从基因工程微生物改进的黏康酸生产的制作方法
本申请要求2016年3月2日提交的美国临时申请序列号62/302,558的优先权。
发明领域
本发明涉及使用在中心芳香族生物合成途径中进行基因工程改造的生物催化剂生产可再生化学原料的领域。更具体地,本发明提供了使用遗传修饰的生物催化剂改善从可再生碳源生产黏康酸的方法。
背景技术:
己二酸是主要的商品化学品,用于生产尼龙6,6和聚氨酯。己二酸目前衍生自石化原料。目前己二酸的合成对环境有害,释放出亚硝酸(xie等,2014)。或者,己二酸可以通过化学氢化由黏康酸的三种异构体(顺式,顺式;顺式,反式;反式,反式异构体)中的任何一种制成。希望通过用微生物发酵从可再生资源中生产黏康酸,然后通过氢化过程产生己二酸,因为这种产生己二酸的途径比传统的石化途径更环保(niu,drathsandfrost,2002;frostanddraths,1997)。许多其他化学品可以通过一种或多种黏康酸异构体的化学转化来制备,包括但不限于1,6己二醇、3-己烯二甲酸、1,6-己二胺和对苯二甲酸。
国际专利申请公开号wo2011/017560要求保护具有黏康酸途径的生物催化剂和利用这些生物催化剂来生产黏康酸的方法。简而言之,这件公开的专利申请公开了四种不同的生产黏康酸的途径。第一种用于黏康酸生产的途径以琥珀酰-coa和乙酰-coa起始。第二种用于黏康酸生产的途径以丙酮酸和丙二酸半醛起始。第三种用于黏康酸生产的途径以丙酮酸和琥珀酸半醛起始。第四种用于黏康酸生产的途径以赖氨酸起始。在此专利申请中提出的所有这些用于黏康酸生产的途径都是基于计算机建模,而这些生物催化剂究竟能否产生为具备商业上可接受的黏康酸生产力和产率还有待观察。
在科学文献(niu等人,2002;frostanddraths,1997)和专利文献(us5,487,987;us5,616,496;wo2011/085311a1)中描述了使用基因工程大肠杆菌系统生产顺式,顺式-黏康酸的发酵途径。然而,用于黏康酸生产的现有技术方法具有显著的缺点,例如昂贵的培养基组分(芳香族氨基酸和维生素)和化学诱导剂,以及低于工业生产所需的产率。最近的美国专利申请公开号us2015/0044755(其全部内容通过引用并入本文)提供了用于黏康酸生产的改进的生物催化剂,涉及相关基因的组成型表达、改进的异源基因和新的“渗漏”aroe酶。本发明提供了美国专利申请公开号us2015/0044755中描述的黏康酸生物催化剂的进一步改进。
发明概述
本发明提供从非芳香族碳源开始生产顺式,顺式-黏康酸的基因改造的微生物,非芳香族碳源如,糖和碳水化合物,包括但不限于葡萄糖,蔗糖,甘油和纤维素水解产物。
在本发明的一个实施方案中,芳香族氨基酸生物合成的负调节物的活性受基因操纵。在本发明的一个方面,负调节物tyrr的活性通过控制编码tyrr蛋白的tyrr基因的表达而实质降低。在本发明的另一方面,负调节物tyrr的活性通过将来自微生物的染色体dna的tyrr基因缺失或失活而完全消除。
在本发明的另一个实施方案中,通过遗传操作克服某些代谢物对芳香族氨基酸途径中的某些酶的反馈抑制。在大多数野生型大肠杆菌菌株中,在芳香族氨基酸途径开始时起作用的脱氧阿拉伯-庚酮糖酸7-磷酸合酶(“dahp合酶”)作为三种不同的同工酶发生,已知这三种不同的同工酶由三种不同的基因即arog,arof和aroh编码。由这三种基因中的每一种编码的蛋白质受到芳香族氨基酸途径的一种或多种代谢物的反馈抑制。在本发明的一个方面,野生型arog基因被经修饰的arog基因取代,该经修饰的arog基因编码arog蛋白,该蛋白对微生物细胞内芳香族氨基酸途径的一种或多种代谢物的反馈抑制具有抗性。这种反馈抗性形式的arog蛋白称为“arogfbr”。在本发明的另一个方面,野生型arof基因被arof基因取代,后者编码arof蛋白,该arof蛋白对微生物细胞内芳香族氨基酸途径的一种或多种代谢物的反馈抑制具有抗性(aroffbr)。在本发明的另一个方面,野生型aroh基因被aroh基因取代,后者编码aroh蛋白,该aroh蛋白对微生物细胞内芳香族氨基酸途径的一种或多种代谢物的反馈抑制具有抗性(arohfbr)。在本发明的另一方面,选择用于商业生产顺式,顺式-黏康酸的生物催化剂可具有dahp合酶的一种以上反馈抗性同工酶。
在本发明的另一个实施方案中,增强了微生物细胞内中心芳香族生物合成途径中涉及的一种或多种酶的活性。在本发明的一个方面,通过遗传操作实现参与芳香途径和/或黏康酸途径操作的一种或多种酶的活性的增强。在本发明的一个优选方面,编码蛋白质arof,arog,aroh,arob,tkta,talb,aroz,qutc,qa-4,asbf,quic,aroy,rpe,rpi,pps,cata和catx或其同源物或类似物的一种或多种基因的表达被增强,导致所述蛋白质的活性增加。rpe是核酮糖-5-磷酸差向异构酶,rpi是核酮糖-5-磷酸异构酶,pps是磷酸烯醇丙酮酸合成酶(neidhardt和curtiss,1996)。如果宿主菌株是酵母,例如酿酒酵母(saccharomycescerevisiae),或丝状真菌,例如,粗糙脉孢菌(neurosporacrassa),那么催化莽草酸途径中的反应的几种酶可以组合成一种称为arolp的大蛋白质或多肽,其在酿酒酵母的情况下由aro1基因编码。aro1p结合了arob,arod,aroe,arok(或arol)和aroa的功能。因此,出于本发明的目的,arolp和aro1或其部分可用作arob,arod,aroe,arok和/或aroa的替代或补充。
在本发明的另一个实施方案中,通过在磷酸戊糖途径的操作中过表达酶来增强细菌细胞内通过赤藓糖-4-磷酸的通量。在本发明的一个方面,通过遗传修饰增强转醛醇酶,例如由talb或tala基因编码的转醛醇酶的表达。在本发明的另一个方面,通过遗传操作增强编码转酮醇酶的基因,例如tkta基因的表达。在本发明的另一方面,通过遗传操作增强了编码核酮糖-5-磷酸差向异构酶和核酮糖-5-磷酸异构酶之一或两者的基因的表达。
在本发明的另一个实施方案中,通过遗传操作增加芳香族氨基酸途径起作用所必需的磷酸烯醇丙酮酸(pep)库。在本发明的一个方面,通过消除和/或补充葡萄糖摄取的pep依赖的磷酸转移酶系统(pts)与葡萄糖摄取的pep独立的系统来降低pep库的使用的竞争。在本发明的另一个方面,基于galp的糖摄取系统被灭活,目的是在微生物细胞内保存atp。在本发明的另一个方面,在pts系统和基于galp的糖摄取系统(δpts/δgalp)的功能中有缺陷的微生物细胞中,通过引入编码glf(促进葡萄糖扩散的蛋白质)的外源基因或编码glf和glk(葡糖激酶)蛋白的外源基因来实现糖摄取。在本发明的另一个实施方案中,通过增加编码pep合成酶的基因(例如pps)的表达来增加pep的可得性。
在本发明的另一个实施方案中,3,4-二羟基苯甲酸脱羧酶(aroy)的活性增强。在该实施方案的一个方面,通过遗传操作增强aroy的表达。在本发明的另一方面,通过遗传操作增加选自包含ubix,kpdb,elw,kox,lpl及其同源物的作为aroy的辅助蛋白的蛋白质的表达,导致3,4-二羟基苯甲酸脱羧酶活性的增加。
在本发明的另一个实施方案中,通过降低或消除磷酸烯醇丙酮酸羧化酶(ppc)活性来增加pep的可得性。在本发明的一个方面,丙酮酸羧化酶(pyc)活性增加和/或取代ppc活性,特别是当ppc活性被消除时。
附图简述
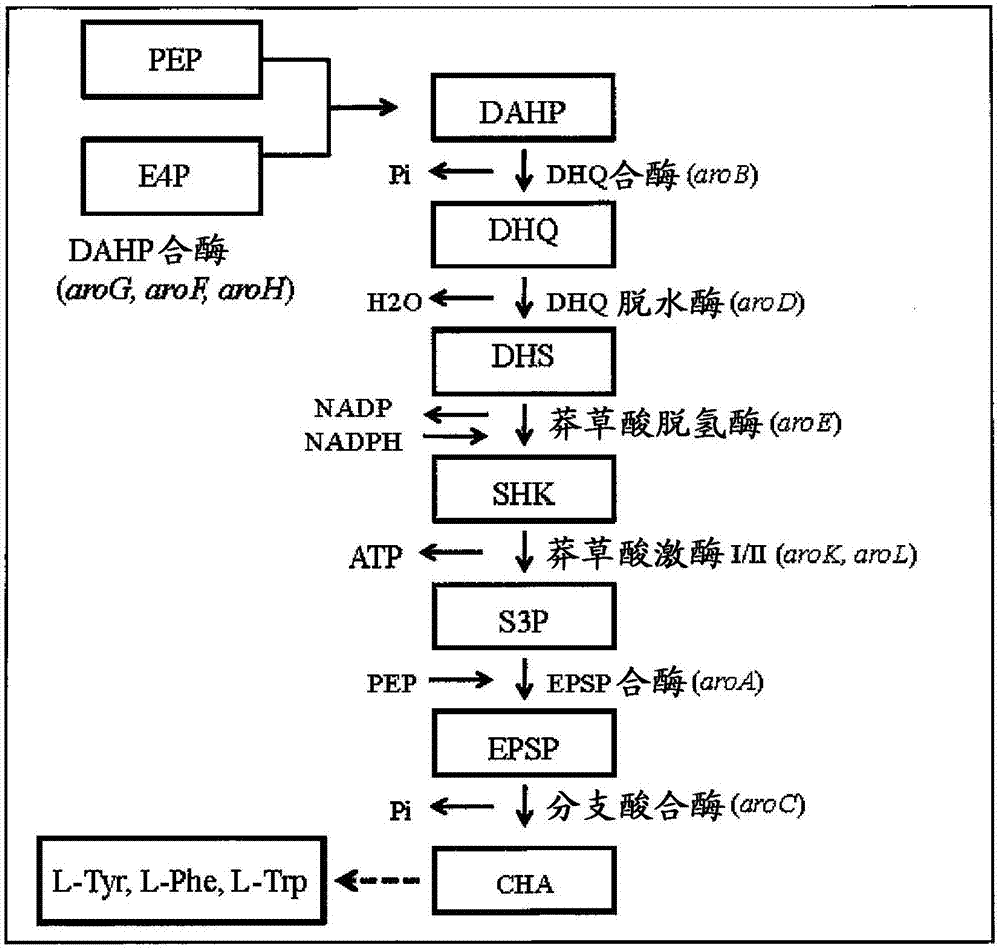
图1.大肠杆菌中用于芳香族氨基酸生物合成的途径。
图2.大肠杆菌中黏康酸生物合成的途径。
图3.将3-脱氧-阿拉伯-庚酮糖酸7-磷酸(dhap)转化为黏康酸的反应步骤。图4.显示用于全部黏康酸和生化中间体的hplc分析的标准品的色谱。
图5.显示用于黏康酸异构体的hplc分析的标准品的色谱。
图6.在用质粒pcp32amp、pcp14和pcp54转化的大肠杆菌菌株myr34中生产dhs的滴度。大肠杆菌的myr34菌株具有aroe基因缺失。质粒pcp32amp表达编码dahp合酶的arog基因。质粒pcp14表达编码dhq合酶的arob基因。质粒pcp54表达arob和arog基因二者。
图7.在用质粒pcp32amp和pcp54转化的大肠杆菌菌株myr34和myr170中生产dhs的滴度。大肠杆菌的myr34菌株具有aroe基因缺失。myr170菌株具有aroe基因缺失和在宿主染色体dna的ack基因座处整合的处于p15启动子控制下的arob基因的第二拷贝。质粒pcp32amp表达编码dahp合酶的arog基因。质粒pcp54表达arob和arog基因二者。
图8.在仅用质粒pmg37或用pmg37和pcp32amp质粒二者转化的大肠杆菌菌株myr34和myr170中生产顺式,顺式-黏康酸的滴度。大肠杆菌myr34菌株具有aroe基因缺失。myr170菌株具有aroe基因缺失和在宿主染色体dna的ack基因座处整合的处于p15启动子控制下的arob基因的第二拷贝。质粒pcp32amp表达编码dahp合酶的arog基因。质粒pmg37表达编码在黏康酸途径中起作用的蛋白的aroz、aroy和catax基因。
图9.在仅用表达arog基因的质粒(pcp32amp)或同时表达arog和tkta基因的质粒(pcp50)转化的大肠杆菌myr170菌株中生产dhs的滴度。myr170菌株具有aroe基因缺失和在宿主染色体dna的ack基因座处整合的处于p15启动子控制下的arob基因的第二拷贝。
图10.来自用质粒pcp32amp和pcp50转化的大肠杆菌myr34和myr170菌株的dhs产率。dhs产率计算为每克消耗的葡萄糖所产生的dhs克数。质粒pcp32amp表达arog基因,而pcp50表达arog和tkta。细菌菌株myr34具有aroe基因缺失。大肠杆菌myr170菌株衍生自myr34且具有在染色体dna上的ack基因座处整合的额外的arob基因。
图11.来自用质粒pcp32amp和pcp50转化的大肠杆菌myr170和myr261菌株的dhs滴度。质粒pcp32amp表达arog基因,而pcp50表达arob和tkta基因。myr170菌株具有aroe基因缺失和在宿主染色体dna的ack基因座处整合的处于p15启动子控制下的arob基因的第二拷贝。大肠杆菌myr261菌株衍生自大肠杆菌myr170菌株。大肠杆菌myr261菌株具有在染色体dna的poxb基因座处整合的tkta基因的第二拷贝及其天然启动子。
图12.大肠杆菌菌株myr170、myr261和myr305中的黏康酸和乙酸生产,这些菌株转化有表达编码莽草酸生物合成途径中的dahp合酶的arog的质粒pcp32amp和表达编码在黏康酸途径中起作用的蛋白的aroz、aroy和catax基因的质粒pmg37。myr170菌株具有aroe基因缺失和在宿主染色体dna中ack基因座处插入的处于p15启动子控制下的arob基因的额外拷贝。myr261和myr305是myr170菌株的衍生物。myr261具有在宿主染色体dna上的poxb基因座处整合的tkta基因的额外拷贝,而myr305在宿主染色体dna上的poxb基因座处具有缺失。
图13.由大肠杆菌菌株myr34产生的内源性dhs向黏康酸的转化。大肠杆菌菌株myr34在编码莽草酸脱氢酶的aroe基因中具有缺失。因此,存在dhs的积累。当菌株myr34用表达编码在黏康酸途径中起作用的蛋白的aroz、aroy和catax基因的质粒转化时,dhs转化成黏康酸。然而,当用无任何外源基因的空质粒载体(pcl1921)转化myr34菌株时,没有出现dhs转化成黏康酸。
图14.aroz类似物将dhs转入黏康酸途径中的能力的比较。将三种不同的aroz类似物,即来自不动杆菌属物种adp1的quic、来自苏云金芽孢杆菌(bacillusthuringiensis)的asbf和来自粗糙脉孢菌的qa-4克隆到还分别自p15和λpr启动子表达catax和aroy基因的低拷贝质粒中p26启动子下。通过转化使这三种不同的质粒构建体在myr34中表达,并测量所产生的黏康酸的量。
图15.将单拷贝的catax、aroy和quic整合到大肠杆菌myr170菌株染色体(δaroe,δack::p15-arob)中,产生myr352(seqidno.41)。还用携带黏康酸途径运作所需的所有基因的低拷贝质粒pmg37转化myr170,得到myr219菌株。用yep24(中拷贝的空载体)或pcp32amp(中拷贝的arog表达质粒)或pcp50(中拷贝arog和tkta表达质粒)转化myr352和myr219,并使用hplc方法对产生的pca、儿茶酚和黏康酸的量进行定量。
图16.通过增加catax的表达来去除myr352中的儿茶酚积累。对myr352转化仅表达aroy的质粒(pmg27)或仅表达quic的质粒(pmg39)或表达所有三种黏康酸途径基因即catax、aroy和quic的质粒(pmg37)或仅表达黏康酸途径中的两种基因即catax和aroy的质粒(pmg33)。仅catax的过表达足以防止儿茶酚积累。
图17.利用不同系统输入葡萄糖的菌株的生长。ptshi和galp(myr31)的缺失导致在基本葡萄糖培养基中生长不足,而安装glf和glk基因(myr217)挽回生长。对照菌株myr34是δaroe,否则则是野生型。向培养基中添加三种芳香族氨基酸和三种芳香族维生素以允许营养缺陷型菌株的生长。
图18.大肠杆菌myr34和myr217菌株中的dhs生产。当用导致dhs生产的质粒转化时,利用glf-glk用于葡萄糖输入的myr217较之利用磷酸转移酶系统(pts)的myr34的转化体产生更高滴度的dhs。
图19.在7升发酵罐中以大肠杆菌myr428菌株进行的黏康酸生产。用质粒pcp32amp和pmg37转化具有δaroeδacka::p15-arobδpoxb::tkta基因型的大肠杆菌myr261菌株以产生大肠杆菌myr428菌株。
图20.经遗传工程改造以产生黏康酸的大肠杆菌菌株myr993和两种myr993衍生菌株myr993δubix和myr993δubid中的黏康酸和pca产生。通过用编码卡那霉素抗性的盒替换ubix基因的编码区,从myr993菌株衍生myr993δubix大肠杆菌菌株。通过用编码卡那霉素抗性的盒替换ubid基因的编码区,类似地从myr933菌株衍生myr993δubid大肠杆菌菌株。
图21.ubix的各种同源物的相对活性的测量。从pca的脱羧作用测量ubix同源物的活性,如通过290nm处吸光度(a290)的降低所测量的。在该研究中使用五种不同的ubix同源物,即kpdb,ubix,elw,kok和lpl。
图22.具有低或高水平kpdb基因表达的大肠杆菌菌株中的黏康酸和pca产生。将没有外源kpdb基因的大肠杆菌菌株myr1305用作亲本菌株,并用具有从p26启动子或大肠杆菌pgi启动子表达的kpdb基因的低拷贝质粒转化。预期来自p26启动子的基因表达处于相对低的水平,而预期来自pgi启动子的基因表达处于相对高的水平。
图23.大肠杆菌菌株myr1674及其衍生物myr1772中的黏康酸产生。通过用pr-pyc基因替换ppc的编码区和启动子,自myr1674衍生myr1772。pr是来自大肠杆菌噬菌体λ的强右向启动子的缩写。
优选实施方式详述
本文引用的所有专利,专利申请,出版物,序列和其他公开材料均通过引用并入本文。
如本专利申请中所用,短语“例如”或“如”意指对于当下主题存在多于一种的方法、途径、方案或物质组成,且给出的实例并不意在局限于该实例。
术语“异源的”是指并非在生物体中天然或原生发现、但可以通过基因改造如通过转化、交配或转导而引入到生物体中的基因或蛋白。异源基因可以整合(插入)到染色体中或包含于质粒上。术语“外源的”是指出于提高、降低或消除活性的目的通过基因改造如通过转化、交配、转导或诱变已经引入到生物体中或在生物体中改变的基因或蛋白。外源基因或蛋白可以是异源的,或者其可以是对于宿主生物体为原生但通过一种或多种方法例如突变、缺失、改变启动子、改变终止子、重复或者在染色体或质粒中插入一个或多个额外拷贝而改变的基因或蛋白。因此,例如,如果在染色体中在与原生位点不同的位点处插入arob基因的第二拷贝,第二拷贝将是外源性的。
如本发明中所用的术语“微生物”包括可通过发酵方法用于顺式,顺式-黏康酸的商业生产的细菌、古细菌、酵母、藻类和丝状真菌。术语“基因工程微生物”是指在自然界中不存在但使用本专利申请中描述的一种或其他遗传修饰产生的微生物。
对于命名法,基因或编码区通常以斜体的小写字母命名,例如“aroz”,而由基因所编码的酶或蛋白可以以相同的字母命名但第一个字母大写且不斜体,例如“aroz”。酶或蛋白也可以用更加描述性的名称指代,例如,aroz也可以指代3-脱氢莽草酸脱水酶。编码具有特定催化活性的酶的实例之一的基因或编码区可以因历史不同起源或因为基因来源于不同种类而具有数种不同的名称。例如,编码来自炭疽芽孢杆菌的3-脱氢莽草酸脱水酶的基因可以命名为asbf而不是aroz,来自构巢曲霉的相同基因可以命名为qutc,来自粗糙脉孢菌的相同基因可以命名为qa-4,并且来自acinetobacterbaylyi的相同基因可以命名为quic。
