,它们被排除在下游数据分析之外。基于参考基因组比对位置,针对每个条形码将具有相同条形码序列的读段1读段分类。计算到下一次比对的读段距离并且沿所述读段距离的读段计数频率绘制于图26中。当带条形码的读段保持原始标记的dna片段的邻接性时,预期邻近读段堆积。来自标记之前原始dna片段的读段距离也将堆积成具有更长距离的读段。预期读段计数频率图的双峰分布,这在图26中准确地观测到。具有约3kb的峰的更短距离的读段的强烈富集证实了成功的条形码读段邻接性。
参考文献
adeya等,2010.genomebiol.11,r119。
aminis等,2014.naturegenetics,46(12):1343-1349。
aut等,2004.emboj.,23:3408-3420。
boekej.d.1989.transposableelementsinsaccharomycescerevisiaeinmobiledna(移动dna中酿酒酵母中的可转座元件),第335-374页,mobiledna(移动dna),d.e.berg和m.m.howe编著。
burtonb.m.和bakert.a.2003.chemistry&biology10:463-472。
caruccio,n.2011.methodsmol.biol.733:241-255。
craign.l.1996.transposontn7(转座子tn7).curr.top.microbiol.immunol.204:27-48。
devine,s.e.和boeke,j.d.1994.nucleicacidsresearch,22(18):3765-3772。
diehlf.等,2005.pnas,102(45):16368-16373。
haapa,s.等,1999.nucleicacidsresearch,27(13):2777-2784。
ichikawah.和ohtsuboe.1990.j.biol.chem.,265(31):18829-32。
kaufmanp.和riod.c.1992.cell,69(1):27-39。
kavanaghi,kiiskinenl.l.以及haakanah.2013.美国专利申请公开us2013/0023423。
klecknern.等,1996.curr.top.microbiol.immunol.,204:49-82。
macosko等,2015.cell,161:1202-1214。
mizuuchim.,bakert.a.以及mizuuchik.1992.cell70,303-311。
lamped.j.,churchillm.e.a.以及robertsonh.m.1996.emboj.,15(19):5470-5479。
ohtsuboe.和sekiney.1996.curr.top.microbiol.immunol.,204:126。
parkb.t.,jeongm.h.以及kimb.h.1992.taehanmisaengmulhakhoechi,27(4):381-9。
savilahtih.,p.a.rice以及k.mizuuchi.1995.emboj.14:4893-4903。
surettem.,buchs.j.以及chaconasg.1987.cell70:303-311。
varmush.和brown.p.a.1989.retroviruses(逆转录病毒),mobiledna(移动dna),bergd.e.和howem.编著,美国微生物学会(americansocietyformicrobiology),华盛顿特区(washingtond.c.)第53-108页。
vosj.c.,baerei.以及plasterkr.h.a.1996.genesdev.,10(6):755-61。
- 还没有人留言评论。精彩留言会获得点赞!