在制造蛋白质期间连续灭活病毒的方法与流程
本发明一般涉及在制造蛋白质期间连续灭活病毒的方法,并且更具体地,涉及包含以下步骤的此方法:(1)以单一预定体积比合并包含蛋白质的组合物和包含病毒灭活试剂的组合物,以获得具有用于病毒的灭活的预定性质的处理组合物;(2)转移处理组合物到处理容器中;(3)在预定条件下在所述处理容器中孵育所述处理组合物;以及(4)将所述处理组合物进行处理后加工操作,其中(i)选择所述单一预定体积比以确保在所述包含蛋白质的组合物中所述蛋白质的预测的浓度范围内保持所述处理组合物的所述预定性质,以及(ii)步骤(1)-(3)如下进行:(a)连续地并且(b)无反馈控制地进行,所述反馈控制通过所述体积比的调节或所述处理组合物的转走(divert)而实施。
发明背景
准备用于生物制药产品如治疗性抗体的包括蛋白质的组合物中可能存在的病毒的灭活是确保生物制药产品会按照预期工作且不会无意中引起疾病或其他危害的质量控制的重要方面。病毒污染可以在制造蛋白质期间通过外源或内源来源发生。鉴于它们的结构和基因组,病毒可能难以检测,并且,一旦存在,由于它们体积小而可能难以物理移除。为了消除病毒污染的可能性,制造蛋白质的工业过程通常包括灭活潜在的病毒污染物的一个或多个步骤。
来自现有技术的典型方法包括将病毒灭活试剂如酸或去污剂添加至包括蛋白质的组合物中,彻底混合,温育特定时间,然后中和或去除病毒灭活试剂,全部以不连续模式进行,即分批模式,以便完成包括蛋白质的组合物中可能存在的病毒的灭活,例如ristoldebartetal.,u.s.pat.no.6,875,848,shadleetal.,u.s.pat.no.5,429,746,和lathametal,u.s.pub.no.2013/0236358教导的。按照这类方法,可能存在的病毒的灭活需要多个不连续步骤和/或延长的温育时间(尽管,在该时间期间包括蛋白质的组合物通常不另外加工),可能增加大量时间至制造蛋白质的总过程。
其他方法包括以连续模式用一剂量的光如单色或多色的光处理包括蛋白质的组合物,例如所述组合物流过薄层辐照器,任选混合以缩小停留时间分布并增加灭活率,以便完成所述组合物中可能存在的微生物的灭活,例如anderleetal.,u.s.pat.no.7,993,580教导的。然而光剂量的控制可能是困难的,因为剂量可以随组合物变化,取决于因素如吸收中的微观异质性和辐照期间组合物的流速,并且可以随时间变化,取决于相应光源的老化和光发射的波动。
其他方法包括在从第一单元操作流至第二单元操作期间例如利用一个或多个串联的静态混合器连续混合包括蛋白质的组合物与病毒灭活试剂如酸或去污剂,以便灭活所述组合物中可能存在的病毒,通过在每个静态混合器之后和ph探针之前具有适当直径和长度的管来改变病毒灭活的停留时间,例如xenopoulos,u.s.pub.no.2015/0064769教导的。然而,这些方法的问题在于,该方法基本上包含基于ph反馈回路的分批滴定的性能,并且依赖于用于在适合于病毒的灭活的低的并且适宜的ph的调节和维持的ph计的适当功能。这使得该方法不稳定并且易受ph的快速和不受控制的波动的影响,这会对病毒的灭活产生负面影响,因此对相应蛋白质的品质产生负面影响。
其他方法包含连续地合并包含生物制品的组合物和包含病毒灭活试剂的组合物的步骤,以获得具有用于病毒的灭活的预定性质的处理组合物,确认该处理组合物具有预定性质,转移处理组合物到包含静态混合器的处理容器中,在所述处理组合物以预定速率流动并接触静态混合器的同时在预定温度下在所述处理容器中孵育处理组合物,并从所述处理容器中收集所述处理组合物,其中所述步骤连续地实施,例如coffman等人wo2015/158776所教导的。然而,这些方法包含一定程度可以消除的控制重担,所述一定程度可以消除的控制重担例如为了反馈控制的目的而实施的确认处理组合物具有预定性质的步骤,如允许不具有预定性质的处理组合物的转走。
因此,存在用于在蛋白质制造期间连续灭活病毒的改进方法的需要。
发明简述
在本公开内容的第一方面,提供了一种在制造蛋白质期间连续灭活病毒的方法。所述方法包含步骤(1)以单一预定体积比,合并(a)包含蛋白质的组合物,和(b)包含病毒灭活试剂的组合物,以获得(c)具有用于病毒的灭活的预定性质的处理组合物。所述方法还包含步骤(2)转移处理组合物到处理容器中。所述方法还包含步骤(3)在预定条件下在所述处理容器中孵育所述处理组合物。所述方法还包含步骤(4)将所述处理组合物进行处理后加工操作。根据所述方法,选择所述单一预定体积比以确保在所述包含蛋白质的组合物中的所述蛋白质的预测的浓度范围内保持所述处理组合物的所述预定性质。同样根据所述方法,步骤(1)-(3)如下进行:(a)连续地并且(b)无反馈控制地进行,所述反馈控制通过所述体积比的调节或所述处理组合物的转走而实施。
不希望受理论束缚,认为通过选择单一预定体积比以确保在所述包含蛋白质的组合物中所述蛋白质的预测的浓度范围内保持处理组合物的预定性质,并且通过连续地并且不施加通过体积比的调节或处理组合物的转走的反馈控制而实施步骤(1)-(3),制造蛋白质期间病毒的灭活能够以稳定的方式实施,不需要在实施病毒的灭活时确认处理组合物表现出预定性质,允许消除与病毒灭活期间的反馈控制相关的控制重担。
在本公开的第二方面,提供用于灭活样品中的一种或多种病毒的方法。所述方法包含在纯化靶分子的过程期间在所述样品从第一单元操作流向第二单元操作时使样品连续地与一种或多种病毒灭活试剂混合。根据所述方法,一种或多种病毒灭活试剂与样品的比例独立于反馈控制机制而确定或不需要反馈控制机制而确定。
不希望受理论束缚,还认为通过在纯化靶分子的过程期间在样品从第一单元操作流向第二单元操作时使样品与一种或多种病毒灭活试剂连续地混合,根据本文公开的原理和方法,包含一种或多种病毒灭活试剂与样品的比例独立于反馈控制机制而确定或不需要反馈控制机制而确定,一种或多种病毒的灭活在足以制造生物制药产品的程度上完成,也允许在一种或多种病毒的灭活期间消除与反馈控制相关的控制负担。
附图简述
当参考附图阅读以下详细描述时,更好地理解所要求保护的方法、装置和系统的所述和其他特征、方面和优点,其中:
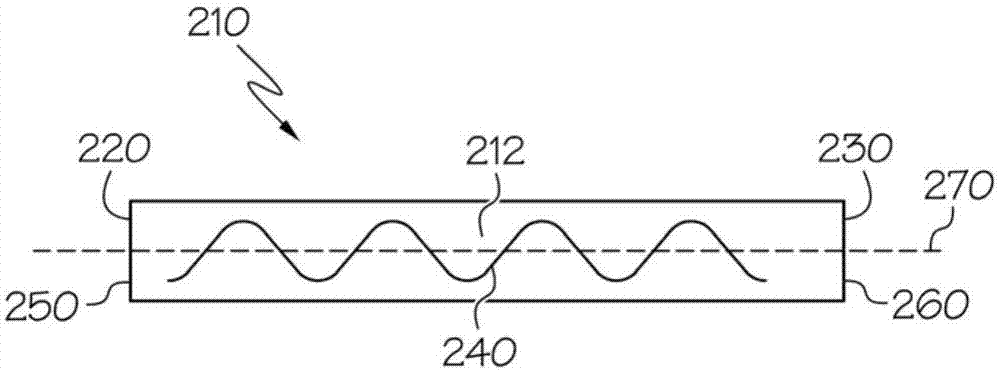
图1是用于在蛋白质制造期间连续灭活病毒的方法中的示例性处理容器210的示意图,其中示例性处理容器210具有线性形状;
图2是用于在蛋白质制造期间连续灭活病毒的方法中的示例性处理容器210的示意图,其中示例性处理容器210具有曲线形状;
图3是用于在蛋白质制造期间连续灭活病毒的方法中的示例性处理容器210的示意图,其中示例性处理容器210具有螺旋形状;
图4是用于在蛋白质制造期间实施连续灭活病毒的方法的示例性系统500的示意图;
图5是显示蛋白a洗脱的分批滴定的结果的蛋白a洗脱谱,其中绿色实线对应于单克隆抗体的浓度(g/l,每柱体积),红色实线对应于蛋白a洗脱的ph(每柱体积)和蓝色实线对应于获得具有ph3.5的处理组合物所需的相对滴定剂体积(每柱体积);
图6是与蛋白a洗脱液合并时达到ph3.5所需的滴定剂的体积(表示为滴定剂溶液体积/洗脱液体积%,表示每体积蛋白a洗脱液的滴定剂溶液的体积,以百分比表示)相对于蛋白a洗脱液中的单克隆抗体的浓度(g/l)的图,针对对应于ph3.3的2m甘氨酸、ph3.4的2m甘氨酸、ph3.3的1.5m甘氨酸、ph3.4的1.5m甘氨酸、ph3.4的1m甘氨酸、ph3.3的1m甘氨酸、ph3.4的0.5m甘氨酸、ph3.3的0.5m甘氨酸、ph3.4的3m甘氨酸和ph3.3的3m甘氨酸的滴定剂溶液(如图例中所示,从上到下排序);
图7是与蛋白a洗脱液合并时达到ph3.5所需的滴定剂的体积(再次表示为滴定剂溶液体积/洗脱液体积%)相对于蛋白a洗脱液中的单克隆抗体的浓度(g/l)的图,针对对应于ph3.4的3m甘氨酸(带短线符号的线)、ph3.3的1.5m甘氨酸(带三角形的线)、ph3.3的2m甘氨酸(带菱形的上面的线,对应于所选滴定剂)和3m甘氨酸ph值为3.3(带菱形的下面的线)的滴定剂溶液;
图8是显示单克隆抗体的偏移测试(excursiontesting)结果的图,表示为ph输出相对于单克隆抗体的浓度(g/l);
图9是滴定剂的偏移测试结果的图,表示为ph输出相对于滴定剂体积(再次表示为滴定剂溶液体积/洗脱液体积%);和
图10是蛋白a洗脱谱,其显示与蛋白a和连续的低ph病毒灭活串联的100l反应器的结果,其中蓝色实线对应于于280nm以mau测量的蛋白a洗脱液中单克隆抗体的相对浓度(每次),红色虚线对应于蛋白a洗脱液的ph(每次),和红色实线对应于最终病毒灭活处理组合物的ph(每次)。
发明详述
参考其中示出实例实施方案的附图,此后会更详细地描述要求保护的方法的方面。只要可能,相同的参考数字在整个图中用来指相同或相似部分。但是,要求保护的方法可以以许多不同形式体现,并且不应当理解为受限于本文所示的实施方案。提供这些实例实施方案以使本公开充分且完整,并且这些实施方案会向本领域技术人员充分传达要求保护的方法的范围。