“质粒”意指实质地小于染色体、与微生物的一个或多个染色体分开且独立于一个或多个染色体进行复制的环状或线性dna分子。“质粒”可以以每细胞约一拷贝或以每细胞多于一个的拷贝存在。将质粒维持在微生物细胞中通常需要抗生素选择,但也可使用营养缺陷型的互补。
如本发明中所用的术语“染色体”或“染色体dna”在细菌细胞的环境下是实质地大于质粒且不需要任何抗生素选择的环状dna分子。
术语“表达盒”或“盒”意指这样的dna序列,其可以是染色体或质粒的一部分,并且其含有至少启动子和编码酶或其它蛋白的基因或区域,从而使编码区由启动子表达,且含有该dna序列的宿主细胞产生该酶或蛋白。“表达盒”可以是至少部分合成的或通过基因工程方法构建,从而使编码区从并非与编码区天然关联的启动子表达。任选地,“表达盒”可含有转录终止子,其可以是或者可以不是与编码区天然关联的终止子。“表达盒”可以具有针对多于一个蛋白的编码区,在这种情况下其可称为操纵子或合成操纵子。
术语基因或编码区的“过表达”意指致使在相同或相似的生长条件下由该基因或编码区编码的酶或蛋白以高于宿主微生物野生型形式中所见水平的水平在宿主微生物中产生。这可以通过例如一种或多种以下方法而实现:1)安装更强的启动子,2)安装更强的核糖体结合位点,如5’-aggagg的dna序列,使其位于翻译起始密码子上游约四至十个碱基的位置,3)安装终止子或更强的终止子,4)改善对编码区中一个或多个位点处的密码子的选择,5)改善mrna稳定性,和6)提高基因的拷贝数,通过在染色体中引入多个拷贝或是在多拷贝质粒上放置盒。由过表达的基因产生的酶或蛋白被称为“超量产生的”。“过表达的”基因或“超量产生的”蛋白可以是对于宿主微生物原生的基因或蛋白,或者其可以是已通过基因工程方法从不同的生物体移植到宿主微生物中的基因或蛋白,在这种情况下酶或蛋白以及编码该酶或蛋白的基因或编码区被称为“外来的”或“异源的”。外来的或异源的基因和蛋白按定义是过表达和超量产生的,因为它们在未经改造的宿主生物体中不存在。
第一基因、dna序列或蛋白的术语“同源物”是执行与所述第一基因、dna序列或蛋白相似的生物学功能,且经用于序列比较的blast计算机程序使用默认参数所测定(altschul等人,1990;altschul等人,1997)与所述第一基因或蛋白具有至少25%的序列同一性(当比较蛋白序列或比较从基因序列得到的蛋白序列时),并允许缺失和插入的第二基因、dna序列或蛋白。大肠杆菌arog基因的同源物的实例会是来自鼠伤寒沙门氏菌(salmonellatyphimurium)的arog基因。
通过同源性相关非常远的两种酶或蛋白质可以实现相同的生物化学功能,但彼此之间仅具有相对弱的同源性。例如,来自大肠杆菌k-12的fuma延胡索酸酶(genbanknp_416129)与来自肉毒梭菌的延胡索酸酶(genbankgae03909.1)在其重叠区域上具有约26.9%的同源性,并且与来自pyrococcussp.st04的延胡索酸酶的β亚基(genbankakf23146.1)在其重叠区域上具有约25.1%的同源性。作为另一个实例,均将dhs转化为pca的克雷伯氏菌属aroz和粗糙脉孢菌qa-4酶具有29.3%的同一性。因此,对于代谢途径的基因工程,异源酶或蛋白质的重要特征是由该酶或蛋白质而不是源生物体或精确氨基酸序列进行的功能或反应,我们定义“同源”酶或蛋白质或“同源物”包含在其氨基酸序列的同一性中为25%或更高的任何一对酶或蛋白质,允许空位,例如通过使用lasergene12(dnastar,madison,wi)megalign程序,用lipman-pearson方法(ktuple=2,空位罚分=4,空位长度罚分=12)使用默认参数进行比对所示。
第一基因、dna序列或蛋白的术语“类似物”是执行与所述第一基因、dna序列或蛋白相似的生物学功能,且经用于序列比较的blast计算机程序所测定(altschul等人,1990;altschul等人,1997)与所述第一基因、dna序列或蛋白具有少于25%的序列同一性(当比较蛋白序列或比较从基因序列得到的蛋白序列时),并允许缺失和插入的第二基因、dna序列或蛋白。肺炎克雷伯氏菌aroz蛋白的类似物的实例会是来自构巢曲霉的qutc蛋白,因为两种蛋白都是催化3-脱氢莽草酸脱水酶反应的酶,但在两种酶或其各自的基因之间没有显著的序列同源性。本领域中有见解的科学家将会知晓,在许多不同的生物体中可以找到许多具有特定生物学功能的酶和蛋白,如dahp合酶或3-脱氢莽草酸脱水酶,它们或是作为同源物或是作为类似物,并且因为这些酶或蛋白家族的成员共有相同的功能,故尽管它们可能在结构上稍有不同或实质不同,但在许多情况下,使用现有的基因改造方法,相同家族的不同成员可以用于执行相同的生物学功能。因此,例如,aroz酶和qutc酶催化相同的反应,dhs脱水酶,这样任一者在合适的环境下都会导致顺式,顺式-黏康酸产生,并且选哪一个来使用最终可通过选择在相似的发酵条件下产生更高滴度的顺式,顺式-黏康酸的一个来作出选择。
术语“非芳香族碳源”或“非芳香族化合物”指可作为碳源和/或能量用于供养本发明的微生物的含碳化合物,其中该化合物不含有与苯相关的六元环。非芳香族碳源的实例包括葡萄糖、木糖、乳糖、甘油、醋酸、阿拉伯糖、半乳糖、甘露糖、麦芽糖或蔗糖。“芳香族化合物”是含有一个或多个与苯相关的六元环的化合物。芳香族化合物的实例是儿茶酚或1,2-二羟基苯。使用葡萄糖作为非芳香族碳源来源选择用于生产黏康酸的微生物可以进一步使用专利文献美国专利号8,871,489和美国专利申请公开号us2013/0337519a1和us2014/0234923a中提供的基因工程技术工程化以使用其他类型的非芳香族碳源,例如甘油,蔗糖和木糖。
术语“强组成型启动子”指这样的dna序列,其通常位于由rna聚合酶转录的dna序列或基因的上游(当以习惯的5’至3’方向描述时在基因的5’侧),并且其导致所述dna序列或基因通过rna聚合酶转录以容易经由任何适当的测定法步骤直接或间接检测到的水平表达。适当的测定法步骤的实例包括1)定量逆转录酶+pcr,2)对于所编码酶的酶测定法,3)考马斯蓝染色的蛋白凝胶,或4)作为所述转录的结果间接产生的代谢物的可测量的产生,且这样的可测量的转录的发生不论特定调节转录水平的蛋白、代谢物或诱导物化学品存在与否。不是“强组成型启动子”的启动子的实例是大肠杆菌的plac启动子,因为其在没有乳糖或诱导物iptg的情况下受阻遏物的阻遏。通过使用领域内熟知的方法,可以使用“强组成型启动子”取代原生的启动子(否则天然存在于dna序列或基因上游的启动子),得到如下的表达盒,所述表达盒可以置于质粒中或染色体中,且以高于原生启动子水平的水平提供期望dna序列或基因的表达水平。强组成型启动子可以是种或属特异性的,但来自细菌的强组成型启动子常常可以在远缘相关的细菌中良好发挥功能。例如,来自枯草芽孢杆菌(bacillussubtilis)或来自通常在枯草芽孢杆菌上生长的噬菌体的启动子可以在大肠杆菌中良好发挥功能。“强组成型启动子”与诱导型启动子如ptac实质地不相同,诱导型启动子已在现有技术中用于生产顺式,顺式-黏康酸且对于期望的功能水平通常需要昂贵的化学品或其它环境变化(niu等人,2002)。强组成型启动子的实例是来自枯草芽孢杆菌噬菌体sp01的p15、p26,和大肠杆菌噬菌体λpr。
“突变”是dna序列中的任何变化,使其与相关的野生型序列不同。“突变”可以包括dna序列中的单碱基变化,缺失,插入,置换,移码,倒位,复制或任何其他类型的变化。通常,“突变”是指对功能具有负面影响或降低基因或基因产物活性的变化,然而,在本文中,术语“突变”也可以指增加基因或基因产物活性的变化。例如,arog基因中的反馈抗性突变在诸如苯丙氨酸的抑制剂存在下增加arog的活性。用不同的,更强的启动子取代启动子,也导致可以增加基因或基因产物活性的突变。“无效突变”是一种突变,例如大多数或全部基因的缺失,是一种有效消除基因功能的突变。“突变体”是包含一个或多个突变的菌株或分离物。
顺式,顺式-黏康酸(本文中简称为“黏康酸”)的生物学产生基于来自芳香族氨基酸途径的碳的重新定向。芳香族氨基酸和维生素的天然生产需要代谢物赤藓糖-4-磷酸(e4p)和磷酸烯醇丙酮酸(pep)。芳香族氨基酸合成中的第一个步骤由3-脱氧-阿拉伯-庚酮糖酸7-磷酸(dahp)合酶催化。在大肠杆菌中,该步骤可以通过三种不同的同工酶arog,arof或aroh进行。这些酶中的每一种都通过阻遏蛋白tyrr在转录水平上调节,并且在蛋白质水平上分别通过抑制途径的末端产物苯丙氨酸、酪氨酸和色氨酸来调节。黏康酸的产生从芳香族氨基酸途径的中间体脱氢莽草酸(dhs)进行,并且需要表达三种异源酶:脱氢莽草酸脱水酶(aroz),3,4-二氢苯甲酸脱羧酶(aroy)和儿茶酚1,2-双加氧酶(cata)。该途径如图1和2所示。
“黏康酸途径”或“黏康酸途径”是指从dhs到pca到儿茶酚到顺式,顺式-黏康酸的生化途径,“黏康酸途径基因”是编码催化黏康酸途径中的步骤的酶,或编码用于增强所述酶之一的活性的辅助功能的基因,例如aroz,aroy,cata,catx和qutc(图3)。dhs是3-脱氢莽草酸的缩写,pca是原儿茶酸的缩写。“黏康酸质粒”是含有一个或多个黏康酸途径基因的质粒。
本发明中使用的遗传操作集中于许多微生物细胞中存在的芳香族氨基酸和芳香族维生素(或维生素样)生物合成的共同途径,如图1所示。如图1所示的芳香族氨基酸生物合成的共同途径可以称为“莽草酸”或“莽草酸”途径,“分支酸”或“分支酸”途径,或“中心芳香族”或“中心芳香族生物合成”途径。
关于微生物的基因工程用于产生芳香族氨基酸苯丙氨酸,酪氨酸和色氨酸有大量已发表的工作(us4,681,852,us4,753,883,us6,180,373,欧洲专利申请86300748.0)。产生芳香族氨基酸的方法包括使用反馈抗性酶(arof,arog,phea,tyra)的多种组合,转录抑制的失调(tyrr-),增加启动子强度(ptac,plac)和增加一种或多种基因(tkta)的拷贝数。可以遵循上述遗传修饰的许多特定组合以获得适合于黏康酸生产的生物催化剂。
根据国际专利申请公开号wo2013/116244(其通过引用整体并入本文)中的公开内容,基因工程微生物不需要包含任何外源质粒以产生黏康酸,尽管它们具有实现所需表型所必需的某些外源或异源基因。在本发明的优选实施方案中,引入微生物中的外源基因稳定整合到染色体dna中。由于外源基因的这种染色体dna整合,完全消除了使用抗生素或其他选择性方法来维持携带外源dna的质粒的需要。此外,不需要化学诱导剂的强启动子用于表达从碳源(例如葡萄糖)到顺式,顺式-黏康酸的途径操作所必需的基因。
当外源编码序列整合到染色体dna中时,它整合在一个基因座上,据报道其缺失不会引起任何不利影响。例如,大肠杆菌中物理位置0039的编码区,也称为ydem,注释为脂蛋白,并且已被证明在缺失时没有不利影响。类似地,大肠杆菌中物理位置2160处的编码区(也称为nlpa)被注释为基本sam结构域蛋白,并且已经证明在缺失时没有不利影响。在本发明中,将pr-catax的拷贝插入大肠杆菌细菌的物理位置0039,并将pr-arogfbr的拷贝插入大肠杆菌细菌的物理位置2160。在本文所述的其他实例中,在有利于敲除或缺失的基因中进行插入,例如编码不需要的功能的基因,例如ptsi基因或tyrr基因。
对于许多微生物特别是大肠杆菌,芳香族氨基酸生物合成途径是熟知的(neidhardt和curtiss,1996)。在野生型细胞中,该途径受反馈抑制和转录阻遏的紧密调控。第一个关键步骤由脱氧-阿拉伯-庚酮糖酸7-磷酸(dahp)合酶催化,对其而言存在由arof、arog和aroh编码的三种同工酶。三种同工酶arof、arog和aroh受芳香族氨基生物合成途径的产物即酪氨酸、苯丙氨酸和色氨酸的反馈抑制。对arof、arog和aroh的反馈抗性突变体是已知的(hu等人,2003;lutke-eversloh和stephanopoulos,2007)。本发明的一个方面涉及利用arof、arog和aroh基因的反馈抗性等位基因以表达抵抗芳香族氨基酸生物合成途径的产物的反馈抑制的arof、arog和aroh酶蛋白。抗反馈抑制的arof,arog和aroh酶蛋白被称为aroffbr,arogfbr和arohfbr。
芳香族途径中涉及的数种操纵子的转录受tyrr基因所编码的阻遏物或trpr基因所编码的阻遏物或二者的调控(neidhardt和curtiss,1996)。特别重要的是当tyrr蛋白与一种或多种芳香族氨基酸结合时其对arog和arof转录的负调节。本发明的一个方面涉及通过从宿主细菌菌株的染色体中消除tyrr或trpr基因而去除这些基因的负调节。
本发明教导了适用于黏康酸生产的生物催化剂中遗传元件的某些组合,例如但不限于,超量产生的反馈抗性的arog、超量产生的反馈抗性的arof、过表达的tkta、过表达的tala、染色体上整合的用于从强组成型启动子表达aroz、aroy和catax(或其类似物或同源物)的表达盒,以及渗漏aroe等位基因(我们将其定义为编码aroe酶的基因,该aroe酶对于芳香族氨基酸和维生素的原养型,但不导致不想要的芳香族化合物的显著分泌)的多种组合。
本文公开的菌株构建的所有具体实例基于野生型大肠杆菌c菌株(atcc8739)或大肠杆菌w菌株(atcc9637)。然而,在这一点上应该认识到,本文公开的遗传元件的表达盒或适当的类似物和同源物可以组装在任何其他合适的微生物中,例如任何其他合适的大肠杆菌菌株和其他种类的细菌,古细菌,酵母,藻类和丝状真菌,其可通过发酵过程用于商业生产黏康酸。
在大肠杆菌中,从葡萄糖的芳香族氨基酸生物合成途径始于磷酸戊糖途径(ppp)的非氧化性分支。非氧化戊糖磷酸途径中的四种关键酶是转酮醇酶,转醛醇酶,核酮糖-5-磷酸差向异构酶和核酮糖-5-磷酸异构酶。这些酶催化导致由己糖或戊糖形成赤藓糖-4-磷酸(e4p)的反应。为了增加e4p在大肠杆菌中的可得性,可以过表达编码转酮醇酶的tkta基因(niu等,2002)。类似地,还预期过表达转醛醇酶基因在一些情况中提高e4p的可得性(bongaerts等人,2001)。在本发明的另一方面,通过导致转酮醇酶和转醛醇酶的酶活性增加的基因操纵来增强转酮醇酶和转醛醇酶基因的表达。在本发明的另一方面,通过超量产生核酮糖-5-磷酸差向异构酶和核酮糖-5-磷酸异构酶来提高经由ppp的非氧化分支的通量。
共同芳香族氨基酸途径中的第一关键步骤和最紧密调节的反应是经dahp合酶(由arog、arof和aroh编码)的磷酸烯醇丙酮酸(pep)和e4p的缩合以产生脱氧阿拉伯-庚酮糖酸7-磷酸(dahp)。大肠杆菌消耗的d-葡萄糖部分通过ppp且部分通过糖酵解被带入芳香族生物合成。当通过用提高拷贝数而使表达增强的质粒转化来扩增转酮醇酶(tkta)和dahp合酶(arog)的同工酶时,进入芳香族途径的葡萄糖流大大提高(niu等人,2002)。在本发明的优选方面,出于扩大转酮醇酶和dahp合酶的酶活性的目的,将外源arog和tkta基因整合到染色体dna中。
在本发明的另一实施方案中,通过增加dahp合成可用的pep,通过减少pep向其他途径的通量,来提高微生物细胞中经由pep的通量。许多属细菌细胞在利用磷酸转移酶系统(pts)的跨细胞膜葡萄糖转运中消耗pep,其中对于每分子跨细菌外膜转运的葡萄糖消耗一个pep分子。通过用非pep依赖性的(pep独立的)葡萄糖摄取机制取代或补偿pep依赖性的pts,可以增加微生物细胞内芳香族氨基酸生物合成途径可用的pep池的大小。例如,用于糖摄取的pts系统可以由基于galp的糖摄取系统或基于glf/glk蛋白的糖转运蛋白系统进行取代或补偿(chandran等人,2003;yi等人,2003)。在本发明的优选方面,除了出于保存微生物细胞内的pep池的目的将用于糖摄取的pts系统缺失外,还出于保存微生物细胞内的atp的目的将基于galp的糖摄取系统失活。在pts系统和基于gal-p的糖摄取系统(δpts/δgalp)的运作中都有缺陷的微生物细胞中,可通过引入编码glf(葡萄糖易化扩散蛋白)的外源基因,或编码glf和glk(葡糖激酶)蛋白二者的外源基因来实现糖摄取。如本发明所用,术语功能性的葡萄糖易化扩散蛋白是指任何glf蛋白以及在功能上等同于glf且作用于通过易化扩散将糖转运到微生物细胞中的任何其它蛋白。在本发明的一个方面,将编码葡萄糖易化蛋白glf的基因引入δpts/δgalp微生物细胞中,且转运到微生物细胞中的葡萄糖通过内源葡萄糖激酶而磷酸化。在本发明的另一方面,将编码glf和glk蛋白二者的基因引入到δpts/δgalp微生物细胞中。在本发明的优选方面,引入到微生物细胞中的外源glf和glk基因被整合到宿主染色体dna中。
在本发明的另一实施方案中,当生长用的碳源和能量需要糖异生(例如如果碳源是乙酸或琥珀酸)时,可通过提高细胞内已存在的羧化酶(例如pep羧化激酶,其由大肠杆菌中的pck编码)的活性或通过引入外源的羧化酶而增加pep池。在优选的实施方案中,引入的编码羧化酶的外源基因稳定整合到宿主染色体中。编码羧化酶的基因可以源自多种多样的微生物种类。编码羧化酶的基因还可经历基因操纵,从而使得用于顺式,顺式-黏康酸生产的生物催化剂内的羧化酶的表达显著增加。
pep是芳香途径的两种主要代谢物之一,并且ppc活性的减少或消除保留了芳香途径的pep和黏康酸产生。ppc催化形成草酰乙酸的添补反应,草酰乙酸是tca循环中的中间体。ppc活性对于野生型大肠杆菌和一些其他生物体在基本培养基中是必不可少的,但在其他培养基中则不存在。一些生物,如缺乏ppc的酵母,利用pyc补充草酰乙酸。ppc活性的降低或缺失可以通过pyc活性,通过提供从丙酮酸代替pep到草酰乙酸的替代途径而补充,这导致pep对于黏康酸生产以及生产其他化合物(例如芳香族化合物)的可得性增加,所述其他化合物的生产需要pep作为中间体。在本文公开的至少一个实例中,用pyc取代ppc可以减少从pep到oaa的通量,这反过来为中心芳香族途径保留了pep。
在本发明的另一实施方案中,通过降低或消除利用pep作为底物的丙酮酸激酶如pyka和pykf的活性而增加微生物细胞内的pep池。
从dahp,芳香族氨基酸途径经由多个中间体至分支酸(cha),其是三种芳香族氨基酸即l-酪氨酸(l-tyr)、l-苯丙氨酸(l-phe)和l-色氨酸(l-trp)的生物合成的分支点。
在共同芳香族氨基酸途径的初始阶段,3-脱氢奎尼酸(dhq)合酶(arob)从dahp移除磷酸基,导致dhq的形成。酶dhq脱水酶(arod)从dhq中移除水分子,导致3-脱氢莽草酸(dhs)的形成,其随后被莽草酸脱氢酶(aroe)还原成莽草酸(shk)。莽草酸激酶i/ii(arok,arol)将莽草酸磷酸化成莽草酸3-磷酸(s3p)。s3p与pep进行缩合,导致5烯醇式丙酮酰莽草酸-3-磷酸(epsp)的形成。epsp的形成由epsp合酶(aroa)介导。epsp的磷酸基由分支酸合酶(aroc)移除,导致分支酸(cha)的形成。
如图2中所示,芳香族氨基酸途径可以因aroe基因突变在3-脱氢莽草酸(dhs)转化成莽草酸(shk)的水平被阻断,导致dhs的积累(niu等人,2002)。引入外源aroz基因起到将dhs转化成原儿茶酸(pca)的作用。pca随后通过由aroy酶介导的脱羧反应转化成儿茶酚。儿茶酚通过cata基因产物的作用最终转化成顺-顺黏康酸(ccmua)。ccmua可以受马来酰乙酰乙酸异构酶的作用而产生反式-反式黏康酸(ttmua)。从dhs到ccmua和/或ttmua的生物合成途径被称为黏康酸途径。负责将dhs转化成ccmua的三种不同基因可以从多种微生物种类获得并引入到选择用于黏康酸生产的微生物如大肠杆菌中。在本发明的优选实施方案中,编码黏康酸途径所涉及蛋白的外源基因被整合到宿主染色体dna中。
在将芳香族氨基酸途径重定向到顺式,顺式-黏康酸生产的过程中,aroe基因突变是关键性的。aroe基因可以完全失活,导致芳香族氨基酸生物合成的完全阻断,如使用描述用于黏康酸生产的大肠杆菌wn1/pwn2.248菌株所进行的(niu等人,2002)。采用wn1/pwn2.248大肠杆菌菌株和相关菌株的重大缺点在于,由于aroe基因完全失活,该菌株成为上述的芳香族酸如苯丙氨酸、酪氨酸和色氨酸以及芳香族维生素或维生素类化合物的营养缺陷型。结果,在其生长以生产顺式,顺式-黏康酸的过程中,该菌株需要外源添加这些化合物(或共同中间体,如莽草酸),由此使得利用该菌株进行的顺式,顺式-黏康酸商业生产的成本实质增加。克服对芳香族氨基酸的外部来源的这种依赖性的新方法是使用具有aroe渗漏突变的菌株。渗漏aroe突变体将允许有限的碳流向莽草酸,同时积累显著量的dhs,其随后可用于经由aroz酶的作用转化成pca。由此,使用aroe的渗漏突变体形式将消除对外源芳香族氨基酸的依赖性,同时仍然将碳流转向顺式,顺式-黏康酸。
用于合成dhs转化成顺式,顺式-黏康酸所必需的aroz、aroy和cata蛋白的编码基因可以源自许多微生物种类的任一种。在一个实施方案中,这些外源基因整合到正在开发的生物催化剂的宿主染色体中。在优选的实施方案中,这些外源基因在生物催化剂内的表达由组成型启动子驱动而不需要任何诱导物。
中间体原儿茶酸的生物合成需要3-脱氢莽草酸脱水酶(aroz;ec4.2.1.118)这种酶。在本说明书中,“aroz”应指任何催化3-脱氢莽草酸脱水酶反应的酶。在现有技术中,该酶从肺炎克雷伯氏菌菌株a170-40(atcc25597)的aroz基因表达(niu等人,2002;draths和frost,1995)。然而,aroz的比活在生物体之间广泛变化,从0.1至261微摩尔/min/mg(wheeler等人,1996;fox等人,2008;pfleger等人,2008),因此,通过从比肺炎克雷伯氏菌具有更高的比活的生物体,例如贝氏不动杆菌、构巢曲霉(wheeler等人,1996)(现在也称为构巢裸胞壳(emericellanidulans))、或粗糙脉孢菌(rutledge,1984;stroman等人,1978)、或鹅柄孢壳菌(podosporaanserina,也称为podosporapauciseta)(hansen等人,2009)表达aroz基因,其也称为asbf(fox等人,2008;pfleger等人,2008)、qutc(wheeler等人,1996)、qa-4(rutledge,1984)和quic,可以具有显著的改进。
作为一个特别的实例,来自粗糙脉孢菌的编码3-脱氢莽草酸脱水酶的qa-4基因的编码序列可以通过数种已知方法中的任一者获得,例如全基因dna合成、cdna克隆或通过基因组dna克隆与pcr或合成dna接头合成的组合。由于qa-4基因中没有内含子,可以通过pcr从基因组dna获得编码区(rutledge,1984)。qa-4酶的蛋白序列(seqidno.4)和原生基因的dna序列(seqidno.5)是已知的。
备选地,可以从构巢曲霉针对3-脱氢莽草酸脱水酶构建表达盒。来自构巢曲霉的qutc酶的编码序列可以通过数种已知方法中的任一者获得,例如全基因dna合成、cdna克隆或通过基因组dna克隆与pcr或合成dna接头合成的组合。qutc的蛋白序列(seqidno.6)和不含内含子的原生基因的dna序列是已知的(seqidno.7;genbank检索号m77665.1)。表达盒可以通过dna合成,或通过基因组克隆与pcr的组合得到,从而使qutc酶能在大肠杆菌中准确产生。通过在大肠杆菌中从强组成型启动子表达qutc的编码序列,可以从整合到染色体中的一个或两个拷贝的基因获得充分表达,省去了可能导致不稳定性的并且如现有技术中所公开的在多拷贝质粒上维持多于两个拷贝的表达盒的需求(niu等人,2002)。上述方法通常可以用于获得编码期望的酶的dna序列,并且该编码序列随后可用于构建设计为在大肠杆菌中或另一合适的微生物宿主生物体中发挥功能的表达盒。
aroz的比活性也可以通过利用现有技术的蛋白序列(niu等人,2002)通过构建改进的表达盒来加以改进,例如,其中在编码区之前已安装更强的启动子和/或核糖体结合位点(rbs),如实施例4中所描述的。
来自肺炎克雷伯氏菌菌株a170-40的编码aroz(3-脱氢莽草酸脱水酶)的aroz基因可以按现有技术中所描述的来获得。该基因的dna序列和周围的dna可以通过领域内熟知的方法确定。可利用原生的dna序列将本发明的异源基因例如aroz建立到表达盒中,或者其可用针对预定宿主生物体密码子优化的序列合成。可以按记载的(draths和frost,1995)从任何其它含有活性aroz基因的微生物克隆aroz基因,所述微生物为例如肺炎克雷伯氏菌菌株342、不动杆菌属物种adp1(贝氏不动杆菌adp1)、苏云金芽孢杆菌、构巢裸胞壳、解淀粉欧文氏菌(erwiniaamylovora)、恶臭假单胞菌w619、粗糙脉孢菌(neurosporacrassa)、构巢曲霉(aspergillusnidulans)和许多其它菌。
中间体儿茶酚的生物合成需要原儿茶酸脱羧酶(aroy;ec4.1.1.63)这种酶。在本说明书中,“aroy”应指任何催化原儿茶酸脱羧酶反应的酶。在现有技术中,该酶从在多拷贝质粒上的肺炎克雷伯氏菌菌株a170-40(atcc25597)的aroy基因表达(niu等人,2002)。然而,再一次地,可以通过从整合到宿主生物体的染色体中的一个或两个拷贝的表达盒产生充足的酶来获得方法改进。这可以通过从以下的生物体获得aroy基因来实现,所述生物体天然产生较之现有技术的肺炎克雷伯氏菌aroy酶具有更高比活的aroy酶,或通过提高肺炎克雷伯氏菌aroy的表达水平而实现,所述提高表达水平是通过构建例如使用强组成型启动子和/或强rbs的表达盒而进行,如以上对实施例4描述的。来自肺炎克雷伯氏菌菌株a170-40的aroy的蛋白序列在seqidno.8中给出。对应的基因aroy可以如上所述进行克隆(draths和frost,1995),或者基于蛋白序列,其可以用针对预定宿主生物体优化的密码子合成。
aroy基因可以从含有同源物或类似物的任何其它微生物获得,例如肺炎克雷伯氏菌菌株nctc418(atcc15380)、肺炎克雷伯氏菌342和arxulaadeninivorans(sietmann等人,2010)。来自肺炎克雷伯氏菌342的aroy基因的dna序列和周围的dna在seqidno.9中给出。
顺式,顺式-黏康酸生物合成的最后步骤需要儿茶酚1,2-双加氧酶(cata;ec1.13.11.1)这种酶。在本说明书中,“cata”应指任何催化儿茶酚1,2-双加氧酶反应的酶。在现有技术中,该酶从多拷贝质粒上的乙酸钙不动杆菌菌株adp1的cata基因表达(niu等人,2002)。来源菌株,乙酸钙不动杆菌菌株adp1显然已重命名为不动杆菌属物种adp1和贝氏不动杆菌adp1(neidle和ornston,1986;barbe等人,2004;deberardinis等人,2008)。在该现有技术实例中,cata基因从ptac启动子表达,该启动子需要乳糖或iptg(异丙基硫代半乳糖苷)作为诱导物。这些化合物对于用于商业发酵而言太过昂贵,因此再一次地,需要通过将表达盒整合到染色体中对方法进行显著改进,既为了消除对昂贵诱导物的需求,也为了产生更稳定的菌株。这可以通过构建利用强组成型启动子、强rbs、和/或更稳定的mrna的cata基因的表达盒而实现,如以上其他实施例中所述。
来自贝氏不动杆菌adp1的cata基因的dna序列和周围序列在seqidno.10中给出。来自相同菌株的cata的蛋白序列在seqidno.11中给出。在优选的实施方案中,cata的表达盒含有一个或两个额外的天然存在于cata下游的开放阅读框,以提高cata基因的表达水平(schirmer和hillen,1998)。许多其它生物体可以是cata基因的来源,例如小田假单胞菌(pseudomonasarvilla)、荧光假单胞菌(pseudomonasfluorescens)(nakazawa等人,1967;kojima等人,1967)、链霉菌属物种菌株2065(streptomycessp.strain2065)(iwagami等人,2000)、钩虫贪铜菌335t(cupriavidusnecator335t)和许多其它菌(perez-pantoja等人,2008)。
为了提高朝向顺式,顺式-黏康酸的碳流,除了通过利用渗漏aroe突变体减小从dhs到莽草酸(shk)的碳流,还有必要阻断从芳香族氨基酸途径分支出的某些其它途径。一些细菌,例如不动杆菌属和假单胞杆菌属中的一些细菌,含有名为poba的基因,其编码将dhs转化成没食子酸的对羟基苯甲酸水解酶这种酶。尽管大肠杆菌中未曾发现poba同源物或类似物,但经改造以生产dhs的大肠杆菌菌株分泌可测量量的没食子酸(li和frost,1999),因此有可能这种酶确实存在于大肠杆菌中。此外,源自dhs的pca可以通过由poba基因所编码的对羟基苯甲酸水解酶(poba)酶的作用转化成没食子酸。由此产生的没食子酸可以在随后被转化成邻苯三酚。阻断所选的生物催化剂中通向没食子酸和邻苯三酚的碳流以提高顺式,顺式-黏康酸的一种方式是通过基因操纵而阻断或减小对羟基苯甲酸水解酶(poba)蛋白的活性。相似地,dhs的前体——dhq也可以受aroe所编码的莽草酸脱氢酶的作用,导致奎宁酸的产生。在本发明的实施方案中,对渗漏aroe突变体酶额外选择或筛选其在将dhq转化成奎宁酸方面的失能或降低的能力。
生产反式,反式-黏康酸代替顺式,顺式-黏康酸有数个优点。在使用乙烯用于生产对苯二甲酸的狄尔斯–阿尔德反应(dielsalderreaction)中对反式,反式-黏康酸的偏好高于顺式,顺式-黏康酸。具有经基因操纵的芳香族途径的生物催化剂产生顺式,顺式-黏康酸,所述顺式,顺式-黏康酸可以在细胞外利用化学转化方法转化成反式,反式-黏康酸。另一方面,通过将马来酰乙酰乙酸异构酶或相似的异构酶引入到生物催化剂中,可以在细菌生物催化剂中将顺式,顺式-黏康酸转化成反式,反式-黏康酸。
在一个实施方案中,本发明提供了增强参与3,4-二羟基苯甲酸(原儿茶酸或pca)向儿茶酚转化的aroy蛋白活性的遗传方法。aroy蛋白的活性已被确定为葡萄糖向黏康酸的生物转化的主要限制和瓶颈(horwitzetal2015;weberetal2012;curranetal2013;sonokietal2014)。aroy属于一类非氧化性脱羧酶,广泛存在于许多细菌中,并利用多种底物(lupa等,2005)。编码非氧化性脱羧酶的这些基因的许多被组织成编码b,c和d型基因的三种基因操纵子。“c”型基因编码脱羧酶如aroy,而b和d型基因的特定功能尚不清楚,尽管它们有时被证明是实现c型脱羧酶的完整活性所必需的(lupaetal2005;jimenezetal2013;linetal2015;sonokietal2014)。weber等(2012)将来自肺炎克雷伯菌或seditntibacterhydroxybenzoicus的b,c和d基因克隆到高拷贝数酵母载体prs4k-hkt7中,并在外部添加pca的培养基中进行发酵以确定存在来自这些基因簇的pca脱羧酶活性;然而,没有任何努力来确定由c基因编码的pca脱羧酶是否依赖于这些基因簇中的b或d基因,也没有任何尝试来确定包含b,c和d基因的这些基因簇的表达是否能够使用非芳香族碳源如葡萄糖来增强黏康酸的生产。
在本文公开的具体实例中使用的编码aroy的基因来自肺炎克雷伯氏菌,但该aroy基因的局部基因结构未显示编码b或d型酶的转录连接的基因。以前的研究表明,当从木质素相关的芳香化合物生产黏康酸时,包含来自肺炎克雷伯菌(来自4-羟基苯甲酸脱羧酶的操纵子的一部分)的另一种b型基因kpdb可以增加aroy活性(sonoki等2014)。然而,木质素是化学品的复杂混合物,其需要昂贵的下游纯化过程以产生纯的产品,例如黏康酸。johnson等(2016)已经表明,在表达来自阴沟肠杆菌(enterobactercloaceae)的aroy(pca脱羧酶)的恶臭假单胞菌菌株中,来自阴沟肠杆菌的与kpdb具有89.3%序列同一性的ecdb蛋白的共表达增加了黏康酸的产生,在54小时发酵结束时使用1.44g/l至4.92g/l的葡萄糖作为碳源;然而,仍然发现这种基于恶臭假单胞菌的系统中的黏康酸产率极低(0.077mol/mol),因此使得这种基于恶臭假单胞菌的系统不适合商业规模应用。因此,仍然需要从廉价、更纯的非芳香族碳源如碳水化合物和其它非芳香族化合物(见上文)中生产黏康酸的改进方法。最近的研究表明,kpdb的同源物ubix产生异戊二烯化的黄素单核苷酸辅因子,并且该辅因子支持ubid在泛醌形成中的脱羧活性(white等人2015;payne等人2015)。尽管ubix及其同源物的先前表征将该蛋白质称为4-羟基-3-聚异戊二烯基苯甲酸脱羧酶,羟基苯甲酸脱羧酶,亚基b蛋白,酚酸脱羧酶或苯基丙烯酸脱羧酶,但这些蛋白质现在被更准确地注释为黄素异戊二烯基转移酶。
黏康酸生产的其他限制可归因于前体代谢物pep和e4p的限制。消除或降低这些代谢物消耗的变化已在本发明中显示出改善产物形成。例如,通过用替代系统(来自运动发酵单胞菌的glf-glk)替换天然葡萄糖转运,即磷酸转移酶系统(pts),已经改善了pep的可得性。大肠杆菌天然使用pts需要利用pep进行葡萄糖的转运和磷酸化。glf-glk系统分别利用易化扩散子(diffuser)和葡糖激酶加atp来运输和磷酸化葡萄糖。已显示使用glf-glk代替pts可提高几种芳香族产物的产率和滴度。pep是多种生物反应的底物。除了pts的葡萄糖转运外,pep是丙酮酸激酶的底物,导致atp和丙酮酸的产生,提供糖酵解中产生的一半atp。已经证明,编码丙酮酸激酶(pyka或pykf)的基因的失活增加了芳香途径中产物的产量(escalante等人,2010)。pep也是通过磷酸烯醇丙酮酸羧化酶(ppc)产生草酰乙酸的底物。在大肠杆菌中,这是必需基因(baba等人,2006),并且ppc缺失的菌株不能在基本培养基上生长。还有其他生物不含磷酸烯醇丙酮酸羧化酶,而是用丙酮酸羧化酶(pyc)从丙酮酸补充草酰乙酸,例如,酿酒酵母。已经研究了在大肠杆菌中添加丙酮酸羧化酶,但仅用于改善琥珀酸生产(lin等人2004;vemuri,eiteman和altman2002),不用于任何芳香族产物。