提供一种在制造蛋白质期间连续灭活病毒的方法。如上文提到的,准备用于生物制药产品的包括蛋白质的组合物中可能存在的病毒的灭活是质量控制的一个重要方面。蛋白质可以是例如治疗性蛋白,如抗体、抗体片段、抗体衍生物、细胞因子、生长因子、激素、酶或凝血因子等,或者疫苗蛋白,如抗原蛋白等。蛋白质可以通过生命系统如细胞、组织或生物体制备,例如通过哺乳动物细胞、植物细胞或细菌细胞等制备。蛋白质可以通过均质方法制备,例如基于使用搅拌槽式生物反应器、气升式生物反应器或波浪式生物反应器的悬浮培养,或者通过异质方法制备,例如基于微载体系统、填充床生物反应器或中空纤维生物反应器的贴壁培养,以不连续模式进行,例如分批培养或补料分批培养,或者以连续模式进行,例如灌注连续培养,并且以任何合适的规模进行,例如实验室、试验或生产规模。病毒可以是可以感染细菌的病毒(即“细菌噬菌体”,也称作“噬菌体”),或者可以感染人和/或动物,例如准备施用蛋白质的个体人或动物,等等。病毒可以从外源性来源引入包括蛋白质的组合物,例如通过疏忽未能保持无菌,或者从内源性来源(例如用来制备蛋白质的生命系统)引入包括蛋白质的组合物。
所述方法可以用来确保灭活在制造蛋白质期间例如基于病毒污染可能已存在的病毒。就可能存在多种不同类型的病毒和/或给定类型病毒的多种活性颗粒而言,所述方法可以用来灭活多种不同类型和/或给定类型的多种活性颗粒。因此,例如,所述方法可以用来确保最终包括蛋白质的生物制药制品不会包括可接受限度以上的任何量的任何类型的病毒活性颗粒,例如生物制药制品将不含病毒活性颗粒。
所述方法包含步骤(1)以单一预定体积比合并(a)包含蛋白质的组合物,和(b)包含病毒灭活试剂的组合物,以获得(c)具有用于病毒的灭活的预定性质的处理组合物。
包括蛋白质的组合物可以是例如直接源自生物反应器的组合物,例如用于通过生命系统如哺乳动物细胞培养制备蛋白质的生物反应器。包括蛋白质的组合物可以是例如获得自以连续模式(例如灌注连续培养)操作的生物反应器的组合物,因此可以包括细胞培养基(已经被哺乳动物细胞培养物的细胞在一定程度上利用)和分泌自细胞的蛋白质。包括蛋白质的组合物还可以是例如间接源自生物反应器的组合物,例如在一个或多个加工操作之后,如过滤、沉淀和/或色谱分离等其他步骤,以便在使包括蛋白质的组合物进行连续灭活病毒的方法之前去除一些或所有不需要的碎片、化合物和其他物质。在这种情况中,所述加工操作可以对应于预处理加工操作,意味着如本文所公开的用于病毒的灭活的所述步骤将在所述加工操作之后实施。
因此,所述包含蛋白质的组合物可以从预处理加工操作中获得。所述预处理加工操作可以对应于用于获得所述包含蛋白质的组合物的加工操作,如通过与不需要的杂质、化合物和/或其他物质(如因蛋白质最初是从生物反应器中获得的而与蛋白质一起存在)分离。所述预处理加工操作可以包含例如过滤、沉淀和/或层析分离,以及其他加工操作。因此,所述包含蛋白质的组合物可以包括,例如,包含所述蛋白质的滤液(如对于预处理加工操作为过滤)、包含所述蛋白质的沉淀物(如对于预处理加工操作为沉淀)、和/或层析洗脱液(如对于预处理加工操作为层析分离),以及其他类型的组合物。
特别是关于层析分离,预处理加工操作可以包括例如免疫球蛋白结合蛋白亲和层析。免疫球蛋白结合蛋白亲和层析可用于纯化免疫球蛋白,包括例如抗体、抗体片段或抗体衍生物,以及其他免疫球蛋白。适用于免疫球蛋白结合蛋白层析的免疫球蛋白结合蛋白包括,例如,蛋白a、蛋白g、蛋白a/g和蛋白l。蛋白a特别是可以以天然和重组形式获得,并且对免疫球蛋白g的可结晶片段(也称为fc)具有高亲和力。可以实施免疫球蛋白结合蛋白层析,例如,基于所述免疫球蛋白结合蛋白固定到固体支持物(诸如,例如交联珠状琼脂糖、聚丙烯酰胺或其他多孔固体载体)。因此,例如,预处理加工操作可以包括例如蛋白a抗体亲和层析、蛋白g抗体亲和层析、蛋白a/g抗体亲和层析和/或蛋白l抗体亲和层析。
如上所述,所述方法包含步骤(1)以单一预定体积比合并(a)包含蛋白质的组合物,和(b)包含病毒灭活试剂的组合物,以获得(c)具有用于病毒的灭活的预定性质的处理组合物。
所述处理组合物的所述用于灭活病毒的预定性质可以包括以下至少一种:在3.0至3.8之间的ph或在0.05%至10%(v/v)(v/v指在去污剂的情况下每个最终体积的去污剂体积)之间的去污剂浓度。在3.0到3.8之间的ph值可以引起病毒的灭活,而不损害蛋白质。同样地,在0.05%和10%(v/v)之间的去污剂浓度可以引起病毒的灭活,而不损害蛋白质。在能够开发足以完成处理组合物中病毒的灭活到预期程度的整个工艺的意义上,可以预先确定所述预定性质,其基于制备具有用于灭活病毒的特定性质的处理组合物并测试多种条件以确定和确认其有效性,然后通常在蛋白质的制造期间应用所述工艺。这种应用可以包括将包含蛋白质的组合物和包含病毒灭活试剂的组合物合并以获得具有所述用于病毒的灭活的预定性质的处理组合物,即所述方法可以根据确保存在的病毒的灭活的特定计划实施。因此,在一些实例中,所述处理组合物的所述预定性质包含3.0至3.8之间(如在3.3至3.7之间,或3.4至3.6之间)的ph。在一些实例中,所述处理组合物的所述预定性质包含0.05%至10%(v/v)之间(如0.05%至5.0%(v/v)之间,或0.05%至2.0%(v/v)之间)的去污剂浓度。在一些实例中,所述处理组合物的所述预定性质包含在3.0至3.8之间的ph值以及在0.05%至10%(v/v)之间去污剂浓度,例如,在3.3至3.7之间的ph值并且在0.05%至5.0%(v/v)之间的去污剂浓度,或在3.3至3.7之间的ph值并且在0.05%至2.0%(v/v)之间的去污剂浓度,或在3.4至3.6之间的ph值并且在0.05%至5.0%(v/v)之间的去污剂浓度,或在3.4至3.6之间的ph值并且在0.05%至2.0%(v/v)之间的去污剂浓度。
所述病毒灭活试剂包含以下至少一种,例如,(a)具有pka在2.3至4.2之间的可滴定基团并且不具有pka在4.2至8.5之间的另一可滴定基团的酸,(b)乙酸,或(c)具有吸收峰在230nm和600nm之间的发色基团的非离子型去污剂。
具有pka为2.3-4.2的可滴定基团且没有pka为4.2-8.5的另一可滴定基团的酸表示酸,其具有至少一个pka为2.3-4.2的可滴定基团,并且可以具有pka低于2.3或高于8.5的额外的可滴定基团,但是没有pka为4.2-8.5的另一可滴定基团,每个pka在约20-25℃下确定。
具有2.3-4.2的pka且没有pka为4.2-8.5的另一可滴定基团的有机酸如羧酸或氨基酸是这样的酸。这类有机酸包括例如乳酸,其具有在25℃下pka为3.86的可滴定基团,并且没有pka为4.2-8.5的另一可滴定基团。甲酸也是这样的有机酸,其具有在20℃下pka为约3.74的可滴定基团,并且没有pka为4.2-8.5的另一可滴定基团。抗坏血酸也是这样的有机酸,其具有在约20-25℃下pka为4.17的可滴定基团以及也在约20-25℃下pka为11.6的额外的可滴定基团,并且没有pka为4.2-8.5的另一可滴定基团。甘氨酸也是这样的有机酸,其具有在约20-25℃下pka为约2.34的可滴定基团,并且没有pka为4.2-8.5的另一可滴定基团。因此,例如,病毒灭活试剂可以是具有pka为2.3-4.2的可滴定基团且没有pka为4.2-8.5的另一可滴定基团的酸,选自具有pka为2.3-4.2的可滴定基团且没有pka为4.2-8.5的另一可滴定基团的有机酸,乳酸,甲酸,抗坏血酸,具有pka为2.3-4.2的可滴定基团且没有pka为4.2-8.5的另一可滴定基团的氨基酸,甘氨酸,或者它们的组合。
至少为了以下原因,具有pka为2.3-4.2的可滴定基团且没有pka为4.2-8.5的另一可滴定基团的酸可以用于所述方法。首先,通过具有pka为2.3-4.2的可滴定基团,酸可以将处理组合物充分缓冲在3.0-3.8的ph下而不需要包括大量的酸,例如酸可以以100mm或低于100mm存在于处理组合物中,并且仍提供足够的缓冲能力。这可以确保处理组合物维持在3.0-3.8的ph下,ph足够低以使得能够灭活病毒,但是足够高以避免对蛋白质的危害,例如蛋白的酸变性。第二,通过没有pka为4.2-8.5的另一可滴定基团,后来可以将包括酸的处理组合物中和而不需要滴定另一可滴定基团,因此不需要添加在另一可滴定基团不存在下不需要添加的额外离子。这可以促进病毒灭活之后可能进行的任何离子交换步骤的有效性。
某些具有pka为2.3-4.2的可滴定基团且没有pka为4.2-8.5的另一可滴定基团的特定酸还可以用于额外的原因。例如,乳酸是额外有用的,因为其天然存在于细胞中,因此在制备蛋白质的过程中,它是fda的公认安全(也称作“gras”)的物质,并且它是廉价的。