本专利申请的说明书提供了与构建足以有效生产黏康酸的微生物菌株相关的本发明的数个不同方面。本领域技术人员能够汇编本发明的数个不同方面以构建具有极高效率的用于生产黏康酸的生物催化剂。
实验部分
总论
菌株和接种物制备:本发明中所用的细菌菌株的清单提供于表1和表2中。本发明中所用的质粒的清单提供于表3中。本文公开的菌株构建体的所有具体实例衍生自野生型大肠杆菌c菌株(atcc8739)或大肠杆菌k-12菌株(ymc9或mm294),但本文公开的遗传元件可以组装到任何其它合适的大肠杆菌菌株中,且本文公开的表达盒或遗传元件的适当类似物和同源物可以组装到任何其它的合适微生物中,如可通过发酵方法用于商业生产顺式,顺式-黏康酸的其它种类的细菌、古生菌、酵母、藻类和丝状真菌。
大肠杆菌c能在am1矿物培养基中发酵10%葡萄糖。am1培养基含有2.63g/l(nh4)2hpo4、0.87g/lnh4h2po4、1.5mmmgso4、1.0mm甜菜碱、和1.5ml/l痕量元素。痕量元素制备成1000x储液并且含有以下组分:1.6g/lfecl3、0.2g/lcocl2·6h2o、0.1g/lcucl2、0.2g/lzncl2·4h2o、0.2g/lnamoo4、0.05g/lh3bo3、和0.33g/lmncl2·4h2o。用1.0~10.0mkoh或1.0~9.0m氢氧化铵使发酵液的ph维持在7.0。
发酵:通过从经基因改造的并存储于-80℃冰箱中的大肠杆菌菌株的40%甘油储液在新鲜的nbs-2%葡萄糖(jantama等人,2008a)平板上划线接种而开始发酵。通过在琼脂板和液体培养基中包含合适的(一种或多种)抗生素而保留质粒(如果存在的话)。氨苄青霉素(钠盐)以150mg/l使用,壮观霉素hcl以100mg/l使用,四环素hcl以15mg/l使用,且硫酸卡那霉素以50mg/l使用。在24至48小时(37℃)后,将单克隆挑选到摇瓶中25ml的相同培养基中。在于37℃200rpm震荡直至细胞生长至约1.0的od600后,在冰上冷却培养物并添加等体积的无菌80%甘油。然后将2ml的等分物在-80℃冻存以用作发酵用的接种物。术语“滴度”是指每单位体积发酵液产生的发酵产物的量,术语“产率”是指产生的发酵产物与消耗的碳源的比率(g/g或mol/mol)。
细胞生长:通过使用thermoelectronicspectronic20分光光度计测量550nm(od550)或600nm(od600)下的光密度来估算细胞质量。
对莽草酸途径和黏康酸途径中的中间体的分析:利用购自sigma-aldrich的标准品,通过hplc和watersalliance仪器并监测210nm下的吸光度或45℃下的折射率来测定发酵液中所产生的全部黏康酸(包括顺式,顺式-黏康酸和顺式,反式-黏康酸以及其它生化中间体)。柱子是bioradaminexhpx-87h,以8mm硫酸作为流动相在0.6ml/min的流速下在50℃运行40分钟。购买的标准品(sigma-aldrich)的色谱图示于图4。为准备用于hplc,将发酵样品在0.05m磷酸钾缓冲液(ph7.0)中稀释10或100倍,以防止顺,顺-形式的黏康酸异构化为顺,反-形式。
为分离黏康酸的异构体,将如上制备的样品在第二hplc系统中运行。仪器是agilent1200hplc,柱子是agilenteclipsexdb-c18,4.6x150mm,以用磷酸调节至ph3.0的50mmkh2po4于30%甲醇中的流动相在30摄氏度运行。流速为1ml/min,持续4分钟,进行278nm下的吸光度检测。通过将顺式,顺式-黏康酸溶解于水中并允许其在室温下经历约2小时的自发的酸催化的异构化来产生顺式,反式-黏康酸标准品,直至hplc峰已经完全移动至新位置。其它标准品购自sigma-aldrich。显示标准品的色谱图示于图5中。
用于发酵方法的黏康酸生产培养基的组成:每升的发酵培养基含有50ml/l的1mkh2po4、10ml的200g/l柠檬酸+25g/l柠檬酸铁、1.2ml的98%硫酸和一滴antifoam204。将这些组分与足量的水混合以允许有添加以下其它组分的空间。在高压灭菌后,添加以下组分:10、20、30或40ml的50%葡萄糖(以得到5、10、15或20g/l终浓度)、2ml的1mmgso4、1ml的0.1mcacl2、10ml的1000x痕量元素(jantama等人,2008a)、和如果需要,1、2、4或8ml的50g/l苯丙氨酸+50g/l酪氨酸+50g/l色氨酸(以得到0.5、0.1、0.2或0.4g/l终浓度)、10ml的1g/l对羟基苯甲酸+1g/l对氨基苯甲酸+1g/l2,3-二羟基苯甲酸(最后三种化合物被称为芳香族“维生素”或“维生素类化合物”)和必要时的1ml的150mg/ml氨苄青霉素(钠盐)和/或1ml的100mg/ml壮观霉素hcl。芳香族氨基酸和维生素不是需要的并且不用于表达功能性aroe蛋白的菌株。
对于摇瓶,用nbs盐(jantama等人,2008a)加0.2mmops缓冲液(ph7.4)代替上述的预高压灭菌混合物,但葡萄糖和其它添加剂是相同的。对于补料分批发酵,进料瓶含有600g/l的无水葡萄糖,和如果需要,32ml/l的50g/l苯丙氨酸+50g/l酪氨酸+50g/l色氨酸。使用9mnh4oh作为碱来维持发酵培养基的ph。不需要芳香族氨基酸和维生素,并且其不用于表达功能性aroe蛋白的菌株。
在7lnewbrunswickscientificfermentors中进行补料分批发酵,其ph,do,温度,葡萄糖和进料速率由dcu控制器或biocommand软件控制。培养基是50mmk2hpo4,20mmk2so4,3mmmgso4和微量元素。微量元素以50x母液制备,含有下列组分:1.6g/lfecl3,0.1g/lcucl2,0.2g/lzncl2·4h2o,0.2g/lnamoo4,0.05g/lh3bo3,和0.55g/lmncl2·4h2o。将温度保持在37℃,用9n铵水将ph维持在7.0。通气量为0.5vvm并且通过将叶轮的速度从750rpm自动增加到1200rpm,溶解氧(do)保持在30%。培养基中的初始葡萄糖浓度为约20至25g/l。当浓度降至5g/l以下时,将进料葡萄糖溶液加入发酵罐中。初始葡萄糖进料速率为4g/l/hr,并且到48小时时升至7g/l/hr的进料速率,之后将其保持在7g/l/hr。
表达黏康酸途径基因的质粒的构建:将dhs转化成黏康酸所需的三种异源基因单独或组合克隆到低拷贝质粒pcl1921中(lerner和inouye,1990)。pcl1921的dna序列在表7的seqidno.20中给出。简言之,catax、aroy和aroz类似物或同源物的编码序列经密码子优化用于在大肠杆菌中表达并商业合成(geneart,invitrogen)。然后利用携带独特的核糖体结合位点的正向引物和携带各基因独特的终止子序列的反向引物将这些序列经pcr扩增。将所得的pcr片段经限制性酶消化并通过标准分子克隆方法克隆到独特的组成型启动子序列的下游。通过从之前描述过的来源dna序列进行pcr扩增(美国专利申请20090191610;美国专利7244593),之后是限制性消化和标准分子克隆,来克隆启动子序列。启动子-rbs-编码序列-终止子序列在一起组成表达盒。接着将单独的表达盒组合以产生表达一个、两个或所有三个黏康酸途径基因的质粒。
使用以下实施例进一步说明本发明;然而,本文中单独或以其任何组合提供的实施例不应被解释为限制本发明的范围或实施方案。最后提供的权利要求限定了本发明的范围。本领域技术人员可以清楚地理解权利要求所限定的本发明的范围。在不脱离本发明的精神和范围的情况下,本领域技术人员可以对本发明提供的技术方案进行修改或改变。
实施例1
增加arog和arof基因的表达
大肠杆菌的tyrr基因可通过数种熟知方法中的任一种来突变,例如化学或辐射诱变和筛选(例如通过pcr和dna测序)或选择类似物抗性(例如,对4-氟酪氨酸的抗性)、转座子诱变、细菌噬菌体mu诱变或转化。在优选的实施方案中,tyrr基因中的突变是无效突变(导致没有可检测的活性的突变),而在更优选的实施方案中,至少部分tyrr基因缺失。这可以通过例如利用使用线性dna分子的两步转化法(jantama等人,2008a;jantama等人,2008b)来实现。在第一步中,通过双重组和选择氯霉素抗性将camr、sacb盒在tyrr基因座处整合以取代大部分的或所有的tyrr开放阅读框。在第二步中,通过双重组、在富培养基如lb中选择对5%蔗糖的抗性将包括缺失型的tyrr基因的线性dna整合。通过诊断性聚合酶链式反应(pcr)来鉴定和确定正确的缺失。将tyrr缺失的目的是增加arog和arof的表达。实现相似结果的备选方法是用强组成型启动子取代arog和/或arof之前的原生启动子和(如果必要的话),添加转录终止子。关于总体如何实现的更多细节在以下实施例4中给出。
以上描述的用于克服tyrr蛋白对arog和arof活性的抑制的两种途径中后者是优选的,因为tyrr缺失会引起基因如arolm的不希望的过表达(neidhardt和curtiss,1996)。关于总体如何实现的更多细节在以下实施例4中给出。
实施例2
反馈抗性的arog和arof
导致反馈抗性的arog酶(3-脱氧-d-阿拉伯庚酮糖酸-7-磷酸合酶或dahps)的arog基因中的突变是本领域内已知的(shumilin等人,1999;kikuchi等人,1997;shumilin等人,2002)。还已知的是产生、鉴定和表征这些突变的方法(ger等人,1994,hu等人,2003)。优选的突变是引起对苯丙氨酸抑制完全抗性的突变。可以通过数种已知方法中的任一种将任何已知的已公开的反馈抗性突变引入到染色体中或质粒上所包含的arog基因中,其中一个方法实例是诱变pcr,其中期望的突变作为pcr引发寡核苷酸的部分被合成(hu等人,2003)。通过dna测序来证实突变的正确置入。来自大肠杆菌c的野生型arog基因的序列在seqidno.18中给出。优选的突变是将arog的氨基酸150从脯氨酸变成亮氨酸的点突变,例如通过将密码子150从cca变成cta(hu等人,2003)。在更优选的实施方案中,密码子150从cca变成ctg,其是大肠杆菌中的优选密码子。该特别的arog等位基因是优选的,因为所编码的dahp合酶对高达3mm的苯丙氨酸抑制完全抵抗,并且其具有与野生型酶相似的比活性(hu等人,2003)。
可以通过诱变和选择对一种或多种苯丙氨酸类似物(如β-2-噻吩丙氨酸、p-氟代苯丙氨酸、p-氯代苯丙氨酸、o-氟代苯丙氨酸和o-氯代苯丙氨酸)的抗性、接着证明导致抗性的突变与arog基因关联,来得到另外的反馈抗性arog等位基因(ger等人,1994;us专利4,681,852)。与arog的关联可以通过dna测序或苯丙氨酸存在和不存在情况下的酶测定来直接证明(ger等人,1994),或通过噬菌体介导的转导和对arog基因座处或其附近的可供阳性或阴性选择的遗传标记物的选择而间接证明(us专利4,681,852)。这样的遗传标记物可以是arog基因自身中的缺失或点突变,或者是任何合适的紧密关联的基因(如大肠杆菌情况下的nada)中的突变。例如,在大肠杆菌中,在诱变和选择苯丙氨酸类似物抗性后,单个突变体或突变体集合可被用作供体用于经p1介导转导至缺失所有三种dahp合酶基因arog、arof和aroh的原生受体中,并选择在合适的基本培养基上的生长。然后转导子将富集(一个或多个)期望的基因中的突变。备选地,在诱变和选择类似物抗性后,单个突变体或突变体集合可被用作供体用于经p1介导转导至包含nada基因中的无效突变的原生受体菌株中,再次选择在缺乏烟酰胺的合适的基本培养基上的生长。另一方法是在这样的菌株背景中选择抗性突变体,所述菌株背景在arog基因附近(如在nada基因中)含有转座子(如tn10)插入。从类似物抗性突变体p1转导到不包含所述转座子的菌株背景中和选择四环素或其它合适的抗生素抗性将富集期望的arog突变。在所有这些方法中,反馈抗性最终通过酶测定和对基因的dna测序而得以证实。我们将对反馈抑制具有抗性的arog的等位基因称为arog*。
菌株wm191(δtyrr,δarof)衍生自ymc9(atcc33927)。利用两步基因取代法(jantama等人,2008a)置入tyrr和arof的完全缺失(cleandeletion),以得到菌株wm191。接着,从cag12147(cgsc7351,coligeneticstockcenter,yaleuniversity)转导入nada::tn10等位基因,以得到菌株wm189(δtyrr,δarof,nada::tn10)。在lb+四环素hcl(15mg/l)上进行选择。菌株ry890(δtyrr::kan,arof363)通过p1转导以三步从mm294(atcc33625)得到。供体菌株依次为jw1316-1(cgsc9179,coligeneticstockcenter,yaleuniversity)、nk6024(cgsc6178,coligeneticstockcenter,yaleuniversity)和ab3257(cgsc3257,coligeneticstockcenter,yaleuniversity),并且三次选择依次为lb+硫酸卡那霉素(50mg/l)、lb+盐酸四环素(15mg/l)和nbs最低限葡萄糖(jantama等人,2008a)及硫胺素hcl(5mg/l)。
用紫外线对wm189诱变至约20%存活,并铺板于含有o-氟代苯丙氨酸(1mm)、硫胺素(5mg/l)和烟酰胺(1mm)的nbs最低限葡萄糖培养基中(jantama等人,2008a)。将从数个板各自得到的菌落收集到单独的池中,并在各个池上制备p1vir裂解物。使用这些裂解物在lb培养基上将wm191转导成四环素抗性(15mg/l),并将得到的菌落复制铺板到含有1mmo-氟代苯丙氨酸、硫胺素(5mg/l)和烟酰胺(1mm)的nbs最低限葡萄糖培养基上。假定在四环素和类似物上均存活的菌落复制物在arog中含有反馈抗性突变。从5个独立的池中选择八个单独的菌落用于dna测序。通过聚合酶链式反应将arog编码区扩增并测序。示于图4的结果显示,八个菌株各自在其arog基因中包含点突变。一些等位基因与已公开的等位基因相同,但一些是新的。
使用来自以上描述的池之一的p1vir裂解物将ry890(其具有arog野生型等位基因)转导成四环素抗性并通过如上所述的复制铺板转导成对o-氟代苯丙氨酸(0.3mm)抗性。挑取四个克隆,命名为ry893、ry897、ry899和ry901,用于dna测序(表4),再次地,两个等位基因与已公开的等位基因相同,但两个是新的。与后四个菌株同基因但却含有野生型arog基因的菌株ry890通过从cag12147转导被构建为对照。这五株菌在摇瓶中在25mlnbs最低限葡萄糖(15g/l)+硫胺素hcl(5mg/l)和烟酰胺(1mm)中过夜生长。所得的细胞通过离心收集,重悬以用10ml水漂洗,再离心,并重悬于0.5ml的50mm磷酸钾(ph7.0)中。重悬的细胞通过与三滴氯仿一起涡旋而裂解,并使用与文献中所描述的方法类似的方法测定粗裂解物的dahp合酶活性(hu等人,2003),有以下改动。磷酸盐缓冲液是50mm(终浓度),ph7.0,最终的赤藓糖-4-磷酸浓度为2mm,最终的磷酸烯醇丙酮酸浓度为5mm,温育温度为30℃,反应在10分钟时终止。我们将1mu定义为每分钟每毫克蛋白产生1nmdahp的活性。为测试反馈抗性,在有或没有终浓度18mm的苯丙氨酸的存在下对各个粗裂解物进行测定。测定结果示于表5。酶显示出不同的比活性和对苯丙氨酸的抗性,但所有选择的进行测试的突变体形式比野生型对照具有显著更高抗性。
将来自如上所述的ry893、ry899、ry901和ry902的arog等位基因按如下引入黏康酸生产菌株背景中。将来自arog*和arogwt供体菌株的p1vir裂解物用于将myr219(大肠杆菌c,δaroe,δack::p15-arob,pmg37)转导成四环素hcl抗性(15mg/l),以分别得到新菌株ry903、ry909、ry911和ry912。然后使用jw1316-1的p1vir裂解物将这些菌株各自转导成硫酸卡那霉素抗性(50mg/l),以引入δtyrr::kan等位基因,分别得到菌株ry913、ry919、ry921和ry922。始终维持壮观霉素选择以维持黏康酸质粒。所得的四个菌株在37℃在摇瓶中在含有20g/l葡萄糖、0.2mmops缓冲液(ph7.4)、烟酰胺(1mm)、苯丙氨酸(100mg/l)、酪氨酸(100mg/l)、色氨酸(100mg/l)、对羟基苯甲酸(1mg/l)、对氨基苯甲酸(1mg/l)、2,3-二羟基苯甲酸(1mg/l)、酚红(10mg/l)和硫酸铵(1g/l)的补充物的25mlnbs基本培养基(jantama等人,2008a)中生长48小时。通过针对ph7.0标准品目视酚红颜色估计,通过按要求对摇瓶人工添加1ml的1.0mkoh等分物,将ph保持在接近7。产生的黏康酸如上所述通过hplc进行检测,结果显示在表6中。所有三个含有反馈抗性的arog*等位基因的菌株较之含有野生型arog等位基因的同基因菌株产生更多的黏康酸。在本文公开的单独实验中,在多拷贝质粒pcp32amp上含有arog的菌株myr205在摇瓶中产生1.5g/l黏康酸。因此,发明人已经显示,较之含有同基因的arog质粒的菌株,δtyrr和单拷贝染色体arog*的组合可以表现良好地在摇瓶中产生黏康酸。染色体等位基因相较于质粒等位基因的固有的优异遗传稳定性,加之减弱了对于为了维持质粒的选择性培养基的需求,使得本文描述的新菌株更加适合于大规模商业发酵。而且,不需要化学诱导物用于黏康酸途径基因的表达。因此,上述的本发明菌株较之现有技术(niu等人,2002)的菌株而言有改进,所述现有技术菌株全部在不理想的多拷贝质粒上包含用于过表达dahp合酶的基因。
按与以上针对arog描述内容相似的方式,可以在质粒上或在染色体中置入产生抵抗由酪氨酸的反馈抑制的arof或aroh同工酶的突变。优选的突变是将arof的氨基酸148从脯氨酸变为亮氨酸的点突变,例如通过将密码子148从ccg变为ctg(weaver等人,1990),得到命名为arof*的基因。可以按与以上针对arog*等位基因描述内容类似的方式通过对酪氨酸类似物(例如o-氟代酪氨酸、m-氟代酪氨酸、p-氟代苯丙氨酸等)的抗性来分离其它arof*等位基因。可以通过与转座子或卡那霉素抗性插入关联而选择、富集以及转导arof*等位基因,例如,在诸如jw2584(cgsc10051,coligeneticstockcenter,yaleuniversity)的菌株中在紧密连接的δyfir::kan中。
实施例3
从染色体dna缺失aroe和黏康酸生产
在该实施例中研究了过表达多拷贝质粒上的arob和arog以及表达在黏康酸途径中有功能的蛋白的编码基因的作用。在编码莽草酸脱氢酶的aroe基因中含有缺失的菌株myr34被用作这些研究中的亲本菌株。以与上述实施例1中所描述内容相似的方式完成aroe的染色体拷贝的缺失。当用过表达编码在莽草酸途径中有功能的dahp合酶蛋白的arog基因的质粒pcp32amp转化myr34时,dhs的积累有显著提高。当用从组成型启动子表达arob基因的质粒转化myr34时,没有注意到dhs积累的显著提高。然而,当用表达arob和arog基因二者的质粒转化大肠杆菌菌株myr34时,相比使用仅转化有arog的myr34时所观察的情况,dhs的积累有增加,因此表明arob是dhs生产中的第二瓶颈(图6)。
在图7呈现的实验中,检视了整合到宿主染色体dna中的arob基因的额外的拷贝的效果。在衍生自myr34的大肠杆菌菌株myr170中,将处于p15启动子控制下的arob基因的额外拷贝在ack基因座处整合到宿主染色体中。当用pcp32amp质粒转化myr170菌株时,与用相同质粒转化myr34菌株时所检测到的dhs积累相比,dhs积累有轻微的增加。myr170中dhs积累的轻微增加可以归因于整合到宿主染色体dna中的arob基因的额外拷贝。当用表达arob和arog基因二者的pcp54转化myr170菌株时,dhs积累有进一步的增加,意味着arob是dhs生产中的第二瓶颈。
图8提供了使用大肠杆菌菌株myr34和myr170的黏康酸生产结果。在建立了在aroe缺失菌株myr34和myr170中在arob和arog基因的过表达下有dhs积累后,工作致力于观察编码在黏康酸生产途径中有功能的蛋白的“黏康酸途径”基因的表达是否会导致dhs转变成顺式,顺式-黏康酸。在这些实验中,单独用质粒pmg37或用质粒pmg37和pcp32amp二者转化大肠杆菌菌株myr34和myr170。质粒pmg37表达编码在黏康酸途径中有功能的蛋白的aroz、aroy和catax基因。当用质粒pmg37和pcp32amp二者转化这些细菌菌株时,与仅用pmg37质粒转化的这两种菌株中的黏康酸产量相比,myr34和myr170二者中的黏康酸产量增加,这提示在这些菌株中,arob表达是顺式,顺式-黏康酸生产的瓶颈。
实施例4
tkta的过表达
由tkta编码的转酮醇酶是戊糖磷酸途径中的关键酶,并被认为对于黏康酸生产中的关键中间体之一——赤藓糖-4-磷酸的生产是限制性的。已知通过将基因及其原生启动子置于多拷贝质粒上来过表达编码转酮醇酶的tkta(sprenger等人,1995,1995a)能提高进入芳香族途径中的流量(draths等人,1992)。然而,这样的质粒不稳定,并且往往需要抗生素选择以维持。现有技术中的另一方法是向宿主菌株的染色体中添加tkta基因的一个额外拷贝(niu等人,1992)。然而,tkta的一个额外拷贝及其原生启动子并不足以用赤藓糖-4-磷酸饱和芳香族途径,因为其原生启动子并非完全接近于理想。这样,该方法需要实质的改进。
可以例如,通过用强组成型启动子例如来自枯草芽孢杆菌噬菌体spo1的p15或p26启动子(分别为seqidno.1和seqidno.2),或来自细菌噬菌体λ的pr启动子(seqidno.3)来替代染色体中的原生tkta启动子,来获得改进的tkta过表达。这通过如实施例1中描述的两步而实现,不同在于在第一步骤中使用camr,sacb盒来取代原生染色体tkta启动子。在第二步中,通过转化线性dna并进行蔗糖抗性选择而置入强组成型启动子,所述线性dna包含强组成型启动子,其侧翼是在强组成型启动子下游侧的tkta编码区5’端的至少50个碱基和在强组成型启动子上游侧的就在原生tkta启动子上游的同源的至少50个碱基对。自这样的表达盒的改进的表达还通过增加从表达盒转录的mrna的稳定性而实现。mrna稳定性的提高通过在mrna的5’端、mrna的3’端或两端增加茎环结构而实现。茎-环结构往往存在于mrna的末端,其由rho-非依赖性的转录终止子自然终止,但如果不是这样,则可以通过遗传工程的公知方法(连接、pcr等)向dna序列中添加rho-非依赖性的转录终止子。这样的终止子可以由在各重复序列中的4至20个碱基的反向重复序列组成,所述反向重复序列由3个或更多个碱基的“环”隔开,后接富含t(脱氧胸苷)的一个或多个碱基的区域。反向重复序列富含g和c(脱氧鸟苷和脱氧胞苷)。类似地,可以通过就在转录起始点的下游但在核糖体结合位点之前插入含有如上所述的茎-环但没有t-富集的区的dna序列,将茎-环构建到mrna的5’端中。对于此的实例结合p15启动子(seqidno.1)给出。
在过表达tkta基因对经由莽草酸途径的碳流的影响的分析中,大肠杆菌菌株myr170被用作亲本菌株。myr170在编码莽草酸脱氢酶的aroe基因中有缺失并在ack基因座处具有arob基因的额外拷贝。
在图9、10和11所描绘的实验中,使用两种不同质粒,即pcp32amp和pcp50。质粒pcp32amp仅从其原生启动子表达dahp合酶arog基因,质粒pcp50从其原生启动子表达转酮醇酶基因tkta连同arog基因。用pcp32amp和pcp50质粒单独转化具有aroe缺失和在染色体dna的ack基因座处整合的在p15启动子控制下的arob基因的额外拷贝的myr170。如图8所示,与仅表达arog基因的大肠杆菌细胞相比,arog基因连同tkta基因的表达使dhs积累进一步增加。
图10提供了用质粒pcp32amp或pcp50转化的两种不同菌株即myr34、myr170中的dhs产率的数据。具有aroe基因缺失的myr34菌株每消耗1克葡萄糖产生0.1克dhs。当用具有arog基因过表达的pcp32amp质粒转化myr34菌株时,该菌株中的dhs产率增至每消耗1克葡萄糖产生0.15克dhs。myr170具有在ack基因座处插入的arob基因的额外拷贝。由于存在arob基因的该额外拷贝,用pcp32amp转化的myr170菌株中的dhs生产的产率稍高于用pcp32amp转化的myr34菌株中所观察到的dhs产率。因此,myr170中arob的额外拷贝的存在引起增加的经由莽草酸途径的碳流。当用表达arog和tkta基因二者的质粒pcp50转化myr170菌株时,观察到dhs产率的进一步提高。因此,tkta的额外拷贝的存在导致经由莽草酸途径的碳流的增加。更具体而言,存在额外的arob和tkta基因的作用引起对dhs产率的累加效应。
图11中描绘的实验中所使用的myr261被改造为在poxb基因座处将tkta基因的额外拷贝整合到myr170的染色体dna中。myr261菌株中期望的基因取代(poxb::tkta)通过pcr验证。用pcp32amp(arog过表达)质粒或pcp50(arog和tkta过表达)质粒转化myr261。作为对照,用pcp32amp质粒转化myr170。如图11所示的结果所显示的,与在用相同质粒转化的myr170菌株中所观察到的dhs生产的滴度相比,myr261的染色体dna中tkta基因的额外拷贝的存在增加了pcp32amp质粒的dhs生产滴度。当用过表达转酮醇酶的质粒pcp50转化myr26菌株时,myr261菌株中转酮醇酶水平的进一步增加导致dhs生产滴度的进一步提高。由poxb编码的酶poxb,或丙酮酸氧化酶,产生乙酸盐作为反应产物。这样,如本文描述的因tkta插入引起的poxb的缺失去除了乙酸生产的潜在活性途径。类似地,同时插入p15arob并缺失编码acka或乙酸激酶的acka(如在以下实施例12中所描述的)去除了通向乙酸的另一潜在活性途径。乙酸的生产在发酵中通常是不合人意的(jantama等人,2008b)。这样,这些缺失可以用于减少乙酸生产。
图12提供了在用质粒pcp32amp和pmg37转化后大肠杆菌myr170、myr261和myr305菌株中的黏康酸和乙酸生产的滴度。通过从染色体dna中缺失poxb基因从myr170得到myr305,而myr261是myr170衍生菌,其中poxb基因已经通过插入tkta基因的额外拷贝而失活。如上所述,质粒pcp32amp表达编码在莽草酸生物合成途径中起作用的dahp合酶蛋白的arog基因,由于大肠杆菌菌株myr170、myr261和myr305中aroe基因的缺失导致dhs的积累。由于质粒pmg37上黏康酸途径基因即aroz、aroy和catax的表达,dhs被转化成顺式,顺式-黏康酸,如图2所示。在myr261菌株中arob基因和tkta基因的额外拷贝的存在下,黏康酸生产稍有增加,伴随着乙酸积累的降低。
实施例5
tala或talb的过表达
talb基因编码大肠杆菌中主要的转醛醇酶,但tala基因也编码小部分的转醛醇酶。已知转醛醇酶的过表达提高进入芳香族途径的流量(lu和liao,1997;sprenger,1995;sprenger等人,1995b)。在现有技术中,这是通过在多拷贝质粒上从原生启动子过表达tal基因(现在称为talb基因)而实现的(lu等人,1997,sprenger等人,1995b)。然而,这样的质粒并不稳定,并且需要抗生素筛选以维持。因此,需要有改进的方法。可以例如通过用强组成型启动子例如来自枯草芽孢杆菌噬菌体spo1的p15或p26启动子(分别为seqidno.1和seqidno.2),或来自细菌噬菌体λ的pr启动子(seqidno.3)来替代染色体中的原生talb启动子来获得改进的talb表达。这通过如实施例1中描述的两步而实现,不同在于在第一步骤中使用camr,sacb盒来取代原生染色体talb启动子。在第二步中,通过用线性dna转化并进行蔗糖抗性选择而置入强组成型启动子,所述线性dna包含强组成型启动子,其侧翼是在强组成型启动子下游侧的talb编码区5’端的至少50个碱基和在强组成型启动子上游侧的就在原生talb启动子上游的同源的至少50个碱基对。也可以通过相似方法来过表达tala基因,但优选过表达talb基因,因为其编码主要活性(sprenger,1995;sprenger等人,1995b)。对于有关设计为用于过表达的表达盒的构建的更多细节,参见实施例4。
实施例6
aroz、aroy和catax基因的表达
为证实大肠杆菌所产生的内源性dhs转化成黏康酸,将来自不动杆菌属物种adp1的异源基因catax、来自肺炎克雷伯氏菌的aroy、和来自不动杆菌属物种adp1的quic克隆到低拷贝质粒pcl1921中强组成型启动子(分别为p15、pr和p26)下(lerner和inouye,1990),以生成“黏康酸质粒”pmg37。携带空载体(pcl1921)或pmg37的myr34衍生菌在含有2%葡萄糖的摇瓶培养基(补充有芳香族氨基酸和维生素的nbs基本培养基)中在37℃生长17小时。收集上清液并通过hplc分析。与显示dhs积累的myr34/pcl1921相比,myr34/pmg37显示黏康酸的产生(图13)。从后一菌株中没有检测到显著量的dhs或中间体产物如pca和儿茶酚,提示从pmg37表达的异源基因是有功能并且足够的。
实施例7
aroz同源物的比较
比较三种不同的aroz同源物和类似物(图14)将dhs转入黏康酸生产途径的能力。来自不动杆菌属物种adp1的quic、来自苏云金芽孢杆菌的asbf和来自粗糙脉孢菌的qa-4被报告为编码具有aroz样活性的蛋白(elsemore和ornston,1995;fox等人,1995;rutledge,1984)。这些基因各自经密码子优化以用于在大肠杆菌中表达且通过geneart(invitrogen)合成,并克隆到还分别从p15和pr启动子表达catax和aroy基因的低拷贝“黏康酸质粒”中强组成型p26启动子下。myr34/pcl1921、myr34/pmg37(以quic作为aroz的黏康酸质粒)、myr34/pmg47(以qa-4作为aroz的黏康酸质粒)和myr34/pmg70(以asbf作为aroz的黏康酸质粒)在具有含2%葡萄糖、芳香族氨基酸和芳香族维生素的基本培养基的摇瓶中在37℃生长48小时。收集上清液并通过hplc分析。如所预期的,转化空载体的myr34积累dhs且不产生黏康酸。检查的两种aroz同源物和一种类似物在将dhs转向黏康酸生产中是有功能的,但程度不同。表达quic基因的myr34衍生菌最为强烈,显示近乎100%的dhs向黏康酸的转化和微乎其微的dhs存留量。表达真菌aroz同源物qa-4的myr34衍生菌以约80%的dhs向黏康酸的转化和20%的dhs存留紧接其后。最后,表达asbf基因的myr34衍生菌显示仅50%的dhs向黏康酸的转化和50%的dhs存留。汇总而言,在我们的摇瓶测定条件下,与其它aroz同源物相比,quic基因的表达和/或活性看起来是最高的。
实施例8
catax、aroy和quic的染色体整合
可以通过含有仅在adhe基因座处染色体整合的从组成型启动子表达的cata-x、aroy和quic的单拷贝的菌株来生产黏康酸。
高dhs生产菌——myr170(δaroe,δack::p15-arob),是用于在染色体中adhe基因座处整合黏康酸途径基因的宿主菌株(seqidno.41)。对所得的菌株myr352转化质粒yep24(中拷贝,空载体)、pcp32amp(中拷贝,arog从原生启动子表达)或pcp50(中拷贝,arog和tkta从其各自的原生启动子表达),以生成衍生菌株。后两种质粒用于增加dhs产量。菌株在37℃在如上所述的含2%葡萄糖的摇瓶培养基中生长72小时。在72小时收集上清液并通过hplc分析。如所预期的,与空载体对照相比,转化有arog和arog/tkta的myr352衍生菌显示出总产物形成的整体提高(图15)。所有的myr352转化体生产可测量滴度的黏康酸,首次证明黏康酸可以由仅含有整合的“黏康酸途径”基因的菌株生产且不需要补料基因表达的化学诱导物。
任何这些myr352衍生菌中产生的dhs并非全被转化成终产物黏康酸。相反,有显著量的儿茶酚积累(图15),提示当catax在染色体上从单拷贝表达时,其表达或活性是限制性的。由于主要的积累中间体是儿茶酚,可能在用于黏康酸合成的myr352菌株背景中quic和aroy基因表达和/或活性是充足的。
将myr352菌株衍生物与类似的myr219菌株衍生物平行比较。myr219菌株与myr170菌株相同,但含有表达黏康酸途径基因的低拷贝质粒pmg37。因此,myr352与myr219菌株的主要区别涉及到黏康酸途径基因的剂量(分别为1个拷贝vs.约5个拷贝)。与myr352衍生菌株相反,myr219衍生菌株显示出极少的儿茶酚或其它中间体的积累,并成功地生产终产物黏康酸。总的来说,这些结果表明需要提高菌株例如myr352中的catax活性。