在约20至25℃下具有4.74的pka并且不具有另一可滴定基团的乙酸也可用于所述方法中。
具有吸收峰在230nm和600nm之间的发色基团的非离子型去污剂包含,例如,具有芳族基团的聚环氧乙烷去污剂等,其中包含例如triton-x100去污剂等。因此,例如,所述病毒灭活试剂是具有吸收峰在230nm和600nm之间的发色基团的非离子型去污剂,其选自:具有芳族基团的聚环氧乙烷去污剂、triton-x100去污剂及其组合。
出于至少以下原因,所述具有吸收峰在230nm和600nm之间的发色基团的非离子型去污剂可用于所述方法中。首先,如果所述非离子型去污剂以合适的浓度存在于所述处理组合物中,则所述非离子型去污剂的所述不带电荷的亲水基团可用于灭活病毒而不损害蛋白质。第二,非离子型去污剂中具有在230nm和600nm之间的吸收峰的发色基团可用于测量(如在所述包含病毒灭活试剂的组合物中和/或在所述处理组合物中)去污剂浓度,例如基于所述发色基团的紫外吸收,其是浓度依赖性的特征。
因此,例如,所述包含病毒灭活试剂的组合物可以是包含以下至少一种的组合物:(a)具有pka在2.3至4.2之间的可滴定基团并且不具有pka在4.2至8.5之间的另一可滴定基团的酸,或(b)乙酸,或(c)具有发色基团的非离子型去污剂,所述发色基团具有在230nm和600nm之间的吸收峰,例如,所述组合物可以包含一种或多种列举的所述酸、一种或多种列举的所述非离子型去污剂,或其组合。
另外例如,所述病毒灭活试剂可以是具有pka在2.3至4.2之间的可滴定基团并且不具有pka在4.2至8.5之间的另一可滴定基团的酸,其选自:具有pka在2.3至4.2之间的可滴定基团并且不具有pka在4.2至8.5之间的另一可滴定基团的有机酸、乳酸、甲酸、抗坏血酸、具有pka在2.3至4.2之间的可滴定基团并且不具有pka在4.2至8.5之间的另一可滴定基团的氨基酸、甘氨酸,或其组合,并且所述预定性质可以包含在3.0至3.8之间的ph。根据该实例,所述预定性质可以包含在3.3至3.7之间的ph。同样根据该实例,所述预定性质可以包含在3.4至3.6之间的ph。
另外例如,所述病毒灭活试剂可以是乙酸,并且所述预定性质可以包含在3.0至3.8之间的ph。根据该实例,所述预定性质可以包含在3.3至3.7之间的ph。同样根据该实例,所述预定性质可以包含在3.4至3.6之间的ph。
另外例如,所述病毒灭活试剂可以是具有吸收峰在230nm和600nm之间的发色基团的非离子型去污剂,其选自:具有芳族基团的聚环氧乙烷去污剂、triton-x100去污剂及其组合,并且所述预定性质可以包含在0.05%至10%(v/v)之间的去污剂浓度。
此外,如上所述,所述方法包含步骤(1)以单一预定体积比合并(a)包含蛋白质的组合物,和(b)包含病毒灭活试剂的组合物,以获得(c)具有用于病毒的灭活的预定性质的处理组合物。用于灭活存在的病毒的所述病毒灭活试剂的有效性将取决于所述处理组合物中所述病毒灭活试剂的浓度以及其他因素。将病毒灭活到给定程度所需的所述病毒灭活试剂的浓度可以在实际条件下凭经验确定、基于类似条件下的先前经验估计和/或基于理论预测。如将进一步领会的,该浓度还可以用于确保所述病毒灭活试剂以合适的浓度包含在所述包含病毒灭活试剂的组合物中以确保,在考虑所述包含病毒灭活试剂的组合物相对于所述处理组合物的体积的比例贡献时,所述病毒灭活试剂将以有效用于病毒的灭活的浓度存在于所述处理组合物中。例如,考虑对应于列举的所述酸(如乳酸)之一的病毒灭活试剂的使用,如果确定所述酸应以约100mm包含在所述处理组合物中,并且所述处理组合物通过每九个体积的所述包含蛋白质的组合物合并约一体积的所述包含病毒灭活试剂的组合物来制备,则所述包含病毒灭活试剂的组合物可以制备为包含浓度约为1m的所述酸。另外例如,考虑对应于列举的所述非离子型去污剂(如triton-x100去污剂)之一的病毒灭活试剂的使用,如果确定所述非离子型去污剂应以约1.0%(v/v)包含在所述处理组合物中(再次v/v指在去污剂的情况下每最终体积的去污剂体积),且所述处理组合物通过每九个体积的所述包含蛋白质的组合物合并约一体积的所述包含病毒灭活试剂的组合物来制备,则所述包含病毒灭活试剂的组合物可以制备为包含浓度约为10%(v/v)的所述非离子型去污剂。
如上所述,根据步骤(1),将所述包含蛋白质的组合物和所述包含病毒灭活试剂的组合物合并,以获得具有用于病毒的灭活的预定性质的处理组合物。例如,所述合并可以在包含一个或多个混合器的容器内实施,使得所述包含蛋白质的组合物和所述包含病毒灭活试剂的组合物被加入到所述容器中,例如分开地和同时地流过容器(如在压力下),并在流动时混合,例如通过一个或多个混合器。例如,所述混合可以发生一段时间,如1至5分钟,其足够长以确保所述处理组合物混合至均匀,但不会长到进行到病毒的灭活的相当程度。也使用其他方法,如在不使用一个或多个混合器的情况下实施组合,和/或组合实施更长的时间段或更短的时间段,及其他方法。
还如所指出的,根据步骤(1),以单一预定体积比组合所述包含蛋白质的组合物和包含所述病毒灭活试剂的所述组合物。这意味着所述包含蛋白质的组合物和所述包含病毒灭活试剂的组合物以预先确定为适于实施所述方法的所述包含蛋白质的组合物的体积和所述包含病毒灭活试剂的组合物的体积的比例合并,如下面更详细地讨论。
所述方法还包含步骤(2)转移所述处理组合物到处理容器中。所述处理容器可以是适于病毒连续灭活的容器。所述转移所述处理组合物到处理容器中可以例如基于所述处理组合物在压力下流向所述处理容器而发生。
参考图1更详细地考虑所述处理容器,提供示例性处理容器210。示例性处理容器210包含入口220、出口230和静态混合器240,并且具有内部体积212,入口220和出口230定位在示例性处理容器210的主轴270的相对的两端,即入口端250和出口端260,且静态混合器240沿着主轴270位于示例性处理容器210的内部。根据该示例,步骤(2)可以包含转移所述处理组合物到示例性处理容器210中,在入口220处发生转移。如图1所示,处理容器210可以为例如柱或管或其他的形式。分别如图1、图2和图3所示,示例性处理容器210可以具有例如线性、弯曲或螺旋或其他形状,并因此可以具有长轴270,其也是例如线性的、弯曲的或者螺旋的或其他类型。示例性处理容器210可以由例如金属、塑料或其组合或其他材料制成。处理容器可以包含一个静态混合器240、或多个静态混合器240,例如两个、三个、四个或多个,以适于确保有效混合。静态混合器可以是包括例如挡板、孔口、冲击板和/或其他直列式突起的静态混合器类型。静态混合器可以由一种或多种与生物制药加工兼容的材料制成,如与生物制药加工兼容的金属、塑料、橡胶和/或玻璃中的一种或多种。
也能使用其他处理容器。
更详细的考虑转移所述处理组合物到所述处理容器中,所述转移可以从包含一个或多个如上所述的混合器的容器到处理容器,其中所述包含一个或多个混合器的容器和所述处理容器是流体连通的,即连接成使得流体可以在内部流动并且至少单向地从包含一个或多个混合器的容器流向处理容器。可以控制转移速率,如通过泵。
所述方法还包含步骤(3)在预定条件下在所述处理容器中孵育所述处理组合物。在步骤(3)之前在所述处理组合物中病毒的存在程度,如作为多种不同类型的病毒和/或给定类型病毒的多种活性颗粒,基于在步骤(3)期间在所述处理容器中的孵育,所述病毒将至少在一定程度上灭活。
所述预定条件可以是可用于灭活病毒的条件的组合,如所述处理组合物在所述处理容器中孵育的预定温度和预定时间。对于包含所述处理组合物流向所述处理容器的所述处理组合物的孵育,孵育的预定时间能基于所述处理容器中所述处理组合物的流速来体现。
因此,例如,所述处理容器可以保持在预定温度(例如,通过使用加热元件),并且所述处理组合物的流动可以保持在预定速率(例如,通过使用泵)。另外例如,所述预定温度和所述预定速率可以预先确定,使得可以开发足以完成病毒的灭活到预期程度的整个工艺,其基于在特定温度下在所述处理容器中孵育所述处理组合物特定时间并测试多种条件以确定和确认有效性,然后通常在蛋白质制造期间中应用所述工艺,包括控制所述处理容器的温度和控制所述处理组合物通过所述处理容器的流速以确保将所述处理组合物进行足以完成病毒的灭活至预期程度的处理。同样,所述方法可以根据特定计划实施以确保可能存在的病毒的灭活。
另外例如,所述预定条件可以包含在17至40℃之间的预定温度和0.3至3乘以所述处理容器内部体积/小时的所述处理组合物通过所述处理容器的预定流速。通常,如果所述预定温度增加,所述预定速率也可以增加,反之亦然。相反,如果所述预定温度降低,所述预定速率可能需要降低,反之亦然。这是因为所述病毒灭活试剂一般在更高的温度下可以更快地灭活所述处理组合物中的病毒,并因此所述处理组合物可以在所述处理容器中孵育更短的时间,同时仍然完成病毒的灭活到预期程度。例如,所述预定温度可以在18至25℃之间,并且预定速率可以是0.5至1.5乘以处理容器内部容积/小时。另外例如,所述预定温度可以在30至39℃之间,并且所述预定速率可以是0.8至2.0乘以处理容器内部容积/小时。
所述预定条件能够足以在步骤(3)期间通过所述病毒灭活试剂以至少1×101倍的系数使所述处理组合物中的病毒灭活。例如,所述预定条件能够足以在步骤(3)期间通过所述病毒灭活试剂以至少1×102、至少1×103、至少1×104、至少1×105或至少1×106的系数使所述处理组合物中的病毒灭活。在根据该实例,所述预定条件能够在实际病毒污染的条件下,以及在可能存在病毒污染的条件下(无论是否实际存在病毒污染),通过所述病毒灭活试剂使所述处理组合物中的病毒灭活至少至所指示的程度。此外,所述引起病毒的灭活的所述预定条件的所述有效性可以涉及对于特定类型的病毒、多种特定类型的病毒和/或一般或不同范围的病毒的灭活。另外,所述预定条件引起所述病毒的灭活的有效性可以涉及对于在步骤(3)期间进行孵育的所述处理组合物的仅一部分(如在所述处理容器中在所述方法实施的这段时间的一部分期间孵育的所述处理组合物的一部分),或对于在步骤(3)期间进行孵育的所述处理组合物的全部(如在所述方法实施的从开始到结束的整个期间内孵育的所述处理组合物的全部)的灭活。
另外例如,所述处理容器的内部体积可以足够大以确保所述处理组合物中不超过百万分之一在所述处理容器中的停留时间比在步骤(3)期间通过所述病毒灭活试剂以至少1×101系数引起所述处理组合物中的病毒的灭活所需的持续时间短。在这方面,基于具有足够大的内部体积来制造或选择所述处理容器以应对所述处理组合物流过所述处理容器时所述处理组合物的轴向分散(即所述处理组合物沿所述处理容器的主轴的分散),以控制和最小化以比在步骤(3)期间通过所述病毒灭活试剂以至少1×101的系数引起所述处理组合物中所述病毒的灭活所需的更短时间内流过处理容器的所述处理组合物的比例。例如,可以基于内部容积制造或选择所述处理容器,所述内部容积为了应对轴向分散而包含超出理论塞流量的额外体积。所述容器的理论塞流量vh*可以计算为临界保持时间tr和所述容器内组合物的所述体积流速q的乘积。可以估算应对轴向分散必需的所述额外体积,例如,通过使用用于层流或塞流的taylor分散模型、用于湍流的gaussian模型或特定为给定静态混合器开发的模型,或其他方法。因此,例如,所述处理容器的所述内部体积可以足够大以确保不超过百万分之一(如不超过千万分之一、百万分之一或十亿分之一)的所述处理组合物在所述处理容器中的停留时间比在步骤(3)期间通过所述病毒灭活试剂以至少1×101的系数(如至少1×102、至少1×103、至少1×104、至少1×105、或至少1×106的系数)引起所述处理组合物中的病毒的灭活所需的持续时间短。另外例如,所述处理容器的所述内部体积可以足够大以确保所述处理组合物中不超过百万分之一(如不超过千万分之一、百万分之一或十亿分之一)的活性颗粒病毒在所述处理容器中的停留时间比在步骤(3)期间通过所述病毒灭活试剂以至少1×101的系数(如至少1×102、至少1×103、至少1×104、至少1×105、或至少1×106的系数)引起所述处理组合物中的病毒的灭活所需的持续时间短。
如上所述,再次考虑如上所述的示例性处理容器210,当所述处理组合物沿着处理容器210的主轴270流动时,所述处理组合物与静态混合器240接触,如包括接触包含多个静态混合器240的处理容器210中的多个静态混合器240。当所述处理组合物流过处理容器210时,这种接触提供所述处理组合物的连续混合,因此最小化所述处理组合物的轴向分散。因此,在一些实例中,步骤(3)包含在预定温度下在示例性处理容器210中孵育所述处理组合物,同时所述处理组合物以预定速率沿主轴270流动并接触静态混合器240,所述预定温度和所述预定速率的组合足够基于所述预定性质引起所述处理组合物中所述病毒的灭活。
也能使用其他方法。
所述方法还包含步骤(4)使所述处理组合物进行处理后加工操作。类似于所述预处理加工操作,所述处理后加工操作对应于用于通过与不需要的杂质、化合物和/或其他物质(如可以和所述处理组合物中的所述蛋白质一起存在的)分离而获得现已处理的包含所述蛋白质的组合物的加工操作。所述处理后加工操作可以包含,例如,过滤、沉淀和/或层析分离,或其他加工操作。因此,例如,所述处理后加工操作包含以下中的一种或多种:阴离子交换层析、阳离子交换层析或通过病毒过滤介质。
关于阴离子交换层析,如同免疫球蛋白结合蛋白层析一样,阴离子交换层析也可用于纯化免疫球蛋白,包含例如抗体、抗体片段或抗体衍生物,或其他免疫球蛋白。适用于阴离子交换层析的阴离子交换基团包含,例如,二乙基氨基乙基(diethylaminoethyl,也称为deae)基团、聚乙烯亚胺(polyethyleneimine,也称为pei)基团和季铵(quarternaryammonium,也称为q)基团。阴离子交换基团可以用于,例如,基于合适的分辨率和速度纯化蛋白质的最终步骤(也称为精制步骤)。阴离子交换层析也可以,例如,基于固体载体上阴离子交换基团的存在而实施。因此,例如,所述处理后加工操作包含,例如,二乙基氨基乙基阴离子交换层析、聚乙烯亚胺阴离子交换层析或季铵阴离子交换层析。
为了根据步骤(4)使所述处理组合物进行处理后加工操作的目的,可以从所述处理容器中获得所述处理组合物,例如,通过从所述处理容器中收集所述处理组合物。所述收集可以对应于,例如,允许所述处理组合物从处理容器流向其他容器,如用于中和或移除病毒灭活试剂、用于进一步病毒的灭活、用于在将所述处理组合物进行处理后加工操作之前储存、和/或直接将其进行处理后加工操作。
如上所述,再次考虑如上所述的示例性处理容器210,可以在出口230处从示例性处理容器210收集所述处理组合物,并然后根据步骤(4)将其进行处理后加工操作。
根据所述方法,选择所述单一预定体积比以确保在所述包含蛋白质的组合物中的所述蛋白质的预测的浓度范围内保持所述处理组合物的所述预定性质。如上所述,所述包含蛋白质的组合物和所述包含病毒灭活试剂的组合物的体积以在实施所述方法之前确定的包含蛋白质的组合物的体积和包含病毒灭活试剂的组合物的体积的比例组合。具体地,选择所述单一预定体积比以确保在所述包含蛋白质的组合物中的所述蛋白质的预测的浓度范围内保持所述处理组合物的所述预定性质,不需要响应于所述蛋白质的浓度中的变化而改变体积比,并因此不需要使用除单一预定体积比之外的体积比。
令人惊讶的是,已经确定在包含蛋白a洗脱物的单克隆抗体的处理组合物中病毒的灭活所需的病毒灭活试剂的浓度主要由单克隆抗体的浓度驱动。因此,通过测定包含蛋白质的组合物中蛋白质的预测浓度范围(如从免疫球蛋白结合蛋白亲和层析的洗脱液的连续级分中预测的蛋白质浓度范围),其将与包含病毒灭活试剂的组合物合并以获得所述处理组合物,以及通过确保蛋白质的浓度基本上不超过所述范围的上限,可以实施病毒连续灭活的步骤而不需要通过调节体积比或转走处理组合物来实施的反馈控制。
换句话说,所述方法可以通过化学控制实施,意味着,通过使用包含病毒灭活试剂的组合物,以对于进入的蛋白质流,保持处理组合物具有所述病毒灭活试剂的预定浓度和/或在窄窗内的ph。所述方法可以物理地依赖于两种或多种溶液(如包含蛋白质的组合物和包含病毒灭活试剂的组合物)的泵计量,以达到所述病毒灭活试剂的预定浓度和/或ph,而不是依赖于来自探头的活性或动态反馈或基于输出的比例积分衍生物(proportional-integral-derivative,也称为pid)控制回路,即所述处理组合物的监测。所述包含蛋白质的组合物的体积与所述包含病毒灭活试剂的组合物的体积的比例是通过所述过程中关键因素的先验知识而预定的。因此,所述过程的物理控制(如对于泵计量)能预先设定,并且在整个所述方法中能够使用单一体积比,而不管实施所述方法时所述包含蛋白质的组合物中所述蛋白质的浓度的变化。
考虑所述方法的一个实例包括使用包含酸(如有pka在2.3至4.2之间的可滴定基团并且不具有pka在4.2至8.5之间的另一可滴定基团的酸,和/或乙酸)的病毒灭活试剂,以及包括包含蛋白a抗体亲和层析的预处理加工操作,所述方法通过在处理组合物从一个单元串联流向另一个(如从预处理加工操作到后处理加工操作)时在窄且统一的3.3-3.7的ph范围的处理组合物(包含蛋白a洗脱液,与包含病毒灭活试剂的组合物合并),解决了上述问题,从而在不依赖于ph计的情况下准确且有效地实现病毒灭活。所述方法可以包含先验地确定哪种病毒灭活试剂将为给定蛋白质提供最有效的病毒的灭活。然后可以将所述病毒灭活试剂加入到或用其滴定来自蛋白a抗体亲和层析的洗脱液,以获得所述处理组合物。所述处理组合物然后从一个单元操作流向下一个时,保持3.3至3.7的ph范围。所述方法不依赖于ph计或ph反馈回路来控制泵的功能来启动、停止或以其他方式调节包含病毒灭活试剂的组合物的添加速率以与洗脱液合并。相反,基于使用单一预定体积比,所述处理组合物的ph保持在最佳范围内。由于ph计不能实时报告ph,消除对于ph计或ph反馈回路的依赖提高病毒灭活步骤的效率和准确性。
了解通过添加包含病毒灭活试剂的组合物滴定的蛋白质的主要种类有益于先验地预测在低ph下病毒灭活的输出。其它有用的变量包含蛋白质的有效总电荷、滴定时蛋白质的背景缓冲液、强度滴定剂(弱酸)和蛋白质a洗脱液与病毒灭活试剂混合的比例。
由于涉及蛋白a洗脱流中控制动态和变化的系统的复杂性,现有技术滴定以分批或池来自动化。在没有系统的先验知识时,直接在线滴定需要对一个或多个pid回路、ph探头和泵计量的全面验证。此控制是不期望的因为ph探头易受响应延迟、漂移、偏移的影响,特别是在快速反馈环境中。
类似考虑适用于关于使用包括非离子型去污剂的病毒灭活试剂的方法,所述非离子型去污剂具有吸收峰在230nm和600nm之间的发色基团。
因此,在一些实例中,所述包含病毒灭活试剂的组合物具有通过公式1和支持公式2和3确定的ph:
公式1:
公式2:
公式3:
可以认为这些实例包含使用化学控制公式。
同样在一些实施例中,所述包含病毒灭活试剂的组合物至所述包含蛋白质的组合物的恒定%v/v添加被限定为目标ph+/-0.1ph的上限和下限的ph范围内。本文恒定%v/v意指所述包含病毒灭活试剂的组合物和所述包含蛋白质的组合物以在合并过程中不变的体积比合并,所述体积比以每体积所述包含蛋白质的组合物中所述包含病毒灭活试剂的组合物的体积提供,以百分比表示。根据这些实施例,所限定灭活ph的上限等于目的ph+0.1ph。所限定灭活ph的下限等于所述包含病毒灭活试剂的组合物(如滴定剂)的ph。
同样在一些实例中,包含病毒灭活试剂的组合物的%v/v添加是通过公式4和支持公式5至9确定的:
公式4:
a=b/(c-b)
a是病毒灭活试剂的%v/v添加
b是最终灭活试剂体积摩尔浓度
c是储液灭活试剂体积摩尔浓度
确定最终灭活试剂体积摩尔浓度=(最终灭活共轭碱体积摩尔浓度+最终灭活共轭酸体积摩尔浓度)的总和。
公式5:
b=∑(d+e)
d=最终灭活共轭碱体积摩尔浓度
e=最终灭活共轭酸体积摩尔浓度
确定最终灭活共轭碱体积摩尔浓度=总系统共轭碱体积摩尔浓度-总系统非试剂碱体积摩尔浓度。
公式6:
d=f-g
f=总系统共轭碱体积摩尔浓度
g=总系统非试剂碱体积摩尔浓度
确定最终灭活共轭酸体积摩尔浓度=总系统共轭酸体积摩尔浓度-总系统非试剂酸体积摩尔浓度。
公式7:
e=h-g
h=总系统共轭酸体积摩尔浓度
确定非灭活试剂碱体积摩尔浓度等于背景缓冲液共轭碱种类和蛋白质的总电荷体积摩尔浓度之和。
公式8:
g=i+j.
i=背景缓冲液体积摩尔浓度/(1+(1/10^(phproa流-pka背景缓冲液)))
j=蛋白质的总电荷体积摩尔浓度
在给定的ph和体积摩尔浓度下测定总蛋白质电荷是蛋白质的体积摩尔浓度乘以灭活ph下总未被屏蔽电荷。
公式9
j=蛋白质浓度[g/l]/分子量[mw]/(1-蛋白质浓度[g/l]/1000x0.75)*有效mab电荷δ@灭活ph
k=蛋白质质量摩尔浓度
蛋白质浓度等于下游单元操作中遇到的浓度。对于蛋白a流,所述浓度可以是几个因素的结果,包含蛋白a上样挑战、洗脱缓冲液的强度(体积摩尔浓度和ph)、洗脱流速、产物与蛋白a基质的相互作用、以及分散和稀释的洗脱后水平。
l=总未屏蔽蛋白质电荷
l是从内部库预先确定的
有效蛋白质电荷通过“effectofproteinandsolutionpropertiesonthedonnaneffectduringtheultrafiltrationofproteins”glenr.bolton,biotechnol.prog.,2011,vol.27,no.1,第140-152页(measurementofproteinmolecularcharge,第145-146页)中描述的方法凭经验确定。
其中期望的病毒灭活ph=pka病毒灭活试剂+log(总系统共轭碱体积摩尔浓度/总系统共轭酸体积摩尔浓度)
m=n+log(f/g)
m=期望的病毒灭活ph操作温度
n=病毒灭活试剂的pka操作温度
f=总系统共轭碱体积摩尔浓度
g=总系统共轭酸体积摩尔浓度
此外,在一些实例中,包含病毒灭活试剂的组合物的%v/v添加可以交互式求解,例如通过作为所选病毒灭活试剂的%添加、初始蛋白质浓度和主要的背景缓冲液体积摩尔浓度的函数将值收敛于期望的病毒灭活ph。
如上所述,选择所述单一预定体积比以确保在所述包含蛋白质的组合物中的所述蛋白质的预测浓度范围内保持所述处理组合物的所述预定性质,不需要响应于所述蛋白质的浓度中的变化而改变体积比,并因此不需要使用除所述单一预定体积比之外的体积比。以单一预定体积比合并所述包含蛋白质的组合物和所述包含病毒灭活试剂的组合物可以包括以与所述单一预定体积比略有变化的体积比合并所述组合物,如基于所述单一预定体积比变化小于或等于15%、小于或等于10%、小于或等于5%和/或小于或等于1%,取决于例如在合并这些组合物以获得所述处理组合物期间,与所述包含蛋白质的组合物和/或所述包含病毒灭活试剂的组合物的泵送相关的可变性。
同样根据该方法,步骤(1)-(3)如下进行:(a)连续地并且(b)不施加反馈控制地进行,所述反馈控制通过所述体积比的调节或所述处理组合物的转走而实施。
关于连续实施的步骤(1)至(3),这意味着连续实施步骤(1)、步骤(2)和步骤(3),即每个步骤在所述包含蛋白质的组合物、所述包含病毒灭活试剂的组合物和所述处理组合物的不同部分,在至少一段时间内同时实施。例如,步骤(1)、步骤(2)和步骤(3)可以连续实施至少1小时、至少4小时、至少12小时、至少24小时、至少3天、至少10天、或至少30天,或其他时长。还例如,步骤(4)可以同步骤(1)、步骤(2)和步骤(3)连续实施。还例如,步骤(1)、步骤(2)和步骤(3)可以连续实施,不仅在蛋白质的整体制造期间,而且与制造蛋白质期间通过活系统的蛋白质实际生产同时并且连续地进行。
关于步骤(1)至(3)不施加反馈控制地进行,所述反馈控制通过所述体积比的调节或所述处理组合物的转走而实施,这意味着步骤(1)至(3)在没有校正所述处理组合物与病毒灭活的所述预定性质的潜在偏差的情况下进行。因此,步骤(1)至(3)不施加反馈控制地进行,所述反馈控制通过调节所述包含蛋白质的组合物和包含病毒灭活试剂的组合物合并的体积比的而实施。如上所述,所述体积比与所述单一预定体积比相比略有变化,如基于单一预定体积比变化小于或等于15%、小于或等于10%、小于或等于5%或小于或等于1%,取决于例如泵送相关的可变性,但不基于与反馈控制相关的调整。此外,步骤(1)-(3)不施加反馈控制地进行,所述反馈控制通过所述处理组合物从产物流的转走而实施,如从处理容器和/或处理后加工操作转走。可以进行步骤(1)至(3),出于其他原因从产物流的转走所述处理组合物,如由于蛋白质的间歇取样,但不是基于与反馈控制相关的转走。
在一些实施方案中,所述方法还包含确认所述处理组合物表现出所述预定性质,如出于验证的目的,尽管同样不是为了反馈控制。所述确认可以通过使用检测器实施,如ph计、电导率计、温度计、分光光度计装置或光谱装置,其用于测量所述处理组合物的特性(如ph、电导率、温度、分光光度特性或光谱特性)。
更详细地考虑所述蛋白质,所述蛋白质可以是例如上文提到的任何蛋白质,例如治疗性蛋白,如抗体、抗体片段、抗体衍生物、细胞因子、生长因子、激素、酶或凝血因子等,或者疫苗蛋白,如抗原性蛋白等。
例如,所述蛋白质可以是抗体、抗体片段或抗体衍生物。还例如,抗体、抗体片段或抗体衍生物可以选自抗体、单克隆抗体、多克隆抗体、哺乳动物抗体、小鼠抗体、灵长类抗体、人抗体、嵌合抗体、灵长类化抗体、人源化抗体、免疫球蛋白轻链、免疫球蛋白重链、免疫球蛋白轻链和免疫球蛋白重链、抗体片段、抗体衍生物、fab片段、f(ab')2片段、fc片段、fc-fc融合蛋白、fv片段、单链fv片段、单结构域fv片段、四价单链fv片段、二硫键连接的fv片段、双链抗体、三链抗体、四链抗体(tetrabody)、五链抗体(pentabody)、微抗体、微型抗体、免疫球蛋白单可变结构域、免疫球蛋白单可变重链结构域、免疫球蛋白单可变轻链结构域、vhh结构域、人源化的vhh结构域、单结构域抗体、包括以模块形式与另一免疫球蛋白单可变结构域或功能结构域连接在一起的免疫球蛋白单可变结构域的蛋白、包括以模块形式连接在一起的两个或更多个相同的免疫球蛋白单可变结构域的多价蛋白、包括以模块形式连接在一起的两个不同的免疫球蛋白单可变结构域的双异位蛋白、包括以模块形式连接在一起的两个不同的免疫球蛋白单可变结构域的双特异性蛋白、包括以模块形式连接在一起的免疫球蛋白单可变结构域和功能结构域的双功能蛋白、结构域缺失的抗体、抗体片段与另一肽或多肽的融合多肽、fc-肽融合物、fc-毒素融合物、以及抗体片段与支架蛋白的融合物。
如本领域公知的,抗体是特异性地结合至特定底物,即它们的抗原的蛋白,抗体一般享有相似的整体结构,即免疫球蛋白结构,并且每个特定抗体分子具有独特的结构,允许特定抗体特异性地结合至其相应抗原。示例性抗体例如由murphyetal.,janeway’simmunobiology,7thedition,garlandscience,newyork(2008)描述。还如公知的,抗体可以对应于单克隆抗体或多克隆抗体,取决于抗体是怎样产生的,可以对应于哺乳动物抗体、小鼠抗体、灵长类抗体或人抗体,取决于衍生或可以衍生抗体的生物体,可以对应于嵌合抗体、灵长类化抗体或人源化抗体,取决于抗体是否已修饰以使抗体更适合用于特定生物体,并且可以对应于免疫球蛋白轻链、免疫球蛋白重链或者免疫球蛋白轻链和免疫球蛋白重链等,取决于抗体的结构,例如tamashiroetal.,monoclonalantibodies,pp.409-433,inanimalcelltechnology:frombiopharmaceuticalstogenetherapy(eds.castilhoetal.),taylor&francisgroup,newyork(2008)等所述。
还如公知的,抗体片段和抗体衍生物可以制备自抗体。例如,fab片段(也称作抗原结合片段)由通过相邻恒定区维持在一起的免疫球蛋白重链和免疫球蛋白轻链各自的可变区组成。fab片段可以通过蛋白酶消化,例如用木瓜蛋白酶形成自常规抗体,或者可以通过遗传工程形成。相似地,f(ab')2片段包括也通过相邻恒定区维持在一起的两条重链和两条轻链各自的可变区。f(ab')2片段可以通过胃蛋白酶进行蛋白酶切割来制备。
此外,利用遗传工程方法,可以产生仅由重链(vh)和轻链(vl)的可变区组成的缩短的抗体片段,称作fv片段(也称作可变片段)。因为fv片段缺少免疫球蛋白重链和免疫球蛋白轻链的恒定区而因此缺少其半胱氨酸之间的共价键合,所以fv片段常常会被稳定化。例如,使用短肽连接重链和轻链的可变区以稳定fv片段是有利的。短肽可以包括例如10-30个氨基酸,优选15个氨基酸。这样,获得由通过肽接头连接的vh和vl组成的单肽。这种抗体蛋白已知为单链fv(也称作scfv)。示例性的这种scfv-抗体蛋白由hustonetal.,proceedingsofthenationalacademyofsciencesusa85:5879-5883(1988)描述。
此外,最近几年已开发各种策略将scfv制备为多聚体衍生物。特别地,这是为了导致具有改进的药物动力学和生物分布特性以及增加的结合活性的重组抗体。为了实现scfv的多聚化,将scfv制备为与多聚化结构域的融合蛋白。多聚化结构域可以是例如免疫球蛋白g(也称作igg)的ch3区或者卷曲结构(螺旋结构)如亮氨酸拉链结构域。还有这样的策略,其中将scfv的vh/vl区之间的相互作用用于多聚化,例如双链抗体、三链抗体和五链抗体(pentabody)等。
因此,例如,双链抗体是二价同源二聚scfv衍生物。将scfv分子中的接头缩短至5-10个氨基酸导致形成同源二聚体,其中发生链间vh/vl-叠加。可以通过并入二硫键额外地稳定双抗体。示例性双链抗体例如由perisicetal.,structure2:1217-1226(1994)描述。
还例如,三链抗体是三价同源三聚scfv衍生物。其中vh-vl直接融合没有接头序列的scfv衍生物导致三聚体的形成。示例性三链抗体例如由korttetal.,proteinengineering10:423-433(1997)描述。
还例如,微抗体是二价同源二聚scfv衍生物。微抗体由融合蛋白组成,其包含免疫球蛋白,优选igg,最优选igg1的ch3区,作为通过铰链区(例如也来自igg1)和接头区连接至scfv的多聚化区。示例性微抗体抗体蛋白由huetal.,cancerresearch56:3055-3061(1996)描述。
还例如,微型抗体是具有二价、三价或四价结构的scfv衍生物。微型抗体多聚化通过二聚、三聚或四聚卷曲结构进行,例如lovejoyetal.,science259:1288-1293(1993),packetal.,biotechnology11:1271-1277(1993)和packetal.,journalofmolecularbiology246:28-34(1995)公开的。
抗体片段和抗体衍生物还包括例如免疫球蛋白单可变结构域。免疫球蛋白单可变结构域可以是例如免疫球蛋白单可变重链结构域(也称作vh结构域)或免疫球蛋白单可变轻链结构域(也称作vl结构域),如wardetal.,nature341:544-546(1989)描述的。免疫球蛋白单可变结构域还可以是例如vhh结构域,衍生自骆驼重链抗体,如hamers-castermanetal.,nature363:446-448(1993)描述的,优选人源化vhh结构域。免疫球蛋白单可变结构域还可以是例如单结构域抗体。免疫球蛋白单可变结构域还可以是例如包括一个免疫球蛋白单可变结构域的nanobody(r)(ablynxn.v.拥有的商标)治疗性蛋白。
抗体片段和抗体衍生物还包括例如以模块形式与另一免疫球蛋白单可变结构域或功能结构域连接在一起的免疫球蛋白单可变结构域。这类蛋白的实例包括:多价蛋白,其包括以模块形式连接在一起的两个或更多个相同的免疫球蛋白单可变结构域,例如两个或更多个相同的vhh结构域;双异位蛋白,其包括以模块形式连接在一起的两个不同的免疫球蛋白单结构域,例如两个不同的vhh结构域,各自识别相同抗原上的不同表位;以及双特异性蛋白,其包括以模块形式连接在一起的两个不同的免疫球蛋白单可变结构域,例如两个不同的vhh结构域,各自识别不同的抗原。这类蛋白的实例还包括双功能蛋白,其包括以模块形式连接在一起的免疫球蛋白单可变结构域和功能结构域。实例还包括nanobody(r)多价、双异位、双特异性和双功能治疗性蛋白,
抗体片段和抗体衍生物还包括例如抗体片段和支架蛋白的融合物,即包括融合形成单多肽链的抗体片段和支架蛋白的蛋白。在这种情况下,支架蛋白可以是例如通过基因克隆或通过共翻译加工与抗体片段偶联的另一蛋白的任何功能结构域。
考虑其他类型的蛋白质,感兴趣的蛋白可以是例如胰岛素,胰岛素样生长因子,hgh,tpa,细胞因子,如白介素(il),例如il-1、il-2、il-3、il-4、il-5、il-6、il-7、il-8、il-9、il-10、il-11、il-12、il-13、il-14、il-15、il-16、il-17、il-18,干扰素(ifn)α、ifnβ、ifnγ、ifnω或ifnτ,肿瘤坏死因子(tnf),如tnfα和tnfβ、tnfγ,trail,或者g-csf、gm-csf、m-csf、mcp-1或vegf。感兴趣的蛋白还可以是例如促红细胞生成素或任何其他激素生长因子。感兴趣的蛋白还可以是例如darpin。
蛋白质可以通过宿主细胞制备。宿主细胞可以是例如上文提到的任何细胞,例如哺乳动物细胞、植物细胞或细菌细胞等。宿主细胞可以是例如仓鼠细胞,如bhk21、bhktk-、cho、cho-k1、cho-dukx、cho-dukxb1或cho-dg44细胞或者这样的细胞系的衍生物/子代。宿主细胞还可以是例如小鼠骨髓瘤细胞细胞,例如nso和sp2/0细胞或者这样的细胞系的衍生物/子代。宿主细胞还可以是例如这些细胞的衍生物/子代,其他哺乳动物细胞,包括但不限于人、小鼠、大鼠、猴和啮齿动物细胞系,或者其他真核细胞,包括但不限于酵母细胞和昆虫细胞。示例性宿主细胞由léoetal.,animalcells:basicconcepts,pp.13-37,inanimalcelltechnology:frombiopharmaceuticalstogenetherapy(eds.castilhoetal.),taylor&francisgroup,newyork(2008)等描述。
宿主细胞可以在培养基中培养。培养基可以是例如可商购的培养基,如ham'sf12(sigma,deisenhofen,germany)、rpmi-1640(sigma)、达尔伯克改良伊格尔培养基(dmem;sigma)、最低必需培养基(mem;sigma)、iscove's改良达尔伯克培养基(imdm;sigma)、cd-cho(invitrogen,carlsbad,calif.)、cho-s-invitrogen)、不含血清的cho培养基(sigma)以及不含蛋白的cho培养基(sigma)。任何培养基可以在需要时补充各种化合物,其实例包括激素和/或其他生长因子(如胰岛素、转铁蛋白、表皮生长因子、胰岛素样生长因子)、盐(如氯化钠、钙、镁、磷酸盐)、缓冲液(如hepes)、核苷(如腺苷、胸苷)、谷氨酰胺、葡萄糖或其他等效的能源、抗生素、痕量元素。还可以以适当浓度包括任何其他必需的补充物。示例性培养基例如由moraesetal.,culturemediaforanimalcells,pp.111-128,inanimalcelltechnology:frombiopharmaceuticalstogenetherapy(eds.castilhoetal.),taylor&francisgroup,newyork(2008)等描述。
通过宿主细胞表达感兴趣的蛋白可以包括在宿主细胞内转录和/或翻译编码感兴趣的蛋白的核酸序列。感兴趣的蛋白的表达水平可以例如在宿主细胞中存在的编码感兴趣的蛋白的相应mrna的量、宿主细胞中存在的感兴趣的蛋白的量或者从宿主细胞分泌的感兴趣的蛋白的量等的基础上确定。例如,相应mrna可以通过rna印迹杂交、核糖核酸酶rna保护、原位杂交至细胞rna或rt-pcr等其他方法定量,如sambrooketal.,molecularcloning:alaboratorymanual,2dedition,newyork,coldspringlaboratorypress(1989)和ausubeletal.,currentprotocolsinmolecularbiology,(1987-2014)等描述的。还例如,宿主细胞中存在或从其分泌的感兴趣的蛋白的量可以通过各种方法定量,例如通过elisa、、蛋白印迹、放射免疫测定、免疫沉淀、测定蛋白的生物活性、蛋白免疫染色然后facs分析或均相时间分辨荧光(htrf)测定等其他方法,同样如sambrooketal.(1989)和ausubeletal.(1987-2014)等描述的。
在培养基中培养宿主细胞,通过宿主细胞表达蛋白质,可以通过上文提到的用于制备蛋白质的任何方法进行,例如均质方法,例如基于使用搅拌槽式生物反应器、气升式生物反应器或波浪式生物反应器的悬浮培养,或者异质方法,例如基于微载体系统、填充床生物反应器或中空纤维生物反应器的贴壁培养,以不连续模式进行,例如分批培养或补料分批培养,或者以连续模式进行,例如灌注连续培养,并且以任何合适的规模进行,例如实验室、试验或生产规模。示例性方法例如由vélizetal.,bioreactorsforanimalcells,pp.221-258,inanimalcelltechnology:frombiopharmaceuticalstogene描述。
所述包含蛋白质的组合物中的所述蛋白质可以以多种浓度存在。例如,所述包含蛋白质的组合物中所述蛋白质的浓度可以是10至70mg/ml。还例如,所述包含蛋白质的组合物中所述蛋白质的浓度可以是20至65mg/ml、30至60mg/ml、40至55mg/ml和/或约50mg/ml。
还提供根据所述在制造蛋白质期间连续灭活病毒的方法制备的蛋白质。所述蛋白质可以是如上所述的蛋白质。
用于实施在蛋白质制造期间连续灭活病毒的方法的示例性系统500示于图4,其显示了化学控制的病毒灭活流途径。示例性系统500包含(i)蛋白质流输入510(如蛋白质a洗脱流和/或产物流输入),(ii)病毒灭活试剂溶液输入520(如酸/滴定剂/灭活溶液输入),和包含静态混合器240(如低ph静态混合器)的处理容器210。可以使用如上所述的示例性系统500来实施所述方法。根据所述方法,预定速率/%添加的病毒滴定剂添加到主要产物流以严格控制ph和灭活。根据示例性系统500,ph检测器用于测量所述过程,但不用于控制所述过程。因此,ph检测器仅用于监测ph,而不用于控制向示例性系统500添加酸的量或时机。
如所提供的,在蛋白质制造期间连续灭活病毒的方法的优点包含以下:(1)在3.3至3.7的狭窄ph范围内,例如ph3.4至3.6,提供连续的病毒灭活,其为病毒灭活的最佳ph范围;(2)通过数学公式预先确定%病毒灭活试剂;(3)通过恒定体积添加避免ph的动态控制和浓度调节;(4)灭活连续或周期性的加工流;(5)在所述连续和周期性阶段消除静态过程保持和相关的断流;(6)自动流灭活和立即开始病毒灭活;(7)用动态流和溶液浓度和ph处理灭活;(8)灭活任何加工流速;(9)自动匹配灭活与加工流速;(10)灭活蛋白a洗脱液流;(11)避免灭活的过度滴定,保留产物品质;(12)通过将灭活阶段置于系统的周期性部分中,使用连续低ph病毒灭活系统的任何配置;(13)使用相同的方法用于碱添加,如在低ph灭活病毒后并在阴离子交换层析的进一步加工操作之前(因此对应于滴定,而不是灭活);(14)达到下一个单元操作的期望条件;(15)使用相同的方法用于去污剂灭活;(16)最小化制造单克隆抗体的成本、复杂性、时间和设施规模。
还提供用于灭活样品中的一种或多种病毒的方法。所述方法包含在纯化靶分子的过程期间在所述样品从第一单元操作流向第二单元操作时,连续混合所述样品与一种或多种病毒灭活试剂。根据所述方法,一种或多种病毒灭活试剂与样品的比例独立于反馈控制机制或不需要反馈控制机制确定。
用于灭活样品中的一种或多种病毒的所述方法可以根据上面讨论的原理和措施实施。例如,所述样品可以对应于如上所述包含蛋白质的组合物。还例如,可以在如上所述包含病毒灭活试剂的组合物中提供所述一种或多种病毒灭活试剂。还例如,所述第一单元操作可以是如上所述预处理加工步骤。还例如,所述第二单元操作可以是如上所述处理后加工步骤。还例如,可以如上所述提前确定所述一种或多种病毒灭活试剂与所述样品的比例,并因此预先确定。还例如,如上所述,所述一种或多种病毒灭活试剂与所述样品的比例独立于馈控制机制或不需要反馈控制机制(例如ph控制反馈回路)确定。
根据所述方法,如上所述,通过公式1和支持公式2和3以%v/v确定所述一种或多种病毒灭活试剂与样品的比例:
公式1:
公式2:
公式3:
还提供根据用于灭活样品中的一种或多种病毒的所述方法制备的蛋白质。所述蛋白质可以是如上所述的蛋白质。
如所提供的,用于灭活样品中的一种或多种病毒的所述方法的优点类似于如上所述在蛋白质制造期间连续灭活病毒的方法。
实施例
实施例1
集成生物加工提供关于更灵活和更能管理的临床制造的优点。连续/集成加工增加稳健性并降低成本,通过例如,提高自动化、减小设备尺寸和增加树脂利用。
示例性连续制造过程涉及(i)灌注反应器和收获,(ii)连续蛋白a捕获(如涉及两个或更多个柱),(iii)连续低ph病毒灭活,和(iv)精制柱层析(如阴离子交换层析法)。低ph病毒灭活对此过程至关重要。
在本文中,在此公开了用于病毒的连续灭活的化学控制概念。目标是以单一比例开发单一滴定剂溶液,以在整个洗脱过程中保持狭窄低ph窗口。这依赖于泵级稳健性,而不是通过ph响应支配的pid反馈回路。一个关键因素是确定一种合适的滴定剂用作病毒灭活试剂,更具体地说是滴定剂添加到洗脱液中的%v/v、滴定剂的合适浓度、和滴定剂的合适ph,从而获得对应于ph3.4至3.6的病毒灭活的输出。
目前的工业实践依赖于分批蛋白a洗脱和池灭活。在分批过程中,一个柱体积至几个或更多个柱体积的材料从洗脱液合并到容器中。用弱酸手动滴定该池以达到低ph,并因此引发病毒灭活。现有技术通过自动滴定引起低ph灭活。这包含,例如,合并蛋白a洗脱液,以获得包含例如12至15g/l的蛋白质的ph3.8至4.0的蛋白a洗脱池,然后用1-10%的弱酸滴定所述池以达到ph3.5。
为了测试化学控制概念,基于离线分级确定分级滴定剂要求。有趣的是,尽管在洗脱进程中蛋白a洗脱液的ph从约ph7.5降至约ph4.5,但具有最高ph(即约ph7.5)的蛋白a洗脱液的级分并未导致滴定剂最高的需求以达到更低的ph值。相反,令人惊讶的是,对滴定剂的需求是由蛋白a洗脱液级分中的蛋白质浓度驱动的。这示于图5,其阐明所需滴定剂百分比与蛋白质浓度强烈相关。
基于这些结果,出现了模型的关键因素。所述关键因素包含蛋白质变量,包含洗脱期间(或滴定前)的蛋白质峰浓度,以及蛋白质电荷。所述关键因素还包含滴定剂变量,包含对于单克隆抗体稳定性和病毒清除的最小/最大ph,以及对于混合和稀释影响的最小/最大%添加量。其他考虑因素包含层析缓冲液和流速影响,尽管这些是与给定平台相关的常数。其他考虑还包含关于通过阴离子交换层析法进行精制的阴离子考虑。
在考虑作为缓冲液等价物的峰影响时,所述模型预测峰蛋白质洗脱时的蛋白质和电荷大于或等于100mm缓冲液。洗涤缓冲液对应于10mm缓冲液。
考虑到约束内的最坏情况峰浓度,在当前约束下,洗脱最大峰浓度为约50g/l。这基于两种单克隆抗体,如下文更详细讨论的,对应于48至58g/l(10%动态结合能力)。洗脱缓冲液相对于平台保持恒定。基于使用初始0.1mm光程和100+au/cm线性度的flowvpekinetic,在线确定浓度。
鉴定了多种蛋白质的电荷密度谱。有趣的是,如表1所示,与其他蛋白质相比,单克隆抗体的电荷密度相对聚集。
表1.单克隆抗体的电荷密度的相对聚类。
该结果提示,在制备单克隆抗体的过程中通用滴定剂条件对于病毒的连续灭活是可能的。
开发了化学控制方程。根据该公式,可以使用用于弱酸的henderson-hasselbalch方程确定病毒灭活ph的条件,其中(mab(mol/kg)*有效mab电荷δ)作为碱滴定剂。
在线命令包含以下:
蛋白a洗脱流速+滴定剂的恒定%添加=限定的灭活ph。
根据该方法,用于病毒的灭活的ph的上限等于目标ph+0.1ph。下限等于滴定剂ph。
对于示例性单克隆抗体mab2,鉴于%条件、产物g/l、滴定剂摩尔浓度和滴定剂ph,产生操作窗口的模型数据。多种滴定剂溶液的结果示于图6。整个空间的平滑曲线是理想的。由此看出,在50g/l的单克隆抗体浓度下,少数组合解决ph3.5。
考虑滴定剂溶液的子集用于进一步测试。滴定剂溶液子集的结果示于图7。挑选对应于ph3.3的2m甘氨酸,并因此对应于低于目标ph3.5的0.2单位的先导滴定剂溶液。值得注意的是,从图7中可看出,ph3.3的3m甘氨酸的滴定剂溶液也是合适的。有趣的是,尽管基于理论考虑,ph3.3的3m甘氨酸的滴定溶液似乎更好,因为其可以用于在50g/l的单克隆抗体浓度下基于比ph3.3的2m甘氨酸滴定溶液更低的添加体积解决ph3.5,令人惊讶的是,观察到ph3.3的2m甘氨酸滴定溶液相对于ph3.3的3m甘氨酸滴定溶液在添加期间ph和滴定剂浓度的更精确控制,以及因此对蛋白质的不希望的副作用的更低风险,以及相应的系统使用的更大灵活性方面提供优势。因此,由此可看出,ph3.3的2m甘氨酸的先导滴定剂溶液导致良好匹配的添加体积33%(表示为%滴定剂溶液体积/洗脱液体积,意味着每蛋白a洗脱液体积的滴定剂溶液体积,以百分比表示)。
如表2所示,确定所选滴定剂的单克隆抗体电荷影响。
表2.单克隆抗体电荷对所选滴定剂的影响。
该结果表明单克隆抗体的扩散可以通过先导滴定剂的添加体积27-34%来解决(再次表示为%滴定剂溶液体积/洗脱液体积)。由此可看出,直接滴定支持模型输出。该结果表明通用滴定剂是可能的。这允许简单性和稳健性来代替与当前工业实践相关的体积效率低下。
还测试了这种方法对偏移(excursion)的稳健性。如图8所示,单克隆抗体向更高浓度60g/l的偏移导致ph仅增加0.03单位。此外,单克隆抗体向较低浓度的偏移导致ph降至先导滴定剂的ph,即ph3.3。而且,如图9所示,相对于先导滴定剂的添加体积%(如模型泵偏移)的相对添加偏移15%,导致ph值变化为0.02单位。
在以下实施例中更详细地考虑这些和其他结果。
实施例2
低ph连续病毒灭活的一般条件。
为了使低ph连续病毒灭活能用于连续过程,对应于来自蛋白a层析柱的洗脱液的产物使用三通阀在线(inline)与对应于酸(如1.5至3m甘氨酸或乙酸)的病毒灭活试剂混合并通过静态混合器。挑选所述静态混合器尺寸以便能够有效地混合所述酸和产物流。在静态混合器之后可以是足够体积的管,从而为稳健的病毒灭活提供足够的停留时间,例如为60分钟,但也为例如短至1至5分钟。为了结束病毒灭活,所述病毒灭活流使用三通阀与碱(如ph8.0的1至2mhepes或ph11的tris碱)混合并通过静态混合器,以能够混合和以提高所述ph至适合于连续过程中的下一个纯化步骤的ph(如通常ph5至8.1)。
实施例3
在该实验中,将称为mabd的单克隆抗体在洗脱前渗滤到中性ph溶液或蛋白a洗涤溶液中,并用病毒灭活溶液手动滴定。所述实验的目的是定量模型预测在不同蛋白质浓度下需求的%滴定剂的准确程度。
在这些研究中使用的mabd是在化学成分明确的培养基中生长的中国仓鼠卵巢细胞中产生的纯化的人源化igg2。该蛋白质的分子量为150,000道尔顿。从氨基酸序列确定理论等电点,并由等电聚焦凝胶确定实际等电点。使用的材料是蛋白a上样材料或纯化的预制剂材料。
使用具有集成cary50bio,varianinstruments的solovpe,ctechnologies并与集成cary50bio,varianinstruments的flowvpe,ctechnologies串联的uv分光光度计静态测量蛋白质浓度。从理论预测用于从uv吸光度计算蛋白质浓度的消光系数。
用thermo/orion4-starph计以活性温度补偿静态测量蛋白质ph。ph探头是在室温22±3℃下测量的orion8102bnuwprossultraph电极。
系统中的蛋白a洗脱缓冲液由乙酸盐或甘氨酸组成,其在测量的样品中不具有有影响的温度系数ph偏离。低ph灭活试剂由乙酸盐或甘氨酸组成,其在测量的样品中也没有影响温度系数的ph偏离。
测量可变性可以产生自预洗脱蛋白a洗涤溶液,其可由tris或磷酸盐组成。此外,蛋白质结构和滴定氨基酸可显示出温度依赖性偏离。
蛋白a纯化在aktaexplorer100系统上进行,并用整合探头进行ph趋势分析。在新的低循环数树脂上以60和240cm/hr之间的可变流速上样分子至10%动态结合容量(dynamicbindingcapacity,也称为dbc)。使用的蛋白a基质是mabselect基质或mabselectsurelx基质。柱高在19到26厘米之间。用一种或多种洗涤溶液洗涤上样的柱以减少非特异性结合并置柱于具有低滴定需求的溶液中。用ph3.5的25mm乙酸钠或ph3.5的150mm甘氨酸以60-240cm/hr进行洗脱。
用于蛋白a纯化的示例性试验性规模方案如下。对于上样材料,滴度相当于3.2mg/ml。将柱上样至40mg/ml。表3中提供了流速和体积。在蛋白a流洗脱期间,通过aktacviexplorer系统添加另外的流速。%滴定剂添加量为30%(再次表示为%滴定剂溶液体积/洗脱液体积)。添加洗脱流的泵a流速为18.25ml/min。
表3.aktaexplorer100系统试验性蛋白a纯化的流速和体积。
用于静态滴定测量的材料取自如上所述的蛋白a纯化作为池,中和至ph7.5±0.2,并进一步渗滤并通过超滤/渗滤(ultrafiltration/diafiltration,也称为uf/df)浓缩。将所述材料针对10mmtris,10mmnaclph7.5,最终的蛋白a洗涤缓冲液渗滤7个或更多“渗滤体积”。通过solovpe测量,进行最终浓缩步骤以回收>75g/l的材料。测量的材料ph7.5±0.1,表明没有发生由于浓度或缓冲液的ph偏离。
使用quattro流体隔膜泵、biopharm管和30kdmilliporeplctk复合再生纤维素膜手动组装所使用的uf/df系统。
为了试验性和示范目的,以专用的aktaexplorer100系统进行串联低ph病毒灭活,该系统命名为cviexplorer,与proteinaaktaexplorer系统分开。该系统重新管化以通过进样阀接受来自proteinaexplorer系统的主要蛋白质,并通过泵a直接添加流体(灭活试剂)到混合室或t和静态混合器中。以串联akta探头测量所得混合溶液ph趋势。编程cviexplorer系统以用灭活试剂上样灭活流体通路。在检测到uv信号后,引导来自废液的蛋白a流至串联以灭活。使来自泵a的流体等于为灭活预定的%v/v添加量并且相对于蛋白质a流的流速设定泵a。例如,如果用于灭活的确定是病毒灭活试剂的30%v/v添加,那么如果蛋白a流以50ml/min的速度进入,则cviexplorer系统将被编程为使泵a达到灭活试剂以15ml/min进入流中。
用先前描述的具有已知背景缓冲液组合物的渗滤材料进行低ph病毒灭活的静态测量。测量75g/l至0g/l的溶液被逐步用灭活试剂滴定,以在22±3℃下达到ph3.50,并用先前描述的ph计和探头测量。
表4中提供了结果。
表4.mabd的预测的和实际的滴定。
实施例4
在该实验中,将指定为mabb的单克隆抗体在洗脱前渗滤到中性ph溶液或蛋白a洗涤溶液中,并用病毒灭活溶液手动滴定。所述实验的目的是定量模型预测在不同蛋白质浓度下需求的%滴定剂的准确程度。
在这些研究中使用的mabb是在化学定义的培养基中生长的中国仓鼠卵巢细胞中产生的纯化的人源化igg1。该蛋白质的分子量为150,000道尔顿。从氨基酸序列确定理论等电点,并由等电聚焦凝胶确定实际等电点。使用的材料是蛋白a上样材料。
如上所述实施研究。
表5中提供了结果。
表5.mabb的预测的和实际的滴定。
实施例5
在该实验中,mabd以在预设的%体积添加预定的病毒灭活溶液(包含甘氨酸)在线连续灭活。该运行在100l反应器规模下在多柱连续蛋白a捕获中操作,其中周期的洗脱以化学控制概念灭活。
结果示于图10。该结果表明,对于动态蛋白a流,实现和维持用于连续病毒灭活的规格内的输出ph。以一次稀释,使用一种病毒灭活试剂溶液实现该结果,且无ph不足,因此证明稳定性。
这些实施例的结果一起表明能先验地稳健地实现低ph病毒灭活的化学控制。这通过滴定所需的最大电荷的知识来完成。滴定剂与流动相的体积比实现ph目标而不会过度滴定。因此,ph测量能减少到确认性趋势测量而不是主动控制。
对于本领域技术人员显然的是,在不脱离所要求保护的方法的精神和范围的情况下,对所描述的实施例进行多种修改和变化。因此,本发明要求保护的方法旨在覆盖本文所述实施例的修改和变化,只要它们落入所附权利要求及其等同物的范围内。
工业实用性
本文公开的方法可用于在蛋白质制造期间连续灭活病毒,并因此用于改进制备蛋白质的工业方法。
- 还没有人留言评论。精彩留言会获得点赞!