实施例9
catax的表达
myr352菌株中儿茶酚的积累和不足的黏康酸生产是由于catax基因产物的有限剂量和/或活性。如上所述,myr352含有δaroe,δack::p15-arob和强组成型启动子下的染色体整合的catax、aroy和quic基因的单拷贝。用中拷贝的空载体对照(yep24)或arog/tkta表达质粒(pcp50)转化该菌株以增加进入芳香族氨基酸合成途径的碳流并生产大量的dhs。转化的菌株在如上所述的补充有2%葡萄糖的摇瓶培养基中在37℃生长72小时,导致儿茶酚中间体的积累。该结果提示catax活性在myr352中可能不足。为验证该假设,测试一种或多种从低拷贝质粒表达的黏康酸途径基因减弱myr352/pcp50中儿茶酚积累的能力(图16)。具体而言,还用低拷贝的空载体对照(pcl1921)或表达黏康酸生产途径的所有三种基因、两种基因或一种基因的质粒转化myr352/pcp50。在如上所述的摇瓶实验中测定衍生菌株。尽管单独增加aroy(来自pmg27)或单独增加quic(来自pmg39)的剂量并未减少儿茶酚积累,但所有的黏康酸途径基因(来自pmg37)的表达或catax和aroy(来自pmg33)共同的表达导致成功地将儿茶酚转化成黏康酸。此外,单独表达catax(来自pmg31)足以生产黏康酸和防止儿茶酚积累。
实施例10
构建渗漏aroe突变
在用于生产顺式,顺式-黏康酸的现有技术方法中,宿主菌株含有名为aroe353的aroe基因突变,其是无效突变。结果,该菌株需要补料从莽草酸途径制备的芳香族氨基酸(苯丙氨酸、酪氨酸和色氨酸)和芳香族维生素(对羟基苯甲酸、对氨基苯甲酸,和2,3-二羟基苯甲酸)。在商业上有吸引力的方法中补料芳香族氨基酸太过昂贵。这样,现有技术方法需要实质的改进。这可以通过置入渗漏型aroe基因(我们会将其称为aroe*)而实现。渗漏突变通过首先产生使aroe编码序列中的一个氨基酸改变引起无效表型的错义突变而获得。这可以通过任何形式的诱变和筛选对于以上列出的六种芳香族化合物的同时营养缺陷型而实现。优选方法是利用taqdna聚合酶,使用野生型大肠杆菌c基因组dna作为模板,并使用与aroe编码区上游的约1000个碱基对和下游的1000个碱基对杂交的pcr寡核苷酸引物,通过易错pcr诱变来产生突变体aroe基因池。所得的线性dna分子池用于转化产生顺式,顺式-黏康酸并且含有已经取代aroe编码区的整合的camr,sacb盒(对于相关实例参见实施例4)的大肠杆菌c衍生菌,并进行蔗糖抗性选择。然后对转化体筛选已经失去氯霉素抗性并且需要以上列出的六种芳香族化合物的营养缺陷型。挑取数个独立的营养缺陷型并通过在没有六种芳香族化合物的基本葡萄糖板上铺板约107、108、或109细胞(在基本葡萄糖培养基中漂洗)来测试可复原力(revertability)。挑取在板上产生菌落的回复体并进行顺式,顺式-黏康酸产生但没有实质性水平的芳香族氨基酸产生的测试。在这些回复体中将有这样的菌株,所述菌株携带aroe基因中的一个或多个突变,从而使aroe酶提供充足的芳香族氨基酸和维生素用于生长、但又没有这些芳香族化合物的剩余。获得渗漏aroe突变体的另一方法是向顺式,顺式-黏康酸生产菌株中置入经典的可回复的aroe突变体如aroe353和aroe24(均可从yaleuniversity的coligeneticstockcenter,newhaven,ct,usa得到)之一,并如上所述选择回复体。
实施例11
通过易化扩散的葡萄糖输入
芳香族途径的第一个关键步骤中的底物之一是磷酸烯醇丙酮酸(pep)。pep也是用于通过细菌磷酸转移酶系统(pts)输入葡萄糖和一些其它糖类的磷酸盐和能量的来源。因此,当细菌在pts-依赖性的糖上生长时,在pts和芳香族途径之间存在对pep的竞争。这样,可以通过缺失pts并提供可选的糖摄取途径来实现增加芳香族途径流量的显著改进。该问题的一个解决方案是用大肠杆菌galp通透酶取代pts,大肠杆菌galp通透酶是对于葡萄糖摄取起相当好作用的质子同向转运蛋白(us专利6,692,794)。然而,质子同向转运蛋白仍然耗用能量以维持驱动通透酶所必需的质子梯度。这样,对于方法的进一步改进存在需求。
一些糖,例如木糖,可以由从atp(三磷酸腺苷)的水解获得能量的转运蛋白运输。再一次地,如果可以用需要较少能量的转运蛋白来取代能量依赖性的转运蛋白,那么可以获得改进,因为atp中固有的能量得以保存用于其它有益用途。
可以通过利用在糖输入中不耗用能量的易化扩散转运蛋白来获得显著改进(parker等人,1995;snoep等人,1994)。例如,由glf基因编码的来自运动发酵单孢菌(zymomonasmobilis)的葡萄糖易化蛋白可以用于代替或额外加至3-脱氢莽草酸(dhs)生产菌株中的pts(yi等人,2003)。然而,这些菌株仍然至少部分依赖于galp进行葡萄糖输入。由于galp需要质子梯度形式的能量用于葡萄糖输入,有需求提高黏康酸生产菌株的葡萄糖输入效率。
用于glf加上葡糖激酶基因glk(也来自运动发酵单孢菌)的表达的盒可以与强组成型启动子如p26组装。然后可以将该盒在不妨碍期望的化合物(在这种情况下为顺式,顺式-黏康酸)的生产的位置处整合到宿主菌株的基因组中。大肠杆菌染色体中这样的位置的实例是苏氨酸降解操纵子,tdcabcdefg。如果生长培养基不含苏氨酸,则不需要操纵子或该操纵子不表达,由此在该操纵子中插入表达盒不会妨碍代谢。
为实现上述的改进,利用与实施例1中所公开内容相似的方法将一个或多个编码pts功能的基因缺失。例如可以缺失ptsh、ptsi、crr或ptsg中的一个或多个。接着使用美国专利号8,871,489中描述的方法缺失galp。然后可以在两步步骤中置入p26-glf,glk盒,与实施例1中所描述内容相似。在第一步中,利用源自pac21(seqidno.15)的线性dna在tdc操纵子处整合camr,sacb盒,并进行氯霉素(30mg/l)抗性选择。在第二步骤中,利用源自pac19(seqidno.15)的线性dna在tdc操纵子处整合p26-glf,glk盒,进行蔗糖抗性选择并进行氯霉素敏感度筛选,在这种情况下是在基本葡萄糖培养基上的改进生长。
为测试葡萄糖的易化扩散是否能替代大肠杆菌中常规的葡萄糖输入系统,从myr34(δaroe)中缺失ptshi基因和galp基因,然后利用源自pac19的线性dna(seqidno.15)在tdc操纵子处整合p26-glf,glk盒,得到菌株myr217。myr217在补充有所需的三种芳香族氨基酸和三种芳香族维生素样化合物的基本葡萄糖培养基上生长相当好(图17)。然而,含有ptshi和galp缺失但不含有glf,glk盒的菌株myr31并未显示出任何可测量的生长(图17)。因此,易化扩散足以取代我们的菌株背景中的两种常规葡萄糖输入系统。
为测试易化扩散是否可用于生产源自芳香族途径的化合物,用pcp54(arog,arob)和pcp55(arog,arob,tkta)转化myr34和myr217。对这两种菌株比较摇瓶中芳香族中间体3-脱氢莽草酸(dhs)的生产(图18)。在pcp54或pcp55的情况下,利用易化扩散的菌株产生与利用常规葡萄糖输入系统的菌株相比同样多或更多的dhs。dhs生产是经改造的大肠杆菌菌株中黏康酸生产的良好代表,因此我们可以得出结论,葡萄糖的易化扩散对于黏康酸生产是有用的改进。
实施例12
arob基因的过表达
据报道arob基因的表达对于顺式,顺式-黏康酸生产是限速性的(niu等人,2002)。在现有技术中,据称这是通过整合arob基因的第二拷贝与其原生启动子而解决的。然而,这并不足以缓解arob限制,因为arob基因的原生启动子和核糖体结合位点还远不够理想。这样,该方法需要实质的改进。
可以例如通过用强组成型启动子例如来自枯草芽孢杆菌噬菌体spo1的p15或p26启动子(分别为seqidno.1和seqidno.2),或来自细菌噬菌体λ的pr启动子(seqidno.3)来替代染色体中的原生arob启动子来获得改进的arob过表达。这通过如实施例4中描述的两步而实现,不同在于在第一步骤中使用camr,sacb盒来取代原生染色体arob启动子和/或核糖体结合位点。在第二步中,通过用线性dna转化并进行蔗糖抗性选择而置入强组成型启动子,所述线性dna包含强组成型启动子,后接核糖体结合位点和在强组成型启动子的下游侧的自arob编码序列5’端的至少50个碱基(包括atg起始密码子),以及在强组成型启动子上游侧的就在原生arob启动子上游的同源的至少50个碱基对。除了置入更强启动子外或代替置入更强启动子,利用相似方法,可以在arob的atg翻译起始密码子上游的约4至10个碱基对处置入更强的核糖体结合位点例如aggagg。这样的合成盒例如p15-arob盒的拷贝可以在不同于原生arob基因座的基因座处例如在ack基因座处整合到染色体中。如实施例4中同时缺失ack基因并缺失poxb基因可帮助减少发酵过程中不希望的乙酸形成。
实施例13
降低经由戊糖磷酸途径的氧化分支的通量
芳香族途径中第一关键步骤所需的赤藓糖-4-磷酸来源于戊糖磷酸途径(ppp)的非氧化部分。存在两种不同的途径可让碳进入ppp。第一种是从葡萄糖-6-磷酸通过酶葡萄糖-6-磷酸脱氢酶(由zwf基因编码)、6-磷酸葡糖酸内酯酶(由pgl基因编码)和6-磷酸葡糖酸脱氢酶(由gnd基因编码),得到核酮糖-5-磷酸。在这三个步骤的最后,一个碳作为co2损失。这种进入ppp的途径称为ppp的氧化分支。核酮糖-5-磷酸然后通过异构酶、差向异构酶、转酮醇酶和转醛醇酶的作用被转化成多种其它糖磷酸盐。这组起始于核酮糖-5-磷酸的可逆反应被称为ppp的非氧化分支。第二种碳进入ppp的途径是通过果糖-6-磷酸和甘油醛-3-磷酸(二者都来自embden-myerhoff途径,也称为糖酵解),它们可经转醛醇酶和转酮醇酶组合和重排而得到多种其它糖磷酸,其中之一是赤藓糖-4-磷酸。如果碳是通过该第二途径进入ppp,则没有co2损失。为了提高自葡萄糖的顺式,顺式-黏康酸产率,通过阻断ppp的氧化分支从而使所有进入ppp的碳必须经历自果糖-6-磷酸和甘油醛-3-磷酸的非氧化途径,可以防止co2损失。利用与实施例1所公开的用于缺失tyrr基因的内容相似的两步法,通过缺失zwf基因来完成ppp氧化分支的阻断。
实施例14
增加通向芳香族途径和经由pep通向芳香族途径的流量
理想的是确保pep不是顺式,顺式-黏康酸途径上的限速中间体。这是例如,通过增加丙酮酸通过酶pep合成酶向pep的再循环而实现的,这是通过如以上在其它实施例中所描述的整合pps基因的过表达盒而实现的。另一方法是限制丙酮酸激酶的pep消耗,丙酮酸激酶在大肠杆菌中由pyka和pykf基因编码。在这种情况下,方法是降低(一种或多种)酶的活性。这是通过缺失一种或多种编码丙酮酸激酶的基因(如实施例1中和美国专利号9,017,976对于tyrr所描述的),或降低这些基因中的一种或多种的表达强度,如通过突变启动子、核糖体结合位点或编码序列从而使丙酮酸激酶活性的水平降低而实现的。例如,大肠杆菌pyka基因之前的rbs是5’cggagtattacatg。atg翻译起始密码子加下划线。该序列可以突变成cagagtattacatg、caaagtattacatg、caatgtattacatg、caatatattacatg等等,从而通过一次一个碱基变化使rbs序列变得与aggagg的共有rbs更加不同。然后将各个突变的形式引入到染色体中pyka基因座处,取代野生型,并测量黏康酸生产水平的改进。
实施例15
赋予蔗糖上的生长
源自大肠杆菌c的菌株不能以蔗糖作为唯一碳源生长。然而,如国际专利申请公开号wo2012/082720和美国专利申请公开号us2013/0337519(在此整体地并入本文以作参考)所公开的,它们可经基因改造而如此生长。这样,可以改造顺式,顺式-黏康酸生产菌株使其在蔗糖上生长,如以上提及的申请中所公开的那样。
实施例16
改进的顺式,顺式-黏康酸生产者
可以通过一个接一个地安装特征将实施例1-15中所描述的所有特征组合到一株大肠杆菌菌株中。所得的菌株包含改进的顺式,顺式-黏康酸生产者。随后可通过在与第一拷贝的位置隔开的位置,一次一个地整合各个上述过表达盒的第二拷贝,对所得菌株进行再进一步的改进。方便且安全的位置的实例是在就在rrff(其编码核糖体rna)的终止子下游的bsrb1限制性位点。期望的盒作为平端线性dna连接到质粒pmh17f(seqidno.17)中唯一的bsrb1位点。实例是catax表达盒连接以得到名为pcatax的质粒。平行地,camr,sacb盒作为平端片段连接到pmh17f中以得到pmh28f(seqidno.19)。使用通过pcr或通过限制性酶切从pmh28得到的线性dna在rrff位点处保存camr,sacb盒。接着,利用在蔗糖上选择,使用通过pcr或通过限制性酶切从pcatax得到的线性dna在rrff基因座处安装catax盒的第二拷贝。然后将所得的菌株与祖代亲本菌株比较顺式,顺式-黏康酸生产,以确定catax是限制性步骤。通过相似的方法,对来自实施例2-15的各个盒测试限速步骤。如果发现步骤是限速性的,则在染色体的另外其它合适位置处整合相关盒的一个或多个额外的拷贝,引起顺式,顺式-黏康酸生产的更进一步改善,而不需要质粒或可诱导的启动子。
实施例17
通过发酵生产顺式,顺式-黏康酸
可以通过以上实施例1-15中所公开的经基因改造的微生物来生产顺式,顺式-黏康酸。生长培养基可以广泛地不同,并且可以是任何支持微生物充分生长的培养基。优选的培养基是含有矿物盐和非芳香族碳源例如葡萄糖、木糖、乳糖、甘油、乙酸、阿拉伯糖、半乳糖、甘露糖、麦芽糖或蔗糖的基本培养基(关于优选基本生长培养基的实例参见上文)。对于经改造的微生物和生长培养基的各个组合,通过常规实验来确定生产顺式,顺式-黏康酸的合适条件,在所述常规实验中,发酵参数系统性地变化,例如温度、ph、通气速率和用于维持ph的化合物。随着顺式,顺式-黏康酸产生,必须向发酵罐中补料一种或多种化合物以防止ph变得太低。用来中和酸的优选化合物包括碱性盐,如铵、钠、钾、钙、镁的氧化物、氢氧化物、碳酸盐和碳酸氢盐,或者两种或更多种的这类碱性盐的组合。
大肠杆菌myr428菌株在7升发酵罐中的黏康酸生产示于图19。对具有基因型δaroeδacka::p15-arobδpoxb::tkta的大肠杆菌myr261菌株转化质粒pcp32amp和pmg37以产生myr428。myr428在如上所述的7升发酵罐中以葡萄糖补料生长48小时。最终黏康酸滴度为16g/l(参见图19)。
在发酵完成后,通过絮凝、离心和/或过滤来移除细胞,然后通过一个或多个后续步骤例如沉淀、结晶、电渗析、层析(离子交换型、疏水亲和性、和/或基于尺寸的)、微滤、纳滤、反渗透和蒸发的组合从澄清发酵液中纯化顺式,顺式-黏康酸。
实施例18
3,4-二羟基苯甲酸(pca)脱羧酶(aroy)活性的改善
将具有表2中提供的基因型的大肠杆菌菌株myr993用作亲本菌株,以产生在ubix基因或ubid基因中具有缺失的菌株。在构建具有ubix缺失的大肠杆菌菌株中,使用与ubix基因的每个末端具有45bp同源性的引物ms608和ms609扩增卡那霉素抗性盒。在构建具有ubid缺失的大肠杆菌菌株中,使用与ubid基因的每个末端具有45bp同源性的引物ms604和ms605扩增卡那霉素抗性盒。pcr产物经柱纯化(qiaquickpcrpurificationkit,qiagen)并用于使用先前开发的方法转化大肠杆菌菌株myr993(表2)(datsenkoka,wannerbl(2000)one-stepinactivationofchromosomalgenesinescherichiacolik-12usingpcrproducts.procnatlacadsciusa97:6640–6645)以产生具有ubix基因或ubid基因缺失的大肠杆菌菌株(表2-myr993δubix和myr993δubid)。预期缺失菌株在呼吸中受损,因此将葡萄糖添加到lb选择平板中以提供发酵生长。
大肠杆菌菌株myr993,myr993δubix和myr993δubid在250ml摇瓶中以250rpm在37℃下在含有5gk2hpo4,3.5gkh2hpo4,3.5g(nh4)2hpo4,1mmmgso4,0.1mmcacl2,微量元素(1.6mgfecl3·6h2o,0.2mgcocl2·6h2o,0.1mgcucl2·2h2o,0.2mgzncl2,0.2mgna2mso4·2h2o,0.05mgh3bo3,0.55mgmncl2·4h2o),和0.2mmops缓冲液(全部每升)的培养基中作为25ml培养物生长48小时,使用葡萄糖作为碳源。在48小时生长结束时,分析培养物上清液中的黏康酸和pca含量。如图20所示的结果表明,亲本菌株myr993主要在培养基中积累黏康酸与极少量pca,而大肠杆菌菌株myr993δubix仅积累了pca,并且未检测到黏康酸。另一方面,大肠杆菌菌株myr993δubid显示出黏康酸和pca的积累减少。结论是ubix蛋白是aroy(pca脱羧酶)活性所必需的。
实施例19
在体外测定中比较ubix同源物的活性
遵循体外测定以比较ubix和其四种同源物的活性。在该体外测定中,将来自过表达aroy蛋白的大肠杆菌菌株的裂解物与来自表达ubix蛋白或其同源物的另一大肠杆菌菌株的裂解物组合,并测定组合的裂解物消耗作为底物的pca的能力。在产生黏康酸的大肠杆菌菌株中,pca被aroy蛋白脱羧,产生儿茶酚,儿茶酚又被cata蛋白转化为黏康酸。预期aroy蛋白的脱羧活性通过ubix或其一种同源物的存在而增强,并且取决于ubix或其同源物的效率,测定溶液中的pca将以不同的速率被消耗。
在该体外测定中,ubix及其四种同源物即由肺炎克雷伯氏菌(klebsiellapnemoniae)(kpdb)的kpdb基因(seqid42)编码的kpdb,由大肠杆菌w的elw基因(seqid46)编码的elw,由产酸克雷伯氏菌(klebsiellaoxytoca)(kox)的kox基因(seqid48)编码的kox,和由植物乳杆菌(lpl)的lpl基因(seqid50)编码的lpl。最后三个同源物的名称仅仅是为本公开给出的临时名称。aroy,ubix,kpdb,elw,kox和lpl由低拷贝质粒(sc101复制起点)上的强组成型λ噬菌体启动子pr(seqid3)表达。质粒pcat350(seqid55)和pcp165(seqid56)用于基因克隆。在构建aroy质粒时,引物rp712和rp714用于扩增aroy基因,引物ms461和ms346用于扩增pcat350质粒骨架。连接所得的pcr产物以获得过表达aroy蛋白的质粒。在构建kpdb质粒时,引物rp731和rp732用于扩增kpdb基因,引物ms461和ms346用于扩增pcat350质粒骨架。连接所得的pcr产物以获得过表达kpdb蛋白的质粒。在构建ubix质粒时,引物ms669和ms666用于扩增ubix基因,引物ms461和rp607用于扩增pcat350质粒骨架。连接所得的pcr产物以获得过表达ubix蛋白的质粒。在构建elw质粒时,引物ms676和ms680用于扩增elw基因,引物ms461和ms621用于扩增pcp165质粒骨架。连接所得的pcr产物以获得过表达elw蛋白的质粒。在构建kox质粒时,引物ms686和ms684用于扩增kox基因,引物ms461和ms621用于扩增pcp165质粒骨架。连接所得的pcr产物以获得过表达kox蛋白的质粒。在构建lpl质粒时,引物ms692和ms691用于扩增lpl基因,引物ms461和ms621用于扩增pcp165质粒骨架。连接所得的pcr产物以获得过表达lpl蛋白的质粒。所有片段含有20bp的同源性,以能够使用nebuilderhifidna装配克隆试剂盒进行克隆,并克隆到neb5α大肠杆菌细胞(newenglandbiolabs)中。
质粒克隆后,开发了体外酶测定法以证明aroy活性。将1ml过夜lb生长的培养物离心并重悬于200μl细菌蛋白质提取试剂(b-per)(thermofisherscientific)中。在旋转混合器中温育5分钟后,通过在台式离心机中以13,000rpm离心样品除去细胞碎片。将澄清的粗裂解物上清液转移到新管中并储存在冰上。将20μlaroy过表达裂解物与20μlubix或同源裂解物合并到含有100mm磷酸钠缓冲液ph6.4,25mmmgcl2和1mm原儿茶酸(sigma-aldrich)的150μl反应物(最终体积)中。每分钟读取290nm处的吸光度,持续60分钟。通过监测a290处pca的消失来测量aroy活性。ubix及其同源物的相对活性如图21所示。所有ubix同源物均测试了改善的aroy活性,但取决于所测试的特定同源物产生了宽的酶活性变化。使用kpdb实现最高的aroy活性,而从乳杆菌同源物观察到最低的活性。所示的宽范围活性可用于改善黏康酸途径表现,因为最高活性可能并不总是最佳的。
实施例20
kpdb表达水平对aroy活性的影响
已经确定kpdb蛋白的表达在体外测定中增强aroy蛋白的活性,努力确定在产生黏康酸的生物催化剂中kpdb蛋白的表达水平是否会影响黏康酸生产的水平。在该实验中,用多种表达不同水平的kpdb的质粒转化黏康酸产生菌株myr1305。用三种不同的质粒进行myr1305的转化。在实验对照中,用对照质粒pcp140转化myr1305,所述质粒没有任何编码kpdb蛋白的基因。pcp140的dna序列在seqid57中给出。简言之,构建pcp140以表达在p15启动子控制下的大肠杆菌密码子优化的catax,在天然大肠杆菌启动子下的大肠杆菌tkta,在p15启动子下的大肠杆菌arob,在p26下的大肠杆菌arod(seqid2)。用于转化myr1305的第二个质粒pcp169是pcp140的衍生物,其另外具有在p26启动子下的kpdb基因。用于转化myr1305的第三个质粒pcp170是pcp140的衍生物,其另外具有在大肠杆菌pgi启动子下的kpdb(seqid52)。使用p26启动子实现低水平表达,而使用大肠杆菌pgi启动子实现高表达。通过首先使用两组引物(第一片段的pcr引物rp607和rp677和第二片段的pcr引物rp671和rp664)将pcp140质粒扩增成两个片段来构建pcp169和pcp170。两种较小的pcr产物促进比单个大的pcr产物更容易构建质粒。在构建质粒pcp169时,使用引物rp702和rp783扩增p26启动子,并使用引物rp781和rp780扩增kpdb。在构建质粒pcp170中,使用引物rp700和rp784扩增pgi启动子,并使用引物rp779和rp780扩增kpdb。所有pcr产物具有20bp的同源性重叠,以使得能够使用nebuilderhifidna组装克隆试剂盒进行克隆。如图22所示,表达kpdb的菌株比没有任何外源kpdb基因表达的对照菌株产生更高水平的黏康酸。另外,在表达外源kpdb基因的菌株中消除了pca积累。产生的黏康酸水平不随kpdb基因表达的增加而增加,表明即使外源kpdb基因在低表达水平下表达,也实现了饱和水平的活性。
实施例21
ppc突变体的互补和对黏康酸形成的影响
使用具有磷酸烯醇丙酮酸羧化酶(ppc)基因缺失的细菌菌株研究了pep对黏康酸形成的可得性的增加。对大肠杆菌菌株myr1674进行基因工程改造,以用作生产黏康酸的生物催化剂。myr1674能够在含有葡萄糖作为碳源和能源的基本培养基中生长并产生黏康酸。然而,当从myr1674中缺失ppc基因时,得到的菌株myr1674δppc不能在含有葡萄糖的基本培养基中生长,并且仅在富培养基如luriabroth(lb)上存活。通过在大肠杆菌的myr1674δppc菌株中的原始ppc基因座处插入编码丙酮酸羧化酶的pyc基因,可以重新获得myr1674δppc在基本培养基上生长的能力的丧失。
使用引物ms1383和ms1384克隆来自酿酒酵母(saccharomycescerevisiae)的丙酮酸羧化酶(pyc)基因(seqid53),其含有与大肠杆菌ppc启动子和终止子的侧翼同源性。为了促进在基本培养基上的强烈生长,需要强组成型向右λ噬菌体启动子pr。使用引物ms1429和ms1430扩增pr启动子,并使用所得pcr产物代替内源性ppc启动子。δppc::pr-pyc基因座的最终核苷酸序列显示在seqid58中。选择酿酒酵母pyc基因是因为它与任何大肠杆菌基因不密切相关,并且由于不同的密码子选择而预期较低的表达可能有利于保存pep用于黏康酸生产。有许多含有丙酮酸羧化酶的生物,并且可以使用任何具有丙酮酸羧化酶活性的同源物或类似物。通过在原始ppc基因座处整合δppc::pr-pyc而衍生自myr1674的新的菌株myr1772在基本培养基上存活,证实了克隆的酿酒酵母pyc基因的功能性。在摇瓶实验中比较了myr1772及其亲本myr1674菌株产生黏康酸的能力。如图23中所示的结果表明,myr1772菌株比亲本myr1674产生更高滴度的黏康酸产生,证明了用外源丙酮酸羧化酶替代内源磷酸烯醇丙酮酸羧化酶的优势。
实施例22
测量黏康酸生产
将细菌菌株myr814,myr993,myr1536,myr1557,myr1570,myr1595,myr1630,myr1674和myr1772在摇瓶培养物中培养过夜,并测定黏康酸生产的滴度和产量。此外,通过测量600nm处的吸光度确定生长速率,并且表8中提供了各种细菌菌株的相对生长。由“+++”表示的细菌生长表示与野生型大肠杆菌菌株中看到的生长相似的生长。由“+”表示的细菌生长表示不良生长。中间增长由“++”表示。当特定菌株显示出不良生长时,该菌株经过5次过夜转移以改善生长,每次转移产生大约10代或倍增。
将细菌菌株myr814,myr1570,myr1630和myr1674以补料分批模式在7升发酵罐中生长,并测定黏康酸生产的滴度和产量(表9)。测试了黏康酸滴度和产量的细菌菌株产生非常少的副产物。例如,在补料分批发酵72小时后的细菌菌株仅显示0.08g/l的pca和0.07g/l的延胡索酸作为副产物。
参考文献
本文的所有专利、专利申请、出版物、序列和其他已公开的材料均以引用方式并入本文。
美国专利号4,480,034
美国专利号4,535,059
美国专利号4,588,688
美国专利号4,608,338
美国专利号4,681,852
美国专利号4,753,883
美国专利号4,833,078
美国专利号4,968,612
美国专利号5,168,056
美国专利号5,272,073
美国专利号5,487,987
美国专利号5,616,496
美国专利号6,600,077
美国专利号6,180,373
美国专利号6,210,937
美国专利号6,472,169
美国专利号6,613,552
美国专利号6,962,794
美国专利号7,244,593
美国专利号7,638,312
美国专利号7,790,431
美国专利号8,871489
美国专利号9,017,976
美国专利申请公开号us2009/0191610a1
美国专利申请公开号us2010/0314243a1
美国专利申请公开号us2013/0337519a1
美国专利申请公开号us.2014/0234923a1
美国专利申请公开号us2015/0044755a1
美国专利申请公开号us2016/0017381a1
欧洲专利申请号86300748.0
国际专利申请公开号wo2011/017560
国际专利申请公开号wo2011/085311
国际专利申请公开号wo2011/123154
国际专利申请公开号wo2013/116244
altschul,s.f.,gish,w.,miller,w.,myers,e.w.,andlipman,d.j.(1990)basiclocalalignmentsearchtool,jmolbiol215,403-410.
altschul,s.f.,madden,t.l.,schaffer,a.a.,zhang,j.,zhang,z.,miller,w.,andlipman,d.j.(1997)gappedblastandpsi-blast:anewgenerationofproteindatabasesearchprograms,nucleicacidsres25,3389-3402.
aussel,laurent,fabienpierrel,laurentloiseau,muriellelombard,marcfontecave,andfrédéricbarras.2014.“biosynthesisandphysiologyofcoenzymeqinbacteria.”biochimicaetbiophysicaacta-bioenergetics1837(7):1004–11.
baba,tomoya,takeshiara,mikihasegawa,yukitakai,yoshikookumura,mikibaba,kirilladatsenko,masarutomita,barrylwanner,andhirotadamori.2006.“constructionofescherichiacolik-12in-frame,single-geneknockoutmutants:thekeiocollection.”molecularsystemsbiology2:2006.0008.doi:10.1038/msb4100050.
barbe,v.,vallenet,d.,fonknechten,n.,kreimeyer,a.,oztas,s.,labarre,l.,cruveiller,s.,robert,c.,duprat,s.,wincker,p.,ornston,l.n.,weissenbach,j.,marliere,p.,cohen,g.n.,andmedigue,c.(2004)uniquefeaturesrevealedbythegenomesequenceofacinetobactersp.adp1,aversatileandnaturallytransformationcompetentbacterium,nucleicacidsres32,5766-5779.
bird,j.a.andcain,r.b.(1968)cis-cis-muconate,theproductinducerofcatechol1,2-oxygenaseinpseudomonasaeruginosa.biochem.j.109,479-481.
bongaerts,j.,kramer,m.,muller,u.,raven,l.andwubbolts,m.(2001)metabollicengineeringformicrobialproducitnofaromaticacidsandderivedcompunds.met.eng.3,289-300.
chandran,s.s.,yi,j.,draths,k.m.,vondaeniken,r.,weber,w.andfrost,j.w.(2003)phosphoenolpyruvateavailabilityandthebiosynthesisofshikimicacid.biotechnol.prog.19,808-814.
chen,r.,hatzimanikatis,v.,yap,w.m.g.j.,potma,p.w.andbailey,j.e.(1997)metabolicconsequencesofphosphotransferase(pts)mutationinaphenylalanie-producingrecombinatnescherichiacoli.biotechnol.prog.13,768-775.
chen,k.,dou,j.,tang,s.,yang,y.,wang,h.,fang,h.andzhou,c.(2012)deletionofthearokgeneisessentialforhighshikimicacidaccumulationthroughtheshikimateine.coli.bioresourcetechnol,119,141-147.
choi,w.j.,lee,e.y.,cho,m.h.,andchoi,c.y.(1997)enhancedproductionofcis,cis-muconateinacell-recyclebioreactor.j.fermentationandbioengineering.84,70-76.
curran,kathleena.,johnm.leavitt,ashtys.karim,andhals.alper.2013.“metabolicengineeringofmuconicacidproductioninsaccharomycescerevisiae.”metabolicengineering15(1):55–66.
deberardinis,v.,vallenet,d.,castelli,v.,besnard,m.,pinet,a.,cruaud,c.,samair,s.,lechaplais,c.,gyapay,g.,richez,c.,durot,m.,kreimeyer,a.,lefevre,f.,schachter,v.,pezo,v.,doring,v.,scarpelli,c.,medigue,c.,cohen,g.n.,marliere,p.,salanoubat,m.,andweissenbach,j.(2008)acompletecollectionofsingle-genedeletionmutantsofacinetobacterbaylyiadp1,molsystbiol4,174.
draths,k.m.,pompliano,d.l.,conley,d.l.,frost,j.w.,berry,a.,disbrow,g.l.,staversky,r.j.,andlievense,j.c.(1992)biocatalyticsynthesisofaromaticsfromd-glucose-theroleoftransketolase,journaloftheamericanchemicalsociety114,3956-3962.
draths,k.m.,andfrost,j.w.(1995)environmentallycompatiblesynthesisofcatecholfromd-glucose,journaloftheamericanchemicalsociety117,2395-2400.
elsemore,d.a.,andornston,l.n.(1995)unusualancestryofdehydratasesassociatedwithquinatecatabolisminacinetobactercalcoaceticus,jbacteriol177,5971-5978.
escalante,a.,calderon,r.,valdiva,a.,deanda,r.,hernandez,g.,ramirez,o.t.,gosset,g.andboliver,f.(2010)metabolicengineeringfortheproductionofshikimicacidinanevolvedescherichiacolistrainlackingthephosphoenolpyrvate:carbohydratephosphotransferasesystem.microbialcellfactories9,21-33.
escalante,adelfo,rocíocalderón,aracelivaldivia,ramóndeanda,georginahernández,octaviotramírez,guillermogosset,andfranciscobolívar.2010.“metabolicengineeringfortheproductionofshikimicacidinanevolvedescherichiacolistrainlackingthephosphoenolpyruvate:carbohydratephosphotransferasesystem.”microbialcellfactories9(ccm):21.doi:10.1186/1475-2859-9-21.
flores,n.,xiao,j.,berry,a.,bolivar,f.andvalle,f.(1996)pathwayengineeringfortheproductionofaromaticcompoundsinescherichiacoli.naturebiotechn.14,620–623.
fox,d.t.,hotta,k.,kim,c.y.,andkoppisch,a.t.(2008)themissinglinkinpetrobactinbiosynthesis:asbfencodesa(-)-3-dehydroshikimatedehydratase,biochemistry47,12251-12253.
ger,y.,chen,s.,chiang,h.,andshiuan,d.(1994)asingleser-180mutationdesensitizesfeedbackinhibitionofthephyenylalanine-sensitive3-deoxy-d-arabino-hepulosonate7-phosphate(dahp)synthetaseineschericiacoli,jbiochem116,986-990.
grant,d.j.,andpatel,j.c.(1969)thenon-oxidativedecarboxylationofp-hydroxybenzoicacid,gentisicacid,protocatechuicacidandgallicacidbyklebsiellaaerogenes(aerobacteraerogenes),antonievanleeuwenhoek35,325-343.
hansen,e.h.,moller,b.l.,kock,g.r.,bunner,c.m.,kristensen,c.,jensen,o.r.,okkels,f.t.,olsen,c.e.,motawia,m.s.,andhansen,j.(2009)denovobiosynthesisofvanillininfissionyeast(schizosaccharomycespombe)andbaker'syeast(saccharomycescerevisiae),applenvironmicrobiol75,2765-2774.
horwitz,andrewa.,jessicam.walter,maxg.schubert,stephanieh.kung,kristyhawkins,darrenm.platt,aarond.hernday,etal.2015.“efficientmultiplexedintegrationofsynergisticallelesandmetabolicpathwaysinyeastsviacrispr-cas.”cellsystems.1(1):88-96.
hu,changyun,peihongjiang,jianfengxu,yongqingwu,andweidahuang.2003.“mutationanalysisofthefeedbackinhibitionsiteofphenylalanine-sensitive3-deoxy-d-arabino-heptulosonate7-phosphatesynthaseofescherichiacoli.”journalofbasicmicrobiology43(5):399–406.
hu,c.,jiang,p.,xu,j.,wu,y.,andhuang,w.(2003)mutationanalysisofthefeedbackinhibitionsiteofphenylalanine-sensitive3-deoxy-d-arabino-heptulosonate7-phosphatesynthaseofescherichiacoli,jbasicmicrobiol43,399-406.
iwagami,s.g.,yang,k.,anddavies,j.(2000)characterizationoftheprotocatechuicacidcatabolicgeneclusterfromstreptomycessp.strain2065,applenvironmicrobiol66,1499-1508.
jantama,k.,haupt,m.j.,svoronos,s.a.,zhang,x.,moore,j.c.,shanmugam,k.t.,andingram,l.o.(2008a)combiningmetabolicengineeringandmetabolicevolutiontodevelopnonrecombinantstrainsofescherichiacolicthatproducesuccinateandmalate,biotechnolbioeng99,1140-1153.
jantama,k.,zhang,x.,moore,j.c.,shanmugam,k.t.,svoronos,s.a.,andingram,l.o.(2008b)eliminatingsideproductsandincreasingsuccinateyieldsinengineeredstrainsofescherichiacolic,biotechnolbioeng101,881-893.
jiménez,natalia,joséantoniocuriel,inésreverón,blancadelasrivas,androsario
johnson,c.w.,salvachua,d.,khanna,p.,peterson,d.j.andbeckham,g.(2016)enhancingmuconicacidproductionfromglucoseandlignin-derivedaromaticcompoundviaincreasedprotocatechuatedecarboxylaseactivity.metabolicengineeringcommunication.3:111-119.
kaneko,a.,ishii,y.,andkirimura,k.(2011)high-yieldproductionofcis,cis-muconicacidfromcatecholinaqueoussolutionbybiocatalyst.chem.lett.40,381-383.
kikuchi,y.,tsujimoto,k.,andkurahashi,o.(1997)mutationalanalysisofthefeedbacksitesofphenylalanine-sensitive3-deoxy-d-arabino-heptulosonate-7-phosphatesynthaseofescherichiacoli,applenvironmicrobiol63,761-762.
kojima,y.,fujisawa,h.,nakazawa,a.,nakazawa,t.,kanetsuna,f.,taniuchi,h.,nozaki,m.,andhayaishi,o.(1967)studiesonpyrocatechase.i.purificationandspectralproperties,jbiolchem242,3270-3278.
kramer,m.,bongaerts,j.,bovenberg,r.,kremer,s.,muller,u.,orf,s.,wubbolts,m.andraeven,l.(2003)metabolicengineeringformicrobialproductionofshikimicacid.metabol.eng.5,277-283.
lerner,c.g.,andinouye,m.(1990)lowcopynumberplasmidsforregulatedlow-levelexpressionofclonedgenesinescherichiacoliwithblue/whiteinsertscreeningcapability,nucleicacidsres18,4631.
li,k.andfrost,j.w.(1999)microbialsynthesisof3-dehydroshikimicacid:acomparativeanalysisofd-xylose,l-arabinose,andd-glucosecarbonsources.biotechnol.prog.15,876-883.
lin,h.,ravishankarvvadali,r.v.,georgenbennett,g.n.andsan,k.y.(2004)increasingtheacetyl-coapoolinthepresenceofoverexpressedphosphoenolpyruvatecarboxylaseorpyruvatecarboxylaseenhancessuccinateproductioninescherichiacoli.biotechnologyprogress20(5):1599–1604.
lin,fengming,kylel.ferguson,davidr.boyer,xiaoxianinalin,ande.neilg.marsh.2015.“isofunctionalenzymespad1andubixcatalyzeformationofanovelcofactorrequiredbyferulicaciddecarboxylaseand4-hydroxy-3-polyprenylbenzoicaciddecarboxylase.”acschemicalbiology11(4):1137-1144.
lu,j.l.,andliao,j.c.(1997)metabolicengineeringandcontrolanalysisforproductionofaromatics:roleoftransaldolase,biotechnolbioeng53,132-138.
lupa,boguslaw,delinalyon,morelandd.gibbs,rosalinda.reeves,andjuergenwiegel.2005.“distributionofgenesencodingthemicrobialnon-oxidativereversiblehydroxyarylicaciddecarboxylases/phenolcarboxylases.”genomics86(3):342–51.
lutke-eversloh,t.,andstephanopoulos,g.(2007)l-tyrosineproductionbyderegulatedstrainsofescherichiacoli,applmicrobiolbiotechnol75,103-110.
mizuno,s.,yoshikawa,n.,seki,m.,mikawa,t.,andimada,y.(1988)microbialproductionofcis,cis-muconicacidfrombenzoicacid.applmicrobiolbiotechnol.28,20-25.
nakazawa,a.,kojima,y.,andtaniuchi,h.(1967)purificationandpropertiesofpyrocatechasefrompseudomonasfluorescens,biochimbiophysacta147,189-199.
neidhardt,f.c.,andcurtiss,r.(1996)escherichiacoliandsalmonella:cellularandmolecularbiology,vol.22nded.,asmpress,washington,d.c.
neidle,e.l.,andornston,l.n.(1986)cloningandexpressionofacinetobactercalcoaceticuscatechol1,2-dioxygenasestructuralgenecatainescherichiacoli,jbacteriol168,815-820.
niu,w.,draths,k.m.,andfrost,j.w.(2002)benzene-freesynthesisofadipicacid,biotechnolprog18,201-211.
parker,c.,barnell,w.o.,snoep,j.l.,ingram,l.o.,andconway,t.(1995)characterizationofthezymomonasmobilisglucosefacilitatorgeneproduct(glf)inrecombinantescherichiacoli:examinationoftransportmechanism,kineticsandtheroleofglucokinaseinglucosetransport,molmicrobiol15,795-802.
parsek,m.r.,shinabarger,d..l.,rithmel,r.k.andchakrabarty,a.m.(1992)rolesofcatrandcis,cis-muconateinactivationofthecatbcoperson,whichisinvolvedinbenzoatedegradationinpseudomonasputida.j.bacteriol.174,7798-7806.
patnaik,r.andliao,j.c.(1994)engineeringofescherichiacolicentralmetabolismforaromaticmetabolitewithneartheoreticalyield.app.env.microbiol.60,3903-3908.
payne,karla.p.,markd.white,karlfisher,basilekhara,samuels.bailey,davidparker,nicholasj.w.rattray,etal.2015.“newcofactorsupportsα,β-unsaturatedaciddecarboxylationvia1,3-dipolarcycloaddition.”nature522(7557):497–501.
pfleger,b.f.,kim,y.,nusca,t.d.,maltseva,n.,lee,j.y.,rath,c.m.,scaglione,j.b.,janes,b.k.,anderson,e.c.,bergman,n.h.,hanna,p.c.,joachimiak,a.,andsherman,d.h.(2008)structuralandfunctionalanalysisofasbf:originofthestealth3,4-dihydroxybenzoicacidsubunitforpetrobactinbiosynthesis,procnatlacadsciusa105,17133-17138.
perez-pantoja,d.,delaiglesia,r.,pieper,d.h.,andgonzalez,b.(2008)metabolicreconstructionofaromaticcompoundsdegradationfromthegenomeoftheamazingpollutant-degradingbacteriumcupriavidusnecatorjmp134,femsmicrobiolrev32,736-794.
perez-pantoja,d.,donoso,r.,agullo,l.,cordova,m.,seeger,m.,pieper,d.h.,andgonzalez,b.(2011)genomicanalysisofthepotentialforaromaticcompoundsbiodegradationinburkholderiales,environmicrobiol.14.5(2012):1091-1117.
pittard,j.andwallace,b.j.(1966)distributionandfunctionofgenesconcernedwitharomaticbiosynthesisinescherichiacoli.j.bacteriol.91,1494-1508.
polen,t.,spelberg,m.andbott,m.(2013)towardbitechnologicalproducitonofadipicacidandprecursorsfrombiorenewables,j.biotechnol.167(2):75-84.
rutledge,b.j.(1984)molecularcharacterizationoftheqa-4geneofneurosporacrassa,gene32,275-287.
schirmer,f.,andhillen,w.(1998)theacinetobactercalcoaceticusncib8250mopoperonmrnaisdifferentiallydegraded,resultinginahigherlevelofthe3'cata-encodingsegmentthanofthe5'phenolhydroxylase-encodingportion,molgengenet257,330-337.
shumilin,i.a.,kretsinger,r.h.,andbauerle,r.h.(1999)crystalstructureofphenylalanine-regulated3-deoxy-d-arabino-heptulosonate-7-phosphatesynthasefromescherichiacoli,structure7,865-875.
shumilin,i.a.,zhao,c.,bauerle,r.,andkretsinger,r.h.(2002)allostericinhibitionof3-deoxy-d-arabino-heptulosonate-7-phosphatesynthasealtersthecoordinationofbothsubstrates,jmolbiol320,1147-1156.
shumilin,i.a.,bauerle,r.,wu,j.,woodard,r.w.,andkretsinger,r.h.(2004)crystalstructureofthereactioncomplexof3-deoxy-d-arabino-heptulosonate-7-phosphatesynthasefromthermotogamaritimarefinesthecatalyticmechanismandindicatesanewmechanismofallostericregulation,jmolbiol341,455-466.
shumkova,e.s.,solyanikova,i.p.,plotnikova,e.g.andgolovleva,l.a.(2009)phenoldegrdationbyrhodococcusopacusstrain1g.app.biocehm.microbiol.45,43-49.
sietmann,r.,uebe,r.,boer,e.,bode,r.,kunze,g.,andschauer,f.(2010)novelmetabolicroutesduringtheoxidationofhydroxylatedaromaticacidsbytheyeastarxulaadeninivorans,japplmicrobiol108,789-799.
smith,m.r.andratledge,c.(1989)quantitativebiotransformationofcatecholtocis,cis–muconate.biotech.lett.11,105-110.
snoep,j.l.,arfman,n.,yomano,l.p.,fliege,r.k.,conway,t.,andingram,l.o.(1994)reconstructionofglucoseuptakeandphosphorylationinaglucose-negativemutantofescherichiacolibyusingzymomonasmobilisgenesencodingtheglucosefacilitatorproteinandglucokinase,jbacteriol176,2133-2135.
sonoki,tomonori,miyukimorooka,kimitoshisakamoto,yuichirootsuka,masayanakamura,jodyjellison,andbarrygoodell.2014.enhancementofprotocatechuatedecarboxylaseactivityfortheeffectiveproductionofmuconatefromlignin-relatedaromaticcompounds.journalofbiotechnology192(parta):71–77.
sprenger,g.a.(1995)geneticsofpentose-phosphatepathwayenzymesofescherichiacolik-12,archmicrobiol164,324-330.
sprenger,g.a.,schorken,u.,sprenger,g.,andsahm,h.(1995a)transketolaseaofescherichiacolik12.purificationandpropertiesoftheenzymefromrecombinantstrains,eurjbiochem230,525-532.
sprenger,g.a.,schorken,u.,sprenger,g.,andsahm,h.(1995b)transaldolasebofescherichiacolik-12:cloningofitsgene,talb,andcharacterizationoftheenzymefromrecombinantstrains,jbacteriol177,5930-5936.
stroman,p.,reinert,w.r.,andgiles,n.h.(1978)purificationandcharacterizationof3-dehydroshikimatedehydratase,anenzymeintheinduciblequinicacidcatabolicpathwayofneurosporacrassa,jbiolchem253,4593-4598.
tang,j.,zhu,x.,lu,j.andliu,p.(2012)recruitingalternativeglucoseutilizationpathwaysforimprovingsuccinateproduction.appmicrobiolbiotechnoldoi10,1007/s00253-012-434.1.
tateoka,t.,andyasuda,i.(1995)3-dehydroshikimatedehydrataseinmungheanculturedcells,plantcellreports15,212-217.
vemuri,g.n.,m.a.eiteman,ande.altman.2002.“effectsofgrowthmodeandpyruvatecarboxylaseonsuccinicacidproductionbymetabolicallyengineeredstrainsofescherichiacoli.”appliedandenvironmentalmicrobiology68(4):1715–27.
weaver,l.m.,andherrmann,k.m.(1990)cloningofanarofalleleencodingatyrosine-insensitive3-deoxy-d-arabino-heptulosonate7-phosphatesynthase,jbacteriol172,6581-6584.
weber,c.,bruckner,c.,weinreb,s.,lehr,c.,essl,c.andbole,e.(2012)biosynthesisofcis,cis-muconicacidanditsaromaticprecursorscatecholandproteocatechuicacid,fromrenewablefeedstocksbysaccharomycescerevisiae,appenvironmicrobiol.78,8421-8430.
wheeler,k.a.,lamb,h.k.,andhawkins,a.r.(1996)controlofmetabolicfluxthroughthequinatepathwayinaspergillusnidulans,biochemj315(pt1),195-205.
white,markd.,karla.p.payne,karlfisher,stephena.marshall,davidparker,nicholasj.w.rattray,drupadk.trivedi,etal.2015.“ubixisaflavinprenyltransferaserequiredforbacterialubiquinonebiosynthesis.”nature522(7557):502–6.
wu,c-m.,wu,c-c.,su,c-c.,lee,s-n.,lee,y-a.andwu,j-y.(2006)microbialsynthesisofcis,cis-muconicacidformbenzoatebysphingobacteriumsp.mutants.biochem.eng.j.29,35-40.
xie,n.,tang,h.,feng,j.,tao,f.,ma,c.andxu,p.(2009)characterizationofbenzoatedegradationbynewlyisolatedbacteriumpseudomonassp.xp-m2.biochem.eng.j.46,79-82.
xie,n.,hongliang,ri-bohuang,andpingxu.2014.“biotechnologicalproductionofmuconicacid:currentstatusandfutureprospects.”biotechnologyadvances32(3):615–22.
yi,j.,draths,k.m.,li,k.andfrost,j.w.(2003)alteredglucosetransportandshikimatepathwayproductyieldsine.coli.biotechnol.prog.2003,19,1450-1459.
yoshikawa,n.,mizuno,s.,ohta,k.,andsuzuki,m.(1990)microbialproductionofcis,cis-muconicacid.j.biotechno.14,203-210.
- 还没有人留言评论。精彩留言会获得点赞